

Abstract
Donor specific HLA antibodies significantly lower allograft survival, but as yet there are no satisfactory therapies for prevention of antibody-mediated rejection. Intracapillary macrophage infiltration is a hallmark of antibody-mediated rejection, and macrophages are important in both acute and chronic rejection. The purpose of this study was to investigate the Fc-independent effect of HLA I antibodies on endothelial cell activation, leading to monocyte recruitment. We used an in vitro model to assess monocyte binding to endothelial cells in response to HLA I antibodies. We confirmed our results in a mouse model of antibody-mediated rejection, in which B6.RAG1-/- recipients of BALB/c cardiac allografts were passively transferred with donor specific MHC I antibodies. Our findings demonstrate that HLA I antibodies rapidly increase intracellular calcium and endothelial presentation of P-selectin, which supports monocyte binding. In the experimental model, donor specific MHC I antibodies significantly increased macrophage accumulation in the allograft. Concurrent administration of rPSGL-1-Ig abolished antibody-induced monocyte infiltration in the allograft, but had little effect on antibody-induced endothelial injury. Our data suggest that antagonism of P-selectin may ameliorate accumulation of macrophages in the allograft during antibody-mediated rejection.
Keywords: Endothelial cells, HLA antibody, signal transduction, monocytes, P-selectin
Introduction
Donor specific antibodies to HLA class I and II molecules have a deleterious impact on graft survival in solid organ transplantation (1-4), and correlate with chronic rejection (5-8). HLA I antibody binding to vascular endothelial and smooth muscle cells activates cell signaling pathways that are required for increased proliferative capacity, migration, stress fiber formation, and survival (9-17). Endothelial vesicles known as Weibel-Palade bodies (WPb) are mobilized after HLA I crosslinking, leading to adherence of the immature neutrophilic cell line HL60 (18).
Antibody-mediated rejection is a complex process, with characteristic features such as endothelial cell swelling and complement deposition in the vessels of the graft (19). In addition, macrophage infiltration frequently accompanies antibody-mediated rejection (AMR), and correlates with donor specific HLA antibodies and poor graft outcome (20-24). Macrophages comprise a significant proportion of graft infiltrating cells in acute rejection (25-29), and are found in lesions of chronic allograft vasculopathy (CAV) in human biopsies and experimental models (21, 29, 30). Macrophages promote deposition of extracellular matrix by SMC (31), release soluble mediators that cause endothelial cell proliferation (32), and may exhibit direct alloreactivity against the allograft (33, 34). Importantly, depletion of macrophages reduces rejection of murine cardiac allografts (35). Therefore, the understanding of the mechanism(s) that promote monocyte recruitment may lead to novel approaches to reduce allograft rejection.
It is incompletely understood how HLA I antibodies may promote monocyte recruitment into the allograft. In this study, we used an in vitro approach to measure endothelial cell activation and the Fc-independent recruitment of monocytes in response to HLA I antibody. Our results show that HLA I signaling in endothelial cells is sufficient to increase monocyte adhesion mediated by HLA I-induced P-selectin. Using an in vivo model of antibody-mediated rejection, we confirmed that donor specific MHC I antibodies elicit macrophage infiltration into the allograft. Importantly this influx can be blocked by the selectin antagonist rPSGL-1-Ig. This study demonstrates that selectin blockade prevents monocyte adherence in vitro and intragraft macrophage accumulation in vivo in response to MHC I antibodies.
Materials and Methods
Reagents
Pan-HLA I antibody (murine monoclonal W6/32, mIgG2a) was obtained from BioXCell. The F(ab’)2 fragment of W6/32 was generated as previously described (36), or using a kit from Thermo Scientific. Neutralizing goat (Santa Cruz) or sheep (R&D) antibody to P-selectin, or antibody to PSGL-1 (Biolegend, San Jose, CA) were used. Purified polyclonal human IgG was obtained from Fisher Scientific. Calcium inhibitor BAPTA-AM was from Calbiochem. Recombinant soluble PSGL-1 Fc chimera (rPSGL-1-Ig) was provided by Y’s Therapeutics (San Bruno, CA). It contains mutations in the crystallizable fragment (Fc) region to eliminate interactions with complement and FcγRs, and inhibits the initial tethering of leukocytes to selectins (37, 38).
Cell Culture
The use of the human aortic tissue for the research described herein was approved by the OneLegacy Biomedical Review Board under the agreement #RS-02-10-2 and UCLA MTA2009-561. Primary human aortic endothelial cells (HAEC) were isolated from aortic rings and cultured as previously described (11). Endothelial cells were seeded in 24 or 48 well plates, and grown to confluence before use in experiments. The monocytic cell line Mono Mac 6 (39, 40) was cultured in RPMI-1640 supplemented with 10% FBS, sodium pyruvate, nonessential amino acids, antibiotics, and insulin, and maintained at fewer than 106 cells per mL.
Primary Human Monocytes
Human peripheral blood mononuclear cells (PBMC) were collected under our institutional review board-approved study and isolated by Ficoll density centrifugation. Monocytes were enriched using MACS Monocyte Isolation Kit II (Miltenyi). Purity was confirmed by CD14 staining and to be greater than 85%.
Measurement of von Willebrand Factor
Confluent aortic endothelial cells were stimulated with HLA I antibody, thrombin, histamine or phorbol myristate acetate (PMA) in M199 supplemented with 5% fetal bovine serum (assay medium) for one hour. Supernatant was collected and von Willebrand Factor (vWF) was measured using an ELISA kit (Helena Laboratories) according to the manufacturer’s protocol.
Flow Cytometric Analysis
Cell surface expression of HLA I, E-selectin or P-selectin was measured by flow cytometry. Basal expression of HLA I on endothelial cells is shown in Supplemental Figure 1. Endothelial cells were treated with stimuli for the indicated times in assay medium and detached using Accutase (Innovative Cell). To preserve epitopes, trypsin was not used (41). Cells stained with PE-conjugated anti-human P-selectin or anti-E-selectin (BD) were assayed on a FACSCalibur flow cytometer (BD). P-selectin results are expressed as proportion of cells staining positively, normalized to untreated endothelial cells.
Intracellular Calcium Measurement
HAEC were plated onto gelatin-coated glass coverslips, then labeled with 5μM Fura-2 AM for 20min at 37°C. Cells were washed and mounted in a standard cuvette filled with saline (at 37°C) (ANO-2100, Hitachi Instruments). The cuvette was placed in a fluorimeter (F-2000, Hitachi Instruments) with a heated jacket (37°C), and continuously stirred. Samples were taken every 0.5 secs (excitation 340nm, emission 380nm) using associated software (F-2000 Intracellular Cation Measurement System). The ratio of the signals at 340 nm/380 nm provided a monitor of Ca2+ concentration.
Monocyte Adhesion Assay
Preliminary studies determined the optimal concentration of blocking reagents. Adherence of the monocytic cell line Mono Mac 6 or freshly isolated primary monocytes to endothelial monolayers was adapted from previously described methods (42). Due to the established affinity of human FcγRs for murine IgG2a (43, 44), we sought to eliminate antibody-FcγR interactions. Briefly, HAEC were stimulated and carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen)-labeled Mono Mac 6 were added at a ratio of approximately 3 monocytes per endothelial cell for 20min. Nonadherent cells were removed by washing three times. Adherent monocytes were imaged by fluorescence microscopy (Nikon Eclipse Ti) and counted in 8-10 fields per sample using automated software (CellProfiler or Image J) (45). HAEC were pretreated with BAPTA-AM (20μM) for 30min, or rPSGL-1-Ig (20μg/mL) or P-selectin neutralizing antibody (10μg/mL) in the last 10min of stimulation. Monocytes were pretreated with a neutralizing antibody to PSGL-1 (10μg/mL), or with polyclonal human IgG (20μg/mL) to block FcγRs where indicated, for 20min (46).
Mice
Male B6.129S7-Rag1tm1Mom (B6.RAG1−/−, H-2b) and male BALB/c (H-2d) mice aged 7-10 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME, USA), housed under pathogen-free conditions in filter-top cages throughout the experiments and cared for according to The Guidelines for Animal Care. All reagents and experiments in this study were reviewed and approved by the UCLA Animal Research Committee.
Murine heterotopic heart transplantation
BALB/c (H-2d) hearts were heterotopically transplanted into abdominal great vessels of B6.RAG1 KO (H-2b) recipients as previously described (16). Briefly, under phenobarbital anesthesia, the donor aorta was anastomosed to the recipient abdominal aorta and the donor pulmonary artery was joined to the recipient inferior vena cava. Graft function was monitored daily by abdominal palpation. On day 30 post-operatively, the donor and native hearts were recovered. All allografts included in the study were functioning at the time of sacrifice. The base of the heart was fixed in formalin and paraffin-embedded.
Adoptive transfer of monoclonal antibodies
All reagents to which mice were exposed were sterile and azide free. Allograft recipient mice were passively transferred with 30μg (approximately 1.5mg/kg) isotype control antibody (clone MOPC-173, Biolegend) or donor specific anti-MHC I antibodies (anti-Kd mIgG2a, clone SF1-1.1; anti-Dd mIgG2a, clone 34-2-12, Biolegend; diluted in sterile saline) by tail vein injection the beginning day 3 post-transplant, continuing weekly thereafter. 70μg of rPSGL-1-Ig (approximately 3.5mg/kg) was injected intravenously beginning on day 3 after transplant and continuing biweekly. The dose and administration regimen were based on previously published studies detailing the pharmacokinetics and efficacy of rPSGL-1-Ig in mouse (47, 48).
Histologic Analysis of Graft Infiltrating Cells
Histological features of antibody-mediated rejection and immunohistochemical staining in allografts were assessed by a pathologist blinded to the experimental groups. Microvascular changes were assessed, including endothelial cell swelling and increased number of intravascular cells, as previously described (16, 19). Cross sections of formalin-fixed tissue were stained for Mac-2 (Acris) or CD45 (Millipore). A Mac-2 or CD45 score of 0 indicates negative staining, 1 is rare, 2 is rare/focal, 3 is focal staining, and 4 is strongest, diffuse staining.
Initial experiments comparing macrophage infiltration into the allograft between animals receiving 5μg and 30μg of anti-Kd antibody, or biweekly administration of 30μg anti-Kd antibody, revealed no statistically significant difference between the three groups when compared by unpaired T test. Therefore, we combined animals from these groups into a single group, the anti-Kd antibody treated group. Groups treated with control antibody at 5μg and 30μg were merged into one control antibody treated group.
Flow Cytometric Crossmatch
To confirm the presence of circulating donor specific antibodies, blood was collected at the time of sacrifice, and serum was separated using a Capiject tube (Terumo T-MG). BALB/c splenocytes were incubated with 25μL of serum for 30min on ice, then stained with fluorescein isothiocyanate (FITC)-conjugated goat-anti-mouse-Fcγ and anti-mouse CD3-phycoerythrin (PE). MHC I antibody binding to T cells was measured by gating on CD3+ using flow cytometry (FACSCalibur, BD).
Statistical Analysis
In the leukocyte adherence assay, the number of adherent cells was counted in 8-10 random fields for each sample, and the mean number of cells was determined. Statistical significance between controls and samples was determined using one-way ANOVA followed by individual comparisons (student’s T-test).
Results
HLA I antibodies trigger endothelial exocytosis of Weibel-Palade bodies via intracellular calcium
To examine whether HLA I crosslinking on endothelial cells could cause release of preformed von Willebrand Factor (vWF) and P-selectin from Weibel-Palade bodies (WPb), we treated confluent monolayers of human aortic endothelial cells with a monoclonal murine pan-HLA I antibody (W6/32), or with classical agonists thrombin, PMA and histamine. Stimulation of endothelium with positive controls thrombin, PMA or histamine, or with HLA I antibody significantly increased vWF secretion by more than 1.5-fold over untreated control (p=0.025). Negative isotype control murine IgG (mIgG) had no effect (Figure 1a).
Figure 1.




HLA I antibodies trigger Weibel-Palade body exocytosis via intracellular calcium.
(a) Human aortic endothelial cells (HAEC) were treated with isotype control mIgG at 5μg/mL, HLA I antibody (clone W6/32, murine IgG2a) at 5μg/mL, thrombin at 10U/mL, PMA at 200nM, or histamine at 10mM for 1 hr. Supernatant was collected and vWF was measured by ELISA. An experiment showing the average optical density from duplicate measurements +/− SEM (upper panel). Bar graph in lower panel shows mean fold increase in optical density (OD) from 2 (PMA and histamine) or 3 (mIgG, HLA I antibody and thrombin) independent experiments for each condition. * p<0.05, ** p<0.01 versus mIgG.
(b) HAEC were treated with mIgG, anti-CD105 antibody, or HLA I antibody at 5μg/mL for 30min, thrombin at 1U/mL for 15min, PMA at 200nM for 20min, or histamine at 10mM for 10min and detached using Accutase. Cell surface P-selectin was stained with conjugated anti-P-selectin-PE and analyzed by flow cytometry. Bar graph shows mean fold increase in P-selectin positive cells from multiple independent measurements for each condition: mIgG (n=5), anti-CD105 antibody (n=2), HLA I antibody (n=10), thrombin (n=3), PMA (n=6), histamine (n=23). * p<0.05, ** p<0.001 versus untreated.
(c) HAEC were pretreated with BAPTA-AM at 20μM for 30min, then stimulated with HLA I antibody at 5ug/mL for 1 hour (h) and P-selectin was measured as in (b). Bar graph shows summary of data from 6 independent experiments. Black bars show the fold increase in P-selectin expression without inhibitor, and white bars show P-selectin induction with inhibitor.
(d) In order to monitor intracellular Ca2+ concentration, HAEC were loaded with Fura 2-AM and then treated with HLA I antibody at 1μg/mL, thrombin at 0.01U/mL and 1U/mL, or isotype control antibody (mIgG) at 1μg/mL. Intracellular calcium was monitored in real-time using a fluorimeter. Data are presented as the ratio of emission at 340nm/380nm, and are representative of 2 (mIgG and thrombin 0.01U/mL) or 3 (HLA I antibody and thrombin 1U/mL) independent measurements. The time of introduction of reagents is marked with an arrow.
HLA I antibody (5μg/mL) significantly increased the proportion of cells staining positively in flow cytometry for P-selectin by 2.0+/−0.14 fold over control after 30min (Figure 1b). Untreated endothelial cells were negative for P-selectin staining. Dose response and kinetics studies revealed a maximal effect on P-selectin induced by 5μg/mL HLA I antibody at 30 minutes (Supplemental Figure Figure 2a, 2b and 2c). This response was specific to HLA I antibodies, as treatment of endothelial cells with nonbinding mIgG or non-HLA antibody against endoglin (CD105) did not significantly alter cell surface P-selectin (Fig. 1b). In contrast, P-selectin expression was elevated on positive control thrombin, PMA or histamine-treated endothelial cells. HLA class I mediated WPb exocytosis comparable to induction by classical agonists (49-54), which trigger rolling of leukocytes on blood vessels in vivo (55-58). Treatment of EC with HLA class I antibody had no effect on expression of E-selectin over a 48-hr time frame (Supplemental Fig. 3), whereas TNFα treatment increased cell surface E-selectin as previously reported (59, 60).
We postulated that HLA I-induced exocytosis might be dependent on Ca2+ signaling. Pretreatment of endothelial cells with BAPTA-AM to chelate intracellular Ca2+ significantly inhibited HLA I-induced P-selectin induction, from 1.70+/−0.11-fold without inhibitor to 1.15+/−0.37-fold over control (Figure 1c). Likewise, vWF secretion from endothelium in response to HLA I antibodies or thrombin was completely prevented by BAPTA-AM (data not shown). To further confirm that HLA I antibodies increase Ca2+-dependent signaling, we measured [Ca2+]i in real time after HLA I crosslinking. Endothelial cells treated with HLA I antibody exhibited a rapid increase in [Ca2+]i levels. As a positive control, we verified that thrombin treatment rapidly increased free cytosolic Ca2+. In contrast, isotype control mIgG had no effect on [Ca2+]i levels (Figure 1d).
HLA I antibody-induced endothelial P-selectin is necessary and sufficient to promote monocyte adherence in vitro
In order to focus on monocyte recruitment due exclusively to HLA I antibody-provoked endothelial cell signaling and to eliminate interaction of FcγRs with the Fc fragment of antibody, endothelial cells were treated with an F(ab’)2 fragment of HLA I antibody (W6/32) and fluorescently labeled Mono Mac 6 were added. Crosslinking of HLA I on endothelium by HLA I F(ab’)2 significantly increased the number of adherent monocytes by 1.66+/−0.07 fold compared with untreated endothelial cells (p<0.0001) (Figure 2a). Representative micrographs and raw data from one representative experiment are given in Figure 2b. As an alternative approach, we treated endothelial cells with intact HLA I antibody and preincubated Mono Mac 6 with soluble IgG to block FcγRs. Treatment of endothelial cells with HLA I antibody significantly increased monocyte adherence by 1.48+/−0.09 fold (p<0.0001). Thrombin also increased Mono Mac 6 binding, while exposure of endothelial cells to isotype control mIgG did not (Figure 2a).
Figure 2.






HLA I antibody-induced endothelial P-selectin is necessary and sufficient to support increased monocyte adherence in vitro.
(a) HAEC were treated with isotype control mIgG (n=17), HLA I intact antibody at 5μg/mL (n=9) or F(ab’)2 fragment at 10μg/mL for 60min (n=18); or thrombin at 1U/mL for 20min (n=16). Fluorescently labeled Mono Mac 6 were left untreated or incubated with soluble human IgG to block FcγRs. Mono Mac 6 with or without an FcγR blockade were added to stimulated endothelial monolayers, and adherent cells were counted after 20min. Bar graph represents mean fold increase in the number of adherent Mono Mac 6 from multiple independent experiments +/−SEM, normalized to untreated. #, p>0.05; *** p<0.0001 versus untreated.
(b) Representative experiment performed as in (a), with data expressed as average number of adherent cells per field in 10 fields +/− SEM. Representative micrographs illustrate fluorescently labeled monocytic cells adherent to the endothelial monolayer. **** p<0.0001 versus untreated.
(c) rPSGL-1 was added to block endothelial P-selectin, and Mono Mac 6 adherence was assessed with or without inhibitor as in (a). Graph shows mean fold increase in the average number of adherent monocytes per field +/−SEM over 5 independent experiments. Black bars show Mono Mac 6 binding without inhibitor, white bars with inhibitor. ** p<0.01 comparing no inhibitor to inhibitor.
(d) Mono Mac 6 were incubated with anti-PSGL-1 blocking antibody (n=2), or stimulated HAEC were incubated with rPSGL-1 (n=5) or with neutralizing anti-P-selectin antibody (n=3) prior to adherence assay. Graph shows the mean percent inhibition +/− SEM of binding to HLA I F(ab’)2 treated endothelium by each inhibitor.
(e) Experiments were performed as in (a) using human monocytes enriched from peripheral blood and rPSGL-1 was added to block endothelial P-selectin. Data is expressed as average number of adherent monocytes per field in 10 fields +/− SEM, and is representative of three independent experiments using monocytes isolated from three different donors. *** p<0.001 versus untreated. ‡ p<0.0001 versus no inhibitor.
(f) HAEC were pretreated with BAPTA-AM at 20μM for 30min, then stimulated with HLA I F(ab’)2 at 10μg/mL for 45min or thrombin at 1U/mL (n=6). Adherence of Mono Mac 6 was assessed. Data are presented as mean fold increase in adherent cells +/−SEM. Black bars are binding without inhibitor, white bars with inhibitor. ** p<0.01, *** p<0.001 versus comparing no inhibitor to inhibitor.
We examined whether P-selectin was involved in mediating the increased adherence of monocytes. rPSGL-1-Ig was added to block P-selectin. Mono Mac 6 adherence to endothelium was increased by 1.61+/−0.06 fold over control in response to HLA I F(ab’)2, but was significantly reduced to 1.13+/−0.09 fold over control by rPSGL-1 (76.7+/−12.8% inhibition). Mono Mac 6 binding to thrombin-stimulated endothelium was also reduced by the presence of rPSGL-1 (Figure 2c). We substantiated the requirement for P-selectin with neutralizing antibody to P-selectin, or to monocytic PSGL-1, both of which significantly prevented Mono Mac 6 recruitment to HLA I F(ab’)2-treated endothelial cells (Figure 2d). Similar results were obtained using human monocytes enriched from peripheral blood (Figure 2e), where HLA I F(ab’)2 fragment or positive control PMA increased the number of monocytes bound to endothelium by more than 2-fold (p<0.01), while anti-CD105 antibody had no significant effect. Binding of peripheral blood monocytes was significantly inhibited by P-selectin antagonism (Figure 2e).
To determine whether endothelial cells required intact Ca2+ signaling to recruit monocytes after HLA I crosslinking, endothelial cells were pretreated with BAPTA-AM. Activation of BAPTA-AM treated endothelial cells with HLA I F(ab’)2 or thrombin failed to trigger Mono Mac 6 adherence (p<0.001 compared with no inhibitor) (Figure 2f).
Passive transfer of donor specific MHC I antibodies increases macrophage infiltration into the cardiac allograft
To elucidate the role of P-selectin under physiological conditions, we utilized an in vivo model of AMR (16, 61). C57BL/6 RAG1 KO (H-2b) recipients of BALB/c donor (H-2d) hearts were transferred with murine monoclonal IgG2a recognizing donor MHC I molecules (anti-Kd or anti-Dd) or control murine IgG2a (MOPC-173). The treatment regimen and dose of MHC I antibody were based on a previously described mouse model of antibody-mediated rejection (61, 62). Grafts were recovered on day 30 post-transplant and analyzed as outlined in Figure 3a.
Figure 3.
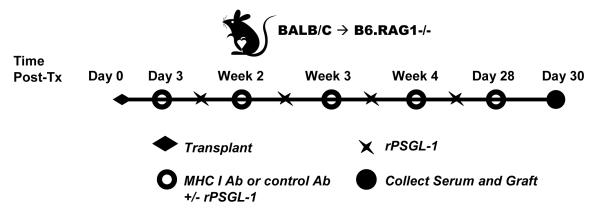


Donor specific MHC I antibodies increase monocyte recruitment into a murine cardiac allograft.
(a) Diagram of antibody administration protocol. BALB/c (H-2d) donor hearts were heterotopically grafted into RAG1 knockout recipients on a C57BL/6 background (H-2b). Animals were passively transferred with a murine isotype control monoclonal IgG2a or monoclonal directed against the donor MHC I locus beginning on day 3 after transplant and continuing weekly thereafter until day 28. Grafts were recovered from recipients while still beating on day 30 post-transplant. Cellular infiltration in the base was assessed by immunohistochemical staining and scored by a pathologist blinded to the experimental groups. The fusion protein rPSGL-1-Ig was administered to a group of animals biweekly beginning on day 3 post-transplant at 70μg per animal.
(b) The presence of circulating donor specific MHC I antibodies in recipient serum was confirmed using flow cytometric crossmatch on BALB/c splenocytes. BALB/c splenocytes were incubated with 25μL of neat serum collected from allograft recipients, and stained with anti-mouse Fcγ-FITC. Cells were stained with anti-CD3-PE and fluorescence was measured by flow cytometry. CD3-positive cells were gated as shown in the representative dot plot, and the median fluorescence in the FL-1 channel was determined. Results are shown as median fluorescence intensity of serum on CD3 positive cells +/−SEM, using sera from multiple animals for each condition: unstained (n=5), control mIgG2a (n=3), anti-Kd mIgG2a (n=7), anti-Dd mIgG2a (n=2), anti-Kd + Dd mIgG2a (n=2).
(c) Native or transplanted hearts were assessed immunohistochemically for Mac-2 and staining was scored by a pathologist blinded to the experimental groups. The box and whiskers graph shows the distribution of scores for each group. Infiltration near the suture site or in the endocardium was disregarded. Global ANOVA, then individual groups. * p<0.05, ** p<0.01, *** p<0.001 versus control mIgG2a group.
We performed flow crossmatching to measure circulating antibodies. The flow cytometric cross-match revealed no binding of sera from control antibody-treated recipients to BALB/c CD3+ splenocytes. In contrast, sera from animals passively transferred with MHC I antibodies bound highly to donor CD3+ splenocytes, and cells stained positively even when serum was diluted to 1:4. These results document the presence of anti-donor antibodies in the circulation of passively transferred cardiac allograft recipients (Figure 3b).
We then characterized the infiltration of macrophages in cardiac allografts in response to MHC I antibody treatment by immunohistochemistry. Less than half (42%) of grafts from animals which received control mIgG2a antibody were positive for macrophages, with a score of 2 or greater, and none (0%) were strongly positive with a score of 3 or 4 (Table 1). Strikingly, 84% of cardiac allografts from animals treated with anti-Kd antibody stained positively for the macrophage marker, with 40% having strong infiltration. Macrophage staining in anti-Kd antibody-treated allografts was significantly greater (p<0.001) when compared with allografts from control antibody-treated animals. We confirmed this phenomenon using a different monoclonal donor specific MHC I monoclonal antibody. Eighty percent of anti-Dd antibody treated allografts scored positively for macrophages, with 40% having strong macrophage infiltration. When allograft recipients were given both anti-Kd and Dd antibodies together, all transplanted hearts had a score of two or greater for Mac-2 macrophage marker (Table 1 and Figure 3c). These results demonstrate that donor specific MHC I antibodies promote macrophage accumulation at the allograft site.
Table 1.
MHC I antibody-induced macrophage accumulation in the allograft.
| Group | Donor | Treatment | Hearts with Mac-2 ≥2 |
% positive |
Hearts with Mac-2 ≥3 |
% positive |
p value |
|---|---|---|---|---|---|---|---|
| 1 | n/a | C57BL/6 Native Heart | 0/13 | 0 | 0/13 | 0 | |
| 2 |
BALB/c
H-2d |
Control mIgG2a | 6/14 | 42 | 0/14 | 0 | |
| 3 |
BALB/c
H-2d |
anti-Kd mIgG2a | 21/25 | 84 | 10/25 | 40 |
0.0002
versus group 2 |
| 4 |
BALB/c
H-2d |
anti-Dd mIgG2a | 4/5 | 80 | 2/5 | 40 |
0.025
versus group 2 |
| 5 |
BALB/c
H-2d |
anti-Kd + anti-Dd mIgG2a |
4/4 | 100 | 1/4 | 25 |
0.012
versus group 2 |
The selectin antagonist rPSGL-1 significantly reduces MHC I antibody-elicited macrophage and CD45+ accumulation in the allograft
We sought to determine whether antagonism of P-selectin could reduce immune cell recruitment in response to MHC I antibodies in vivo. While greater than 80% of anti-Kd antibody treated allografts were positive for Mac-2 staining, only 25% of allografts which received concurrent rPSGL-1 therapy had a Mac-2 score of 2 or higher. Remarkably, rPSGL-1 abolished the strong macrophage infiltration induced otherwise by donor specific MHC I antibodies (from 40% to 0%; Table 2 and Figure 4A and 4C).
Table 2.
rPSGL-1 therapy reduces MHC I antibody-induced macrophage infiltration into the allograft.
| Group | Treatment | Hearts with Mac-2 score ≥2 |
% positive |
Hearts with Mac-2 score ≥3 |
% positive |
p value |
|---|---|---|---|---|---|---|
| 1 | Control mIgG2a |
6/14 | 42.8% | 0/14 | 0% | |
| 2 | Control mIgG2a + rPSGL-1 |
2/5 | 40.0 | 2/5 | 40.0 | |
| 3 | anti-Kd mIgG2a |
21/25 | 84.6 | 10/25 | 40.0 | |
| 4 | anti-Kd mIgG2a + rPSGL-1 |
2/8 | 25.0 | 0/8 | 0 |
0.004 versus
group 3 |
Figure 4.



rPSGL-1-Ig therapy reduces macrophage and CD45+ accumulation in the allograft in response to MHC I antibody. Native or transplanted hearts were assessed immunohistochemically for Mac-2 (a) or CD45 (b) and staining was scored by a pathologist blinded to the experimental groups. Global ANOVA and individual comparisons * p<0.05. *** p<0.001. Graphs show the distribution of scores for each group. Representative micrographs at 100x and 200x magnification of Mac-2 staining and H&E are shown in (c)
We also examined the allografts for total immune cell infiltration by immunohistochemistry. Allografts from control antibody-treated animals were negative for CD45 staining. In contrast, 57% of allografts from animals which received anti-Kd antibody were positive for CD45 (score ≥2), and nearly one third (29%) stained strongly for CD45. Increased CD45 infiltration elicited by MHC I antibody (anti-Kd mIgG2a) was abolished by concurrent administration of rPSGL-1-Fc (p<0.05), as no allografts from animals which received both anti-Kd antibody and rPSGL-1 were positive for CD45 (Table 3 and Figure 4B). Therefore, rPSGL-1 therapy specifically reduced macrophage infiltration and prevented CD45+ immune cell infiltration.
Table 3.
rPSGL-1 therapy reduces CD45+ cell infiltration in donor specific MHC I antibody-treated allografts.
| Group | Treatment | Hearts with CD45 score ≥2 |
% positive |
Hearts with CD45 score ≥ 3 |
% positive |
p value |
|---|---|---|---|---|---|---|
| 1 | Control mIgG2a | 0/2 | 0% | 0/2 | 0% | |
| 2 | anti-Kd mIgG2a | 4/7 | 57 | 2/7 | 29 |
0.048 versus
group 1 |
| 3 | anti-Kd mIgG2a + rPSGL-1 |
0/4 | 0 | 0/4 | 0 |
0.037 versus
group 2 |
The allografts were examined for histological features of antibody-mediated rejection. H&E sections of cardiac allografts showed characteristic histological features of AMR including microvascular changes and endothelial cell swelling (Figure 4c) (16, 19, 20, 27). No hemorrhage or intravascular neutrophils, as commonly seen in severe acute humoral rejection, were observed. An increase in overall cellularity in the interstitium, with cells in a linear arrangement indicative of intracapillary macrophages (19, 27), was found in allografts when compared with native hearts. However, allografts treated with donor specific MHC I antibody had notably greater cellularity and endothelial cell swelling due to intravascular macrophages when compared with control antibody-treated grafts (Figure 4c). While rPSGL-1-Ig therapy decreased cellularity, allografts receiving this treatment had no detectable change in the histological pattern of endothelial cell injury.
Discussion
In this study, we established that MHC I crosslinking on endothelial cells increases cell surface P-selectin, which is required for recruitment of monocytes independently of antibody-FcγR interactions. We also determined that [Ca2+]i signaling downstream of HLA I ligation is necessary for exocytosis, consistent with previous studies (18, 63). Importantly, we confirmed these results in an in vivo system, where donor specific MHC I antibodies increased macrophage infiltration into cardiac allograft, which could be abolished by P-selectin antagonism.
Given the Ca2+ dependence of exocytosis, we postulated that HLA I crosslinking elicited an increase in [Ca2+]i in the endothelial cell. HLA I antibodies (1μg/mL) stimulated a sustained increase in [Ca2+]i, suggesting that Ca2+-dependent pathways are activated shortly after HLA I signaling. Our group has previously demonstrated that HLA ligation rapidly stimulates several intracellular signaling pathways, necessary for proliferation and cytoskeletal changes (14, 64). Increased intracellular Ca2+ (reviewed in (65)) triggers a variety of signaling pathways, including protein kinase C (PKC), calmodulin and myosin light chain kinase (MLCK) (66) which are not yet explored in the context of HLA I signaling. It is interesting that the potent diacylglycerol (DAG) surrogate PMA stimulated P-selectin and increased adherence of monocytic cells, suggesting that DAG/PKC acts in conjunction with Ca2+ signaling in mediating HLA I-triggered events.
Herein, we describe HLA I antibody-elicited recruitment of monocytic cells in vitro, which could be blocked by several strategies of P-selectin antagonism. P-selectin is an important early mediator of the leukocyte recruitment cascade, which initiates the capture of immune cells from circulation and precedes firm adhesion and extravasation events. P-selectin is upregulated on endothelium in acutely rejecting cardiac allografts, and on thickened arterial intima during chronic rejection in rodent models (67, 68). Allografts from murine donors deficient in both intercellular adhesion molecule (ICAM-1) and P-selectin or in all three selectins (ELP-/-) had significantly longer graft survival than their wild-type counterparts (69, 70). rPSGL-1 or SLX (antagonists of selectin) reduce immune cell infiltration, neointimal hyperplasia and rejection in many models (48, 71-76). Currently, clinical trials are underway to investigate the use of rPSGL-1-Ig in humans as a treatment to reduce ischemia/reperfusion injury (77-79). However, the effect of rPSGL-1-Ig on long-term survival and leukocyte recruitment in human transplantation has yet to be revealed.
Our group previously reported increased macrophage infiltration into allografts treated with a single high dose of MHC I intact antibody or F(ab’)2 (16). P-selectin and vWF were increased in acutely rejecting murine allografts from wild-type but not IgKO recipients, suggesting that these mediators are specifically induced in presence of donor specific antibodies (80, 81). Another group demonstrated that HLA I F(ab’)2 caused rapid neutrophil recruitment and acute rejection of human skin grafts on immunodeficient mice (18). We attempted confirm our findings in vivo with an MHC I F(ab’)2 fragment, but were unable to achieve consistently detectable levels of circulating antibody, likely due to their short half-life in vivo (82). Nevertheless, the data reported here show that P-selectin is a critical initiator which facilitates capture of monocytes from the circulation by graft endothelium in a long-term vascularized model of AMR.
We found that overall CD45+ infiltration was reduced by rPSGL-1 therapy. NK cells, neutrophils, monocytes and T cells utilize selectins to adhere to endothelium and transmigrate from the blood into tissue (83-86). Donor specific MHC I antibodies cause platelet activation in skin allograft recipients (80), and attachment of platelets to endothelium can support monocyte tethering through P-selectin (87). Moreover, recent data point to a novel role for NK cells in AMR (61, 62, 88). NK cells adhere to P-selectin during recruitment to atherosclerotic lesions (89). In our in vivo model, it is possible that administration of rPSGL-1-Ig also antagonized platelet or NK cell adhesion. While P-selectin is the preferred ligand of PSGL-1, PSGL-1 can interact with other adhesion molecules, such as E- and L-selectin and von Willebrand Factor (90). We did not observe an increase in E-selectin expression induced by HLA I crosslinking in vitro, but E-selectin may be involved in vivo. In light of the myriad parallel mechanisms which may promote leukocyte recruitment during antibody-mediated rejection, the promiscuity of rPSGL-1-Ig may be viewed as advantageous from the therapeutic standpoint since it can disrupt many of the interactions between leukocytes and endothelium.
Complement and endothelial activation are parallel pathways of AMR. rPSGL-1 therapy did not change the histological pattern with respect to endothelial cell injury (19, 27), emphasizing the complexity of antibody-mediated rejection. Our results suggest that concurrent therapies may be required to inhibit the multiple mechanisms of HLA antibody-mediated graft injury. We did not observe graft failure in this model, consistent with several studies (16, 61, 62), nor did we observe chronic vascular lesions in allografts from animals transferred with MHC I antibody, in disagreement with previous reports (61, 62). This inconsistency may be due to the restricted localization of transplant arteriosclerosis in this model, which is reported to occur only in the proximal coronary arteries.
In conclusion, we have demonstrated that monocyte infiltration is directly triggered by HLA I antibody activation of endothelial cell exocytosis and P-selectin expression. These results strongly suggest that selectin antagonism may reduce the accumulation of macrophages and other recipient immune cells into the allograft during AMR.
Supplementary Material
Acknowledgements
This research was supported by the NIH grant U01AI077821 (E.F.R.); the National Heart, Lung and Blood Institute grant RO1 HL 090995 (E.F.R.) and the Ruth L. Kirschstein National Research Service Award T32HL69766 (to N.M.V.).
Abbreviations
- AMR
antibody mediated rejection
- CAV
chronic allograft vasculopathy
- CFSE
carboxyfluorescein diacetate succinimidyl
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- Fc
crystallizable fragment of immunoglobulin
- FITC
fluorescein isothiocyanate
- HAEC
human aortic endothelial cell
- ICAM
intercellular adhesion molecule
- PKC
protein kinase C
- PMA
phorbol myristate acetate
- vWF
von Willebrand Factor
Footnotes
Disclosure
The authors have no conflict of interest to declare.
References
- 1.Tambur AR, Pamboukian SV, Costanzo M-R, Herrera ND, Dunlap S, Montpetit M, et al. The Presence of HLA-Directed Antibodies after Heart Transplantation Is Associated with Poor Allograft Outcome. Transplantation. 2005;80:1019–1025. doi: 10.1097/01.tp.0000180564.14050.49. [DOI] [PubMed] [Google Scholar]
- 2.Ho EK, Vlad G, Vasilescu ER, de la Torre L, Colovai AI, Burke E, et al. Pre- and posttransplantation allosensitization in heart allograft recipients: Major impact of de novo alloantibody production on allograft survival. Human Immunology. 2011;72:5–10. doi: 10.1016/j.humimm.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Ho EK, Vasilescu ER, Vlad G, Marboe CC, Addonizio LJ, Suciu-Foca N. HLA antibodies in pediatric heart transplantation. Pediatric Transplantation. 2011;15:458–464. doi: 10.1111/j.1399-3046.2010.01435.x. [DOI] [PubMed] [Google Scholar]
- 4.Kimball PM, Baker MA, Wagner MB, King A. Surveillance of alloantibodies after transplantation identifies the risk of chronic rejection. Kidney International. 2011;79:1131–1137. doi: 10.1038/ki.2010.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Cecka JM, Gjertson DW, Ge P, Rose ML, Patel JK, et al. HLA and MICA: Targets of Antibody-Mediated Rejection in Heart Transplantation. Transplantation. 2011;91:1153–1158. doi: 10.1097/TP.0b013e3182157d60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu GW, Kobashigawa JA, Fishbein MC, Patel JK, Kittleson MM, Reed EF, et al. Asymptomatic Antibody-mediated Rejection After Heart Transplantation Predicts Poor Outcomes. The Journal of Heart and Lung Transplantation. 2009;28:417–422. doi: 10.1016/j.healun.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lachmann N, Terasaki PI, Budde K, Liefeldt L, Kahl A, Reinke P, et al. Anti-Human Leukocyte Antigen and Donor-Specific Antibodies Detected by Luminex Posttransplant Serve as Biomarkers for Chronic Rejection of Renal Allografts. Transplantation. 2009;87:1505–1513. doi: 10.1097/TP.0b013e3181a44206. [DOI] [PubMed] [Google Scholar]
- 8.Everly MJ, Everly JJ, Arend LJ, Brailey P, Susskind B, Govil A, et al. Reducing De Novo Donor-Specific Antibody Levels during Acute Rejection Diminishes Renal Allograft Loss. American Journal of Transplantation. 2009;9:1063–1071. doi: 10.1111/j.1600-6143.2009.02577.x. [DOI] [PubMed] [Google Scholar]
- 9.Li F, Zhang X, Jin Y-P, Mulder A, Reed EF. Antibody ligation of human leukocyte antigen class I molecules stimulates migration and proliferation of smooth muscle cells in a focal adhesion kinase-dependent manner. Human Immunology. 2011;72:1150–1159. doi: 10.1016/j.humimm.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziegler ME, Souda P, Jin Y-P, Whitelegge JP, Reed EF. Characterization of the Endothelial Cell Cytoskeleton following HLA Class I Ligation. PLoS ONE. 2012;7:e29472. doi: 10.1371/journal.pone.0029472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Y-P, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed EF. RNA Interference Elucidates the Role of Focal Adhesion Kinase in HLA Class I-Mediated Focal Adhesion Complex Formation and Proliferation in Human Endothelial Cells. The Journal of Immunology. 2007;178:7911–7922. doi: 10.4049/jimmunol.178.12.7911. [DOI] [PubMed] [Google Scholar]
- 12.Jin Y-P, Singh RP, Du Z-Y, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA Class I Molecules on Endothelial Cells Induces Phosphorylation of Src, Paxillin, and Focal Adhesion Kinase in an Actin-Dependent Manner. The Journal of Immunology. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 13.Jin Y-P, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, et al. Anti-HLA class I antibody mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Human Immunology. 2004;65:291–302. doi: 10.1016/j.humimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Jindra PT, Jin Y-P, Rozengurt E, Reed EF. HLA Class I Antibody-Mediated Endothelial Cell Proliferation via the mTOR Pathway. The Journal of Immunology. 2008;180:2357–2366. doi: 10.4049/jimmunol.180.4.2357. [DOI] [PubMed] [Google Scholar]
- 15.Narayanan K, Jendrisak MD, Phelan DL, Mohanakumar T. HLA class I antibody mediated accommodation of endothelial cells via the activation of PI3K/cAMP dependent PKA pathway. Transplant Immunology. 2006;15:187–197. doi: 10.1016/j.trim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen X-D, Gao F, et al. Anti-MHC Class I Antibody Activation of Proliferation and Survival Signaling in Murine Cardiac Allografts. The Journal of Immunology. 2008;180:2214–2224. doi: 10.4049/jimmunol.180.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, et al. Phosphorylated S6 Ribosomal Protein: A Novel Biomarker of Antibody-Mediated Rejection in Heart Allografts. American Journal of Transplantation. 2006;6:1560–1571. doi: 10.1111/j.1600-6143.2006.01355.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proceedings of the National Academy of Sciences. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishbein GA, Fishbein MC. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Hum Immunol. 2012 doi: 10.1016/j.humimm.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Fishbein MC, Kobashigawa J. Biopsy-negative cardiac transplant rejection: etiology, diagnosis, and therapy. Current Opinion in Cardiology. 2004;19:166–169. doi: 10.1097/00001573-200403000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Loupy A, Cazes A, Guillemain R, Amrein C, Hedjoudje A, Tible M, et al. Very Late Heart Transplant Rejection Is Associated with Microvascular Injury, Complement Deposition and Progression to Cardiac Allograft Vasculopathy. American Journal of Transplantation. 2011;11:1478–1487. doi: 10.1111/j.1600-6143.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- 22.Tinckam KJ, Djurdjev O, Magil AB. Glomerular monocytes predict worse outcomes after acute renal allograft rejection independent of C4d status. 2005;68:1866–1874. doi: 10.1111/j.1523-1755.2005.00606.x. [DOI] [PubMed] [Google Scholar]
- 23.Desai HS, Parasuraman RK, Samarpungavan D, Rooney MT, Cohn SR, Reddy GH, et al. Glomerulitis During Acute Cellular Rejection May Be a Surrogate Marker of Vasculitis in Renal Allografts: Better Index for Diagnosis of Vasculitis. Transplantation Proceedings. 2011;43:1629–1633. doi: 10.1016/j.transproceed.2011.01.187. [DOI] [PubMed] [Google Scholar]
- 24.Ashokkumar C, Ningappa M, Ranganathan S, Higgs BW, Sun Q, Schmitt L, et al. Increased Expression of Peripheral Blood Leukocyte Genes Implicate CD14+ Tissue Macrophages in Cellular Intestine Allograft Rejection. The American Journal of Pathology. 2011;179:1929–1938. doi: 10.1016/j.ajpath.2011.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hancock W, Thomson NM, Atkins RC. Composition of interstitial cellular infiltrate identified by monoclonal antibodies in renal biopsies of rejecting human renal allografts. Transplantation. 1983;35:458–63. doi: 10.1097/00007890-198305000-00013. [DOI] [PubMed] [Google Scholar]
- 26.Sun H-j, Zhou T, Wang Y, Fu Y-w, Jiang Y-p, Zhang L-h, et al. Macrophages and T lymphocytes are the predominant cells in intimal arteritis of resected renal allografts undergoing acute rejection. Transplant Immunology. 2011;25:42–48. doi: 10.1016/j.trim.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Michaels PJ, Espejo ML, Kobashigawa J, Alejos JC, Burch C, Takemoto S, et al. Humoral rejection in cardiac transplantation: risk factors, hemodynamic consequences and relationship to transplant coronary artery disease. The Journal of Heart and Lung Transplantation. 2003;22:58–69. doi: 10.1016/s1053-2498(02)00472-2. [DOI] [PubMed] [Google Scholar]
- 28.Matheson PJ, Dittmer ID, Beaumont BW, Merrilees MJ, Pilmore HL. The Macrophage Is the Predominant Inflammatory Cell in Renal Allograft Intimal Arteritis. Transplantation. 2005;79:1658–1662. doi: 10.1097/01.tp.0000167099.51275.ec. [DOI] [PubMed] [Google Scholar]
- 29.Sis B, Grynoch R, Murray AG, Campbell P, Solez K. Antibody-Mediated Rejection With a Striking Interstitial Monocyte/Macrophage Infiltration in a Renal Allograft Under FTY720 Treatment. American Journal of Kidney Diseases. 2008;51:127–130. doi: 10.1053/j.ajkd.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 30.Feng Y, van Eck M, Van Craeyveld E, Jacobs F, Carlier V, Van Linthout S, et al. Critical role of scavenger receptor-BI-expressing bone marrow-derived endothelial progenitor cells in the attenuation of allograft vasculopathy after human apo A-I transfer. Blood. 2009;113:755–64. doi: 10.1182/blood-2008-06-161794. [DOI] [PubMed] [Google Scholar]
- 31.Edwards IJ, Wagner WD, Owens RT. Macrophage secretory products selectively stimulate dermatan sulfate proteoglycan production in cultured arterial smooth muscle cells. Am J Pathol. 1990;136:609–621. [PMC free article] [PubMed] [Google Scholar]
- 32.Schubert SY, Benarroch A, Ostvang J, Edelman ER. Regulation of Endothelial Cell Proliferation by Primary Monocytes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:97–104. doi: 10.1161/ATVBAHA.107.157537. [DOI] [PubMed] [Google Scholar]
- 33.Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, Lakkis FG. An Innate Response to Allogeneic Nonself Mediated by Monocytes. The Journal of Immunology. 2009;183:7810–7816. doi: 10.4049/jimmunol.0902194. [DOI] [PubMed] [Google Scholar]
- 34.Liu W, Xiao X, Demirci G, Madsen J, Li XC. Innate NK Cells and Macrophages Recognize and Reject Allogeneic Nonself In Vivo via Different Mechanisms. The Journal of Immunology. 2012;188:2703–2711. doi: 10.4049/jimmunol.1102997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitchens WH, Chase CM, Uehara S, Cornell LD, Colvin RB, Russell PS, et al. Macrophage Depletion Suppresses Cardiac Allograft Vasculopathy in Mice. American Journal of Transplantation. 2007;7:2675–2682. doi: 10.1111/j.1600-6143.2007.01997.x. [DOI] [PubMed] [Google Scholar]
- 36.Bian H, Harris PE, Reed EF. Ligation of HLA class I molecules on smooth muscle cells with anti-HLA antibodies induces tyrosine phosphorylation, fibroblast growth factor receptor expression and cell proliferation. International Immunology. 1998;10:1315–1323. doi: 10.1093/intimm/10.9.1315. [DOI] [PubMed] [Google Scholar]
- 37.Farmer DG, Shen XD, Amersi F, Anselmo D, Ma JP, Ke B, et al. CD62 blockade with P-Selectin glycoprotein ligand-immunoglobulin fusion protein reduces ischemia-reperfusion injury after rat intestinal transplantation. Transplantation. 2005;79:44–51. doi: 10.1097/01.tp.0000146965.64706.e8. [DOI] [PubMed] [Google Scholar]
- 38.Amersi F, Farmer DG, Shaw GD, Kato H, Coito AJ, Kaldas F, et al. P-selectin glycoprotein ligand-1 (rPSGL-Ig)-mediated blockade of CD62 selectin molecules protects rat steatotic liver grafts from ischemia/reperfusion injury. Am J Transplant. 2002;2:600–8. doi: 10.1034/j.1600-6143.2002.20704.x. [DOI] [PubMed] [Google Scholar]
- 39.Erl W, Weber C, Wardemann C, Weber PC. Adhesion properties of Mono Mac 6, a monocytic cell line with characteristics of mature human monocytes. Atherosclerosis. 1995;113:99–107. doi: 10.1016/0021-9150(94)05434-k. [DOI] [PubMed] [Google Scholar]
- 40.Erl W, Weber PC, Weber C. Monocytic cell adhesion to endothelial cells stimulated by oxidized low density lipoprotein is mediated by distinct endothelial ligands. Atherosclerosis. 1998;136:297–303. doi: 10.1016/s0021-9150(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 41.Mutin G, George F, Leslaule G, Sampol J. Reevaluation of trypsin–EDTA for endothelial cell detachment before flow cytometry analysis. Endothelium. 1996;4:289–295. [Google Scholar]
- 42.Kim DS, Kwon HM, Choi JS, Kang SW, Ji GE, Kang YH. Resveratrol blunts tumor necrosis factor-alpha-induced monocyte adhesion and transmigration. Nutr Res Pract. 2007;1:285–90. doi: 10.4162/nrp.2007.1.4.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lubeck MD, Steplewski Z, Baglia F, Klein MH, Dorrington KJ, Koprowski H. The interaction of murine IgG subclass proteins with human monocyte Fc receptors. J Immunol. 1985;135:1299–304. [PubMed] [Google Scholar]
- 44.Tax WJ, Hermes FF, Willems RW, Capel PJ, Koene RA. Fc receptors for mouse IgG1 on human monocytes: polymorphism and role in antibody-induced T cell proliferation. J Immunol. 1984;133:1185–9. [PubMed] [Google Scholar]
- 45.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan KR, Zhang SL-X, Tan HC, Chan YK, Chow A, Lim APC, et al. Ligation of Fc gamma receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proceedings of the National Academy of Sciences. 2011;108:12479–12484. doi: 10.1073/pnas.1106568108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khor SP, McCarthy K, DuPont M, Murray K, Timony G. Pharmacokinetics, Pharmacodynamics, Allometry, and Dose Selection of rPSGL-Ig for Phase I Trial. Journal of Pharmacology and Experimental Therapeutics. 2000;293:618–624. [PubMed] [Google Scholar]
- 48.Hicks AER, Nolan SL, Ridger VC, Hellewell PG, Norman KE. Recombinant P-selectin glycoprotein ligand directly inhibits leukocyte rolling by all 3 selectins in vivo: complete inhibition of rolling is not required for anti-inflammatory effect. Blood. 2003;101:3249–3256. doi: 10.1182/blood-2002-07-2329. [DOI] [PubMed] [Google Scholar]
- 49.Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94:1147–55. doi: 10.1172/JCI117430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cleator JH, Zhu WQ, Vaughan DE, Hamm HE. Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood. 2006;107:2736–44. doi: 10.1182/blood-2004-07-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhatia R, Matsushita K, Yamakuchi M, Morrell CN, Cao W, Lowenstein CJ. Ceramide triggers Weibel-Palade body exocytosis. Circ Res. 2004;95:319–24. doi: 10.1161/01.RES.0000136519.84279.7a. [DOI] [PubMed] [Google Scholar]
- 52.Setiadi H, McEver RP. Signal-dependent distribution of cell surface P-selectin in clathrin-coated pits affects leukocyte rolling under flow. J Cell Biol. 2003;163:1385–95. doi: 10.1083/jcb.200307178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsushita K, Morrell CN, Lowenstein CJ. Sphingosine 1-phosphate activates Weibel-Palade body exocytosis. Proc Natl Acad Sci U S A. 2004;101:11483–7. doi: 10.1073/pnas.0400185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyazaki Y, Satoh T, Nishioka K, Yokozeki H. STAT-6-mediated control of P-selectin by substance P and interleukin-4 in human dermal endothelial cells. Am J Pathol. 2006;169:697–707. doi: 10.2353/ajpath.2006.051211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ley K. Histamine can induce leukocyte rolling in rat mesenteric venules. Am J Physiol. 1994;267:H1017–23. doi: 10.1152/ajpheart.1994.267.3.H1017. [DOI] [PubMed] [Google Scholar]
- 56.Asako H, Kurose I, Wolf R, DeFrees S, Zheng ZL, Phillips ML, et al. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J Clin Invest. 1994;93:1508–15. doi: 10.1172/JCI117129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kubes P, Kanwar S. Histamine induces leukocyte rolling in post-capillary venules. A P-selectin-mediated event. J Immunol. 1994;152:3570–7. [PubMed] [Google Scholar]
- 58.Zimmerman BJ, Paulson JC, Arrhenius TS, Gaeta FC, Granger DN. Thrombin receptor peptide-mediated leukocyte rolling in rat mesenteric venules: roles of P-selectin and sialyl Lewis X. Am J Physiol. 1994;267:H1049–53. doi: 10.1152/ajpheart.1994.267.3.H1049. [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Wang L, Dou Y, Zhao J, Jiang T, Qiao Z, et al. Testosterone and estradiol modulate TNF-alpha-induced expression of adhesion molecules in endothelial cells. Methods Find Exp Clin Pharmacol. 2002;24:125–30. doi: 10.1358/mf.2002.24.3.802295. [DOI] [PubMed] [Google Scholar]
- 60.Gunawan RC, Almeda D, Auguste DT. Complementary targeting of liposomes to IL-1alpha and TNF-alpha activated endothelial cells via the transient expression of VCAM1 and E-selectin. Biomaterials. 32:9848–53. doi: 10.1016/j.biomaterials.2011.08.093. [DOI] [PubMed] [Google Scholar]
- 61.Uehara S, Chase CM, Kitchens WH, Rose HS, Colvin RB, Russell PS, et al. NK Cells Can Trigger Allograft Vasculopathy: The Role of Hybrid Resistance in Solid Organ Allografts. The Journal of Immunology. 2005;175:3424–3430. doi: 10.4049/jimmunol.175.5.3424. [DOI] [PubMed] [Google Scholar]
- 62.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, et al. Complement Independent Antibody-Mediated Endarteritis and Transplant Arteriopathy in Mice. American Journal of Transplantation. 2010;10:510–517. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. 2005;15:302–8. doi: 10.1016/j.tcm.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 64.Lepin EJ, Jin Y-P, Barwe SP, Rozengurt E, Reed EF. HLA class I signal transduction is dependent on Rho GTPase and ROK. Biochemical and Biophysical Research Communications. 2004;323:213–217. doi: 10.1016/j.bbrc.2004.08.082. [DOI] [PubMed] [Google Scholar]
- 65.Tran Q, Watanabe H. Calcium signalling in the endothelium. Handb Exp Pharmacol. 2006;176:145–87. doi: 10.1007/3-540-32967-6_5. [DOI] [PubMed] [Google Scholar]
- 66.Cioffi DL, Stevens T. Regulation of Endothelial Cell Barrier Function by Store-Operated Calcium Entry. Microcirculation. 2006;13:709–723. doi: 10.1080/10739680600930354. [DOI] [PubMed] [Google Scholar]
- 67.Koskinen PK, Lemstrom KB. Adhesion Molecule P-Selectin and Vascular Cell Adhesion Molecule–1 in Enhanced Heart Allograft Arteriosclerosis in the Rat. Circulation. 1997;95:191–196. doi: 10.1161/01.cir.95.1.191. [DOI] [PubMed] [Google Scholar]
- 68.Yamazaki S, Isobe M, Suzuki J, Tojo S, Horie S, Okubo Y, et al. Role of selectin-dependent adhesion in cardiac allograft rejection. J Heart Lung Transplant. 1998;17:1007–16. [PubMed] [Google Scholar]
- 69.Räisänen-Sokolowski A, Glysing-Jensen T, Russell ME. Donor and recipient contributions of ICAM-1 and P-selectin in parenchymal rejection and graft arteriosclerosis: insights from double knockout mice. The Journal of Heart and Lung Transplantation. 1999;18:735–743. doi: 10.1016/s1053-2498(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 70.Izawa A, Ueno T, Jurewicz M, Ito T, Tanaka K, Takahashi M, et al. Importance of Donor- and Recipient-Derived Selectins in Cardiac Allograft Rejection. Journal of the American Society of Nephrology. 2007;18:2929–2936. doi: 10.1681/ASN.2006111261. [DOI] [PubMed] [Google Scholar]
- 71.Bienvenu J-G, Tanguay J-Fo, ThéorÃat J-Fo, Kumar A, Schaub RG, Merhi Y. Recombinant Soluble P-Selectin Glycoprotein Ligand-1-Ig Reduces Restenosis Through Inhibition of Platelet-Neutrophil Adhesion After Double Angioplasty in Swine. Circulation. 2001;103:1128–1134. doi: 10.1161/01.cir.103.8.1128. [DOI] [PubMed] [Google Scholar]
- 72.Hayward R, Campbell B, Shin YK, Scalia R, Lefer AM. Recombinant soluble P-selectin glycoprotein ligand-1 protects against myocardial ischemic reperfusion injury in cats. Cardiovascular Research. 1999;41:65–76. doi: 10.1016/s0008-6363(98)00266-1. [DOI] [PubMed] [Google Scholar]
- 73.Wang K, Zhou Z, Zhou X, Tarakji K, Topol EJ, Lincoff AM. Prevention of intimal hyperplasia with recombinant soluble P-selectin glycoprotein ligand-immunoglobulin in the porcine coronary artery balloon injury model. Journal of the American College of Cardiology. 2001;38:577–582. doi: 10.1016/s0735-1097(01)01347-x. [DOI] [PubMed] [Google Scholar]
- 74.Zhou Z, Penn MS, Forudi F, Zhou X, Tarakji K, Topol EJ, et al. Administration of Recombinant P-Selectin Glycoprotein Ligand Fc Fusion Protein Suppresses Inflammation and Neointimal Formation in Zucker Diabetic Rat Model. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:1598–1603. doi: 10.1161/01.atv.0000032676.20514.8f. [DOI] [PubMed] [Google Scholar]
- 75.Tsuchihashi S-i, Fondevila C, Shaw GD, Lorenz M, Marquette K, Benard S, et al. Molecular Characterization of Rat Leukocyte P-Selectin Glycoprotein Ligand-1 and Effect of Its Blockade: Protection from Ischemia-Reperfusion Injury in Liver Transplantation. The Journal of Immunology. 2006;176:616–624. doi: 10.4049/jimmunol.176.1.616. [DOI] [PubMed] [Google Scholar]
- 76.Brandt M, Derner G, Boeke K, Phillips ML, Steinhoff G, Haverich A. Anti-rejection prophylaxis by blocking selectin dependent cell adhesion after rat allogeneic and xenogeneic lung transplantation. European Journal of Cardio-Thoracic Surgery. 1997;12:781–786. doi: 10.1016/s1010-7940(97)00251-0. [DOI] [PubMed] [Google Scholar]
- 77.Gaber OA, Mulgaonkar S, Kahan BD, Steve Woodle E, Alloway R, Bajjoka I, et al. YSPSL (rPSGL-Ig) for improvement of early renal allograft function: a double-blind, placebo-controlled, multi-center Phase IIa study. Clinical Transplantation. 2011;25:523–533. doi: 10.1111/j.1399-0012.2010.01295.x. [DOI] [PubMed] [Google Scholar]
- 78.Cheadle C, Watkins T, Ehrlich E, Barnes K, Osama Gaber A, Hemmerich S, et al. Effects of anti-adhesive therapy on kidney biomarkers of ischemia reperfusion injury in human deceased donor kidney allografts. Clinical Transplantation. 2011;25:766–775. doi: 10.1111/j.1399-0012.2010.01365.x. [DOI] [PubMed] [Google Scholar]
- 79.Busuttil RW, Lipshutz GS, Kupiec-Weglinski JW, Ponthieux S, Gjertson DW, Cheadle C, et al. rPSGL-Ig for Improvement of Early Liver Allograft Function: A Double-Blind, Placebo-Controlled, Single-Center Phase II Study. American Journal of Transplantation. 2011;11:786–797. doi: 10.1111/j.1600-6143.2011.03441.x. [DOI] [PubMed] [Google Scholar]
- 80.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, et al. In Vivo Platelet-Endothelial Cell Interactions in Response to Major Histocompatibility Complex Alloantibody. Circulation Research. 2008;102:777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 81.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, et al. Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2000;71:727–736. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 82.Tibben JG, Massuger LF, Boerman OC, Borm GF, Claessens RA, Corstens FH. Effect of the route of administration on the biodistribution of radioiodinated OV-TL 3 F(ab’)2 in experimental ovarian cancer. Eur J Nucl Med. 1994;21:1183–90. doi: 10.1007/BF00182351. [DOI] [PubMed] [Google Scholar]
- 83.Snapp KR, Craig R, Herron M, Nelson RD, Stoolman LM, Kansas GS. Dimerization of P-Selectin Glycoprotein Ligand-1 (PSGL-1) Required for Optimal Recognition of P-Selectin. The Journal of Cell Biology. 1998;142:263–270. doi: 10.1083/jcb.142.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cai YH, Alvarez A, Alcaide P, Duramad P, Lim Y-C, Jarolim P, et al. Abrogation of Functional Selectin-Ligand Expression Reduces Migration of Pathogenic CD8+ T Cells into Heart. The Journal of Immunology. 2006;176:6568–6575. doi: 10.4049/jimmunol.176.11.6568. [DOI] [PubMed] [Google Scholar]
- 85.Jones TR, Shirasugi N, Adams AB, Pearson TC, Larsen CP. Intravital microscopy identifies selectins that regulate T cell traffic into allografts. The Journal of Clinical Investigation. 2003;112:1714–1723. doi: 10.1172/JCI19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu SX, Holland AM, Na IK, Terwey TH, Alpdogan O, Bautista JL, et al. Absence of P-selectin in recipients of allogeneic bone marrow transplantation ameliorates experimental graft-versus-host disease. J Immunol. 185:1912–9. doi: 10.4049/jimmunol.0903148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L. P-selectin and MAC-1 mediate monocyte rolling and adhesion to ECM-bound platelets under flow conditions. Journal of Leukocyte Biology. 1998;64:467–73. doi: 10.1002/jlb.64.4.467. [DOI] [PubMed] [Google Scholar]
- 88.Hidalgo LG, Sis B, Sellares J, Campbell PM, Mengel M, Einecke G, et al. NK Cell Transcripts and NK Cells in Kidney Biopsies from Patients with Donor-Specific Antibodies: Evidence for NK Cell Involvement in Antibody-Mediated Rejection. American Journal of Transplantation. 2010;10:1812–1822. doi: 10.1111/j.1600-6143.2010.03201.x. [DOI] [PubMed] [Google Scholar]
- 89.Sheikh S, Parhar RS, Bakheet R, Saleh S, Collison K, Al-Mohanna F. Immobilization of rolling NK cells on platelet-borne P-selectin under flow by proinflammatory stimuli, interleukin-12, and leukotriene B4. Journal of Leukocyte Biology. 2004;76:603–608. doi: 10.1189/jlb.0204106. [DOI] [PubMed] [Google Scholar]
- 90.Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, et al. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–52. doi: 10.1182/blood-2006-03-010322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.