

Abstract
There is increasing clinical evidence that phospholipid oxidation products (Ox-PL) play a role in atherosclerosis. This review focuses on the mechanisms by which Ox-PL interact with endothelial cells, monocyte/macrophages, platelets, smooth muscle cells and HDL to promote atherogenesis. In the last few years major progress has been made in identifying these mechanisms. It has been recognized that Ox-PL promote phenotypic changes in these cell types that have long term consequences for the vessel wall. Individual Ox-PL responsible for specific cellular effects has been identified. A model of the configuration of bioactive truncated Ox-PL within membranes has been developed that demonstrates that the oxidized fatty acid moiety protrudes into the aqueous phase, rendering it accessible for receptor recognition. Receptors and signaling pathways for individual Ox-PL species are now determined and receptor independent signaling pathways identified. The effects of Ox-PL are mediated both by gene regulation and transcription independent processes. It has now become apparent that Ox-PL affects multiple genes and pathways some of which are pro-atherogenic and some are protective. However, at concentrations that are likely present in the vessel wall in atherosclerotic lesions, the effects promote atherogenesis. There have also been new insights on enzymes that metabolize Ox-PL and the significance of these enzymes for atherosclerosis. With the knowledge we now have on the regulation and effects of Ox-PL in different vascular cell types, it should be possible to design experiments to test the role of specific Ox-PL on the development of atherosclerosis.
Keywords: Ox-PAPC, atherosclerosis, inflammation, phospholipids
Evidence for a role of phospholipid oxidation products in atherosclerosis
The role of phospholipid oxidation products (Ox-PL) in atherogenesis was first suggested by the demonstration that pro-atherogenic activities of low-density lipoprotein (LDL) mildly oxidized by iron, myeloperoxidase or lipoxygenase were present in the fraction containing oxidized phospholipids.1 Subsequently, phospholipid oxidation products were shown to accumulate in hyperlipidemic plasma2, atherosclerotic lesions2 and in several diseases that predispose to heart attack, including lupus and rheumatoid arthritis.3 Oxidized phospholipids are also present in vivo in lipoprotein(a) [Lp(a)], in membranes of apoptotic cells, and cells undergoing oxidative stress.1
Evidence for the importance of these lipids in heart disease and atherosclerosis comes from several types of studies: 1) In a number of studies, the largest of which was the EPIC Norfolk study, levels of Ox-PL in plasma are associated with increased risk of coronary artery disease (CAD).4 It is however important to recognize that these studies employed E06 antibody which recognizes only a subset of Ox-PL. Ox-PL levels in plasma were also shown to increase during regression.5-7 In these papers they postulate that this increase represents a release from the vessel wall and may aid lesion regression. However, studies with statin have not yet shown a relationship between the Ox-PL increase in plasma by statins and the volume of atherosclerotic lesions in the coronary arteries. It will be important to resolve the issue of whether movement of lipids from the vessel wall is truly a measure of regression. 2) Studies of hyperlipidemic plasma from mice and humans suggest a role for phospholipid oxidation products in thrombosis. Hyperlipidemic animals had higher levels of Ox-PL in the plasma, and treatment with Ox-PL increased platelet activation.8 Platelet activation was also increased when using plasma from human donors with the highest levels of Ox-PL.8 3) Studies in mice also suggest a role for Ox-PL in atherosclerosis. Overexpression of one of the enzymes involved in formation of Ox-PL, myeloperoxidase (MPO), increases atherosclerosis9 and knockout of 12/15 lipoxygenase (12/15 LO), another enzyme shown to produce Ox-PL, decreases atherosclerosis.10 Plasma levels of MPO and its products predict an increase in CAD risk in healthy humans.9 4) D4F, a high-density lipoprotein (HDL) mimetic peptide, strongly protects against atherosclerosis in mice, and a major function of D4F is binding to phospholipid and fatty acid oxidation products.11 While these studies are highly supportive of a role for Ox-PL in atherosclerosis, they are not definitive. It will be important to demonstrate that manipulation of the receptors, signaling pathways and metabolism of Ox-PL affect atherosclerosis. Since two major reviews of Ox-PL were published in 2009/10, this review focuses on papers published in the last three years that address the role of Ox-PL in atherosclerosis.
Phospholipid Oxidation Products
There are a large number of phospholipid oxidation products that are formed from a variety of phospholipids containing polyunsaturated fatty acids. More than 30 different bioactive oxidation products have been identified that accumulate in lesions. Representative Ox-PL receiving the most study are shown in (Figure 1). Furthermore, the common and IUPAC names of the sn-2 position of these phospholipids, in addition to references for synthesis/identification and characterization are given in (Table 1). Ox-PL contain saturated fatty acids of different lengths at the sn-1 position, either acyl or alky esters, and contain phosphatidylcholine (PC), phosphatidylserine (PS) or phosphatidylethanolamine (PE) head groups.1, 2 In vitro, Ox-PL have been produced by a variety of methods of oxidation, all of which yield active products. The majority of studies of these molecules have been done on oxidation products of phospholipids with arachidonate or linoleate in the sn-2 position. However, important studies have also examined effects of phospholipids containing docosahexaenoic acid. Some Ox-PL contain fragmentation products of arachidonate or linoleate whereas others contain derivatives of arachidonate and linoleate that have become oxygenated. The structure of the lipid at the sn-2 position plays a major role in receptor recognition of Ox-PL. In general, differences in the sn-1fatty acid group of Ox-PL and have minor effects on action.2 Alkyl and acyl containing phospholipids bind with different affinities to Ox-PL receptors. The head group can mediate a difference in activity, though not all effects of Ox-PL are sensitive to head group changes. One example is that EO6 recognizes PC-containing but not PS- or PE-containing Ox-PL.12 Another example of head group preference is the differences in ability of oxidized PC-containing phospholipids (Ox-PC) and oxidized PS-containing phospholipids (Ox-PS) to block the uptake of apoptotic cells by macrophages.13, 14
Figure 1.
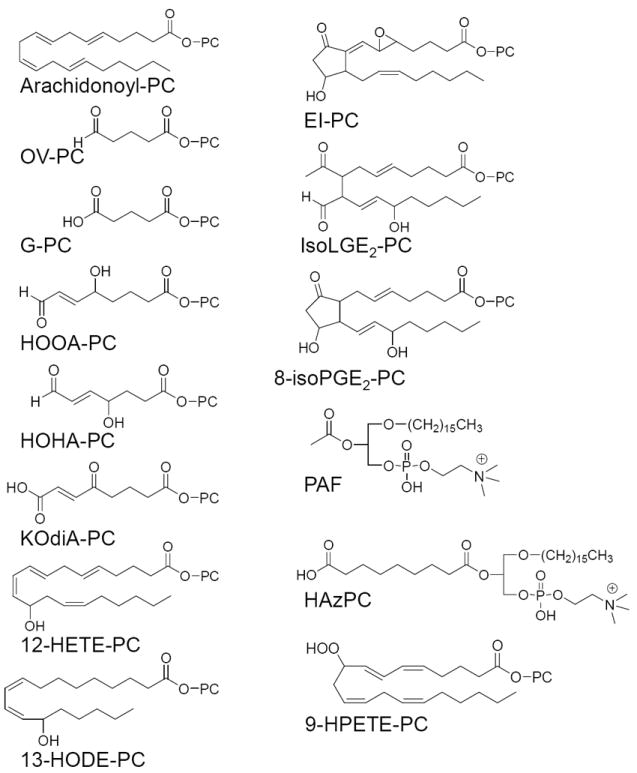
Ox-PL lipids. PC = 1-acyl-2-lyso-sn-glycero-3-phosphatidylcholine. Only the sn-2 position composition is shown for all Ox-PL except those forming an ether bond at the sn-1 position.
Table 1.
Ox-PL sn-2 position common and IUPAC names, references for identification/synthesis and characterization
| Sn-2 Common Name | Sn-2 IUPAC Name | Identified / Synthesized | NMR / MS characterization |
|---|---|---|---|
| OV | 5-oxovaleroyl | 123 | 123, 124 |
| G | glutaryl | 123 | 123 |
| HOOA | 5-hydroxy-8-oxooct-6-enoyl | 125, 126 | 125 |
| HOHA | 4-hydroxy-7-oxohept-5-enoyl | 127, 128 | 127 |
| KOdiA | 7-carboxy-5-oxo-hept-6E-enoyl | 129 | 129 |
| 12-HETE | 12(S)-hydroxyeicosatetraenoyl | 43, 130, 131 | 130, 131 |
| 13-HODE | 13(S)-hydroxy-(9Z,11E)-octadeca-9,11-dienoyl | 132, 133 | 124, 133 |
| EI | 5,6-epoxyisoprostane E2 | 134-136 | 134 |
| LGE2 | levuglandin E2 | 137 | 137 |
| 8-isoPGE2 | 8-isoprostaglandin E2 | 138 | 138 |
| Az | O-(9-carboxyoctanoyl) | 139 | 139 |
| 9-HPETE | 9-hydroperoxyeicosatetraenoyl | 140 | 140 |
Many Ox-PL are active electrophiles and can covalently bind to lysines, forming Schiff bases, or to cysteine or histidine, forming Michael Adducts (Figure 2). These interactions may result in covalent binding or reversible interactions. Some Ox-PL such as 1-palmitoyl-2-(5-hydroxy-8-oxooct-6-enoyl)-sn-glycero-3-phosphocholine (HOOA-PC), 1-palmitoyl-2-(4-hydroxy-7-oxohept-5-enoyl)-sn-glycero-3-phosphocholine (HOHA-PC) and isolevuglandin-PC can interact with proteins and form pyrrole groups (e.g. 2 (w-carboxyethyl)pyrrole (CEP)) which remain attached to the proteins (Figure 2).15, 16 Some of these covalently bound products act as ligands for receptor activation. The extent of covalent binding varies for different Ox-PL and is strongest for isolevuglandin-PC. Covalent binding can also activate cell surface proteins.17
Figure 2.
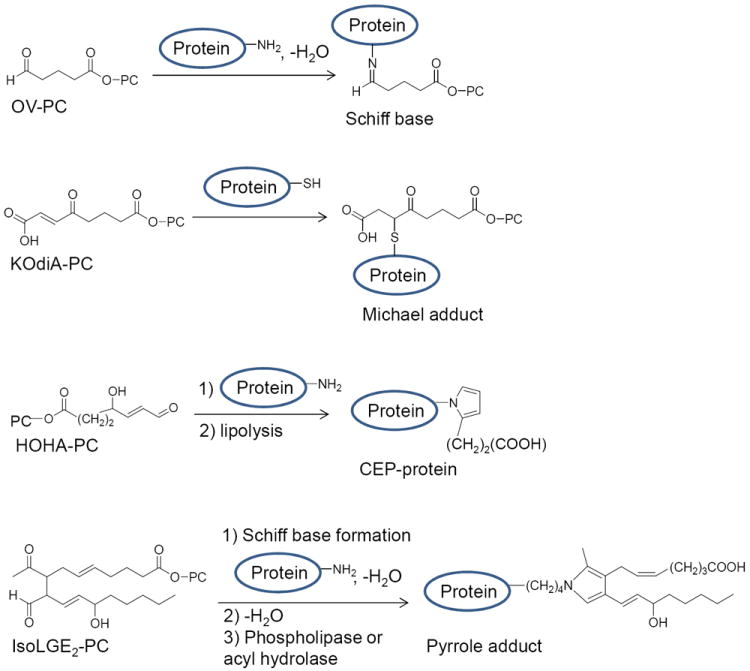
Interaction of Ox-PL lipids with proteins. Examples of three different types of interactions are shown. Only the composition of the sn-2 position is shown.
Summary of Vascular Cell and HDL Effects of Oxidized Phospholipids
Ox-PL have both atherogenic effects on cells and effects which have been shown to reduce atherosclerosis or protect cells against apoptosis (Table 2), but the net effect of Ox-PL on vascular wall cells and on HDL function is pro-atherogenic. This review focuses on effects of Ox-PL on endothelial cells, smooth muscle cells, platelets and macrophages. Although lymphocytes are important regulators of atherosclerosis18 we have not included lymphocytes in this review since there are only a few studies on Ox-PL regulation in lymphocytes. However, we have referred to the effects of antibodies to Ox-PL as inhibitors of atherosclerosis. The effects of these antibodies have recently been reviewed.19
Table 2.
Pro- and anti-atherogenic properties of oxidized phospholipids (Ox-PL)
| Pro-atherogenic: | |||
|---|---|---|---|
| Cell Type | Protein Regulated | Function | Reference |
| HAEC | IL-8, MCP-1, Gro α&β | Inflammation | 21 |
| Mouse macrophage | IL-1β, MIP-1, MIP-2, LTB4, RANTES | Inflammation | 30, 31 |
| Rat aorta SMC | MCP-3, MCP-1, VEGF | Inflammation | 38 |
| HAEC | CS-1 fibronectin | Inflammation | 84 |
| HAEC, HPAEC | Rho, VE-Cadherin | EC Barrier function | 24, 25 |
| HAEC | TF, TM, Serpin2 | Thrombosis | 21, 78, 79 |
| Mouse/human platelets | Hyperreactivity | Thrombosis | 8, 42 |
| Rat SMC | KLF4, collagen, FN | SMC replication/fibrosis | 38, 39 |
| Mouse macrophage | VAV, dynamin | Foam Cell formation | 71 |
| Mouse macrophage | UPR potentiation | Apoptosis | 32 |
| HL60 | Mit integrity | Apoptosis | 75 |
| HUVEC | NfκB/integrin | Angiogenesis | 49 |
| HUVEC | VEGFA | Angiogenesis | 22 |
| Anti-atherogenic or Mixed Effects: | |||
| Cell Type | Protein Regulated | Function | Reference |
| HAEC | Junction proteins | Barrier function | 85, 86 |
| HAEC | HO-1, NQO1, GCLM | Anti-Oxidant | 2, 21 |
| Human macrophage | HO-1, GCLM | Anti-Oxidant | 2, 21 |
| HAEC | UPR activation | Can be anti-apoptotic | 20, 21 |
| HAEC | Inhibitors of DNA replication | Anti-apoptotic | 1, 21 |
| Mouse macrophage | Uptake of apoptotic cells | Inhibit inflammation | 14, 27, 76 |
A. Endothelial cells
A number of papers over the last 12 years have documented the effects of Ox-PL on endothelial cells.1, 2 Taken together these studies suggest that PC-containing Ox-PL (e.g. oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (Ox-PAPC)) is a strong regulator of endothelial cell function. More than 1000 genes are regulated by treatment of human aortic endothelial cells (HAEC) for 4 hours with 40μg/ml Ox-PAPC.20, 21 80% of these genes were also regulated by one Ox-PAPC component, 1-palmitoyl-2-(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphocholine (PEIPC) (1-5μg/ml).21 The major gene ontology categories regulated by Ox-PAPC include sterol synthesis, unfolded protein response (UPR), redox signaling, inflammation, angiogenesis, cell division, endocytosis, filament formation, ecto-protease regulation and thrombosis. Some of these genes can perform protective functions, whereas others are atherogenic. Most regulated genes were in the “protective” categories. Array analysis of endothelial cells from 150 different human donors showed considerable differences between individuals in gene regulation by Ox-PAPC. Longer treatment with Ox-PAPC was shown to induce angiogenesis22 which leads to plaque instability23. Ox-PAPC and PEIPC also cause rapid effects on monolayer permeability, independent of mRNA synthesis. At low concentrations of lipid there was a protective effect on junctions whereas higher concentrations led to a decrease in the endothelial barrier.24, 25 Overall, these studies suggest that at early times of Ox-PAPC treatment the endothelium is subject to oxidative stress, and that this induces pathways to protect against the stress, such as the UPR response, an increase in antioxidant genes and decreased DNA replication. However, at the same time these cells up-regulate genes involved in inflammation, procoagulant activity and angiogenesis and also decrease endothelial barrier function. Taken together these studies suggest that, over the long term, endothelial cells survive Ox-PAPC injury and become inflammatory and angiogenic, contributing to plaque instability. It will be important to compare gene regulation in vitro to that found in endothelial cells of human and mouse lesions. Ambient air pollution has been associated with increased cardiovascular morbidity and mortality, and Ox-PAPC was shown by microarray analysis to synergize with fine particles in air pollutants to activate pro-atherogenic pathways in endothelial cells.26
B. Macrophages
Another cell type where multiple effects of Ox-PL have been observed is macrophages. Early studies from the Witztum group demonstrated that uptake of Ox-LDL led to foam cell formation, and they subsequently identified 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC) as one lipid responsible for foam cell formation.27 The E06 antibody, which binds to a number of short chain Ox-PL, could block foam cell formation by macrophages. Podrez et al.28 demonstrated that at least eight Ox-PL that contain fragmentation products of arachidonate and linoleate interacted with the CD36 receptor on macrophages, resulting in rapid oxidized low-density lipoprotein (Ox-LDL) uptake and foam cell formation. These lipids were enriched in human atherosclerotic lesions.29 Once monocytes enter lesions, they differentiate to macrophages and can produce cytokines. Ox-PAPC induces the production of macrophage cytokines.30 Platelet activating factor (PAF) and PAF-like Ox-PL can also increase synthesis of monocyte chemotactic protein 1 (MCP-1) in several types of leukocytes.31
Ox-PL also have effects on the initiation of apoptosis in macrophages32 and the uptake of apoptotic cells by macrophages.14, 33 Macrophage apoptosis has been shown to play an important role in atherosclerosis. There is evidence that if it occurs at early times, atherogenesis may be inhibited; whereas if apoptosis occurs after foam cell formation, it may increase the formation of the necrotic core and increase atherogenesis.
The differentiation of macrophages and dendritic cells has also been shown to be affected by Ox-PL. Kadl et al34 described a novel macrophage phenotype (named Mox) that develops in response to treatment with Ox-PAPC. Treatment of M0, M1 or M2 macrophage subtypes with Ox-PAPC resulted in the Mox phenotype.34 These Mox cells exhibited a lower capacity for migration, phagocytosis, and production of inflammatory stimuli. They were shown to contain high levels of antioxidant enzymes. These macrophages represent about 30% of CD11B+/F480+ cells in mouse atherosclerotic lesions and may contribute to the buildup of macrophages in lesions.
Dendritic cells in apoE null mice have a diminished ability to promote T-cell responses.35 Oxidized phospholipids were shown to dysregulate the differentiation of dendritic cells from monocytes in culture.36, 37 Ox-PAPC addition to dendritic cell differentiation medium caused the production of dendritic cells that did not express CD1a, CD1b or CD1c; however other dendritic markers were still induced. Thus these Ox-PAPC-treated dendritic cells were not able to present lipid antigens and thus could not mount an antibody response to these antigens. These authors present evidence that Ox-PL inhibits the phosphorylation of Histone H3, which has previously been shown to regulate differentiation of dendritic cells by CD40 ligand.36 Ox-PL also inhibited CD1 expression in dendritic cells treated with mycobacteria37, and CD1 was inhibited in lepromatous lesions which contain high levels of Ox-PL as seen with E06 staining.
C. Smooth muscle cells (SMCs)
Ox-PAPC promotes phenotypic switching in smooth muscle cells, including suppression of contractile SMC differentiation marker genes.38 POVPC and PEIPC, as well as Ox-PAPC, also increased expression of matrix protein type VIII collagen and fibronectin, which increases SMC migration.39 POVPC changed the phosphorylation state of connexin 43, which stimulates SMC replication.40 All of these effects were observed in vitro as well as in vivo and reflect a switch to an atherogenic phenotype. Ox-PAPC was also shown to be involved in smooth muscle cell apoptosis.41 In human lesions extensive SMC death is observed.
D. Platelets and Thrombosis
Thrombotic processes are also induced by Ox-PL. As discussed above, hyperlipidemic serum increases platelet reactivity.8 Several individual Ox-PL, found in hyperlipidemic plasma and which can bind to CD36, were shown to sensitize platelets to adenosine diphosphate (ADP)-induced aggregation. Another study demonstrated the importance of the PAF receptor in the regulation of platelet activation by alkyl Ox-PL.42 Thus more than one receptor appears to contribute to the hyper-aggregation of platelets in hypercholesterolemia. In addition, several groups have shown that platelets actually form 12S-hydroxyeicosatetraenoic acid (12-S-HETE)-containing oxidized phospholipids after exposure to thrombin, ionophore or collagen.43 Furthermore, liposomes containing these phospholipids are able to enhance tissue factor-dependent coagulation.
In microarray studies, Ox-PAPC treatment of HAEC from 150 donors was shown to regulate the levels of major thrombogenic molecules.21 Ox-PAPC significantly downregulated thrombomodulin (TM) expression by 40% while upregulating tissue factor and Serpin B2 by 70%. Taken together, these platelet and thrombotic changes could play a major role in CAD.
E. Ox-PAPC interactions with HDL
In addition to the pro-atherogenic effects of Ox-PL on cells, there is evidence that the binding of Ox-PL to HDL can decrease HDL anti-inflammatory and cholesterol transport functions.44 HDL was shown to be a repository for lipid hydroperoxides.45 Hydroxynonenal (HNE), an oxidized fatty acid, binds to apolipoproteinA I (apoA-I) and regulates HDL’s cholesterol transport and inflammatory functions.44 Since most polyunsaturated fatty acids in living systems are complexed with phospholipids, it was important to examine the binding of Ox-PL to apoA-I. Using a biotinylated-sulfoxide analogue of 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine (PLPC), Szapacs et al. demonstrated binding of several Ox-PL, derived from the biotinylated lipid, to apoA-I at important functional sites in the molecule.46 These included sites involved in lecithin-cholesterol acyl transferase (LCAT) binding and activation. It will be important to also determine the effects of covalent lipid binding on the anti-inflammatory effect of HDL. The importance of oxidized fatty acids and Ox-PL in regulating HDL function is suggested by studies demonstrating that D4F, which binds fatty acid oxidation products whether free or esterified to phospholipids, protects against the pro-inflammatory effects of dysfunctional HDL.11 Overall, Ox-PL can mediate pro-atherogenic changes of all the cell types examined and inhibits the protective functions of HDL.
Identifying the receptors and signaling pathways regulated by Ox-PL
In order to develop strategies for inhibiting the effects of oxidized phospholipids, it is important to identify the most active Ox-PL species and determine their mechanisms of action. There are a number of issues that need to be considered when analyzing effects of Ox-PL. Many studies have been performed using mixtures of oxidized lipids such as mildly oxidized LDL, Ox-PAPC and Ox-PLPC (oxidized 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine). Others have used pure molecular species of Ox-PL. Lipid mixtures have been useful in identifying major processes regulated by Ox-PL, and it is likely that Ox-PL are presented to cells as mixtures. However, in order to identify particular Ox-PL causing a given effect on cells, these lipids must be studied in isolation from other oxidized phospholipids.
Another issue when testing lipid mixtures or individual lipids is how much lipid should be used. This is a difficult issue because, until recently, methods for determining absolute levels of these lipids in vivo were not sufficiently accurate or available. While methods for measuring free Ox-PL have improved, it is important to recognize that many Ox-PL can covalently bind to proteins and the levels of these bound lipids will not be measured by the lipid extraction method. In spite of these problems, several papers give levels that can guide studies of mechanism. In more recent studies the levels of individual Ox-PL in atherosclerotic lesions have been reported to be approximately 1-3μM in human and mouse lesions.47 The values of Ox-PL in lesions may be considerably higher than these values because whole aortic arches were employed in mice and carotid endarterectomy specimens in humans. A large fraction of these tissues are often not actively engaged in atherogenesis. In plasma, levels were approximately 10% of those in lesions.47 In another study the combined values of Ox-PAPC, which bound to CD36 ranged from 5-51μM in plasma.8 In selecting the appropriate concentration of an individual lipid to employ, one must consider that in lesions there may be a number of atherogenic Ox-PL that have similar functions. Therefore it may be necessary to employ higher levels of individual phospholipids to obtain a maximal response. Also, when using mixtures of Ox-PL, such as Ox-PAPC, the concentration to be used will be higher than for individual active Ox-PL because some lipids in the mixture may have low activity.
Two other important issues, the time of treatment and cell type employed, are critical considerations in deciding on an experimental design. While this is true for all cell treatments in general, it is especially true for Ox-PL since they may have opposite effects at different times after treatment and very different effects in different cell types. For example after 4 hours of treatment of endothelial cells with Ox-PL replication is inhibited. However, after 24 hours angiogenesis is stimulated.22 The number of genes regulated by Ox-PAPC after a 4 hour treatment of human aortic endothelial cells is much higher than that seen in human macrophages treated for the same time with the same concentration (Figure 3). Whole gene ontology categories regulated by Ox-PL in endothelial cells are not regulated in macrophages.
Figure 3.
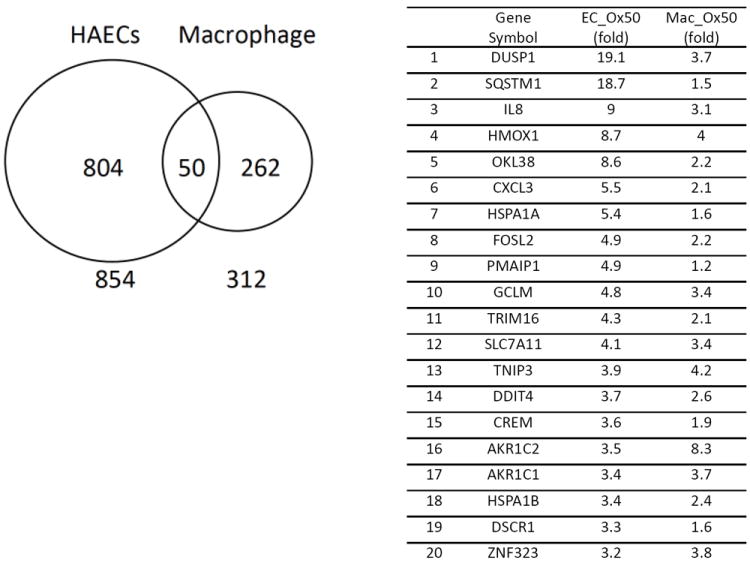
Comparison of the effect of Ox-PAPC treatment (50μg/ml for 4 hours) on gene regulation in human endothelial cells and human macrophages. A list of the top 20 genes in the overlap category is shown.
A. Ox-PL primary interactions: receptor and non-receptor mechanisms
A number of studies have addressed the question of the primary event regulating the interaction of Ox-PL with cells. Ox-PL also interact with soluble receptors and this area has been recently reviewed.19 Cell receptors binding specific Ox-PL or groups of Ox-PL have been identified (CD3613, 28, scavenger receptor B1 (SRB1)48, TLR232, 49, TLR450-52, EP253). The PAF receptor, PAF binding, and the internalization receptor transmembrane protein 30A (TMEM30a) have also been shown to play a primary role in the action of Ox-PL.54, 55 The relative roles of these receptors in the activation of specific pathways may differ. In addition, recent information suggests that non-receptor-mediated events may be causal in the initial response to selected Ox-PL. These include changes to the lipid composition and fluidity of the plasma membrane subsequent to incorporation of Ox-PL56-58 and covalent interaction of Ox-PL with cell surface proteins.17, 32 Identification of the primary event in Ox-PL regulation of cells is an important therapeutic goal.
B. Configuration of the Ox-PL ligand
An important consideration in determining the mechanism by which Ox-PL interact with their receptors is the configuration of the Ox-PL ligand. Cells likely see phospholipid oxidation products in membranes, for example the membranes of apoptotic cells or membrane containing structures like LDL. Studies from the Hazen and Salomon labs59 have produced a model of the configuration of specific phospholipid oxidation products in membranes, referred to as the Lipid Whisker model. For these studies, they employed vesicles of unoxidized PL containing 5-20mol% of specific Ox-PL. They used NMR employing nuclear overhauser effect (NOE) to define the configuration of the specific Ox-PL in membranes. They showed that the addition of polar oxygen atoms to fragmentation products of arachidonate and linoleate reorients the sn-2 acyl chain. This increases the hydrophilicity of this group causing it to protrude into the aqueous phase where the head group is located. This contrasts with the sn-2 chain of the unoxidized parent phospholipid (PAPC), which is buried in the lipid bilayer. The fact that the head group and sn-2 fatty acid oxidation products are both in the aqueous phase may account for the fact that, in some situations, both are important for recognition. The presence of the fragmentation product in the aqueous phase, rather than buried in the lipid bilayer (as seen with PAPC or 1-palmitoyl-2-(13(S)-hydroxy-(9Z,11E)-octadeca-9,11-dienoyl)-sn-glycero-3-phosphocholine (13-HODE-PC)), enables contact between the receptor and the sn-2 position of the ligand (Figure 4). This same sn-2 configuration was seen with PC- and PE-containing Ox-PL. The configuration of Ox-PL containing alkyl fatty acid at the sn-1 position has not been determined but the protrusion of the sn-2 group into the aqueous phase may also be relevant for these lipids.
Figure 4.
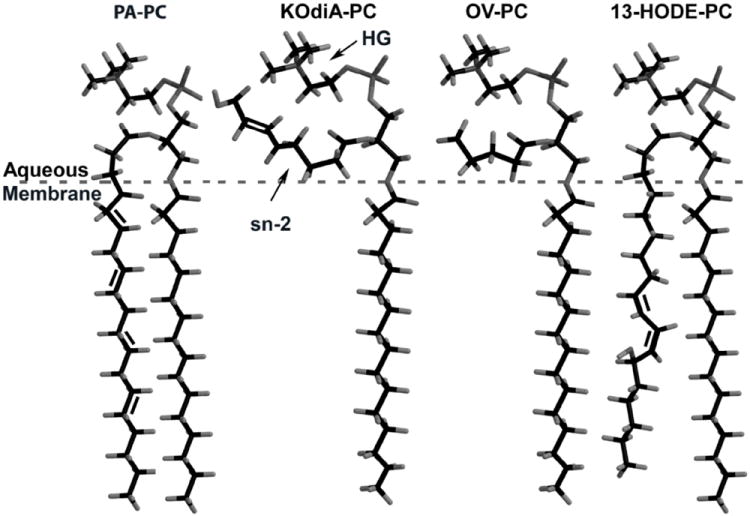
Lipid Whisker Model of the conformation of PAPC, 1-palmitoyl-2-(7-carboxy-5-oxo-hept-6E-enoyl)-sn-glycero-3-phosphocholine (KOdiA-PC), POVPC and 13-HODE-PC within membranes. An arrow points to the sn-3 and head group positions of KOdiA-PC.
C. Cell surface receptor activation by Ox-PL
There are multiple receptors regulating Ox-PAPC action. Some of these receptors appear to act alone while others require the presence of receptor complexes. Most of the initial studies were done in vitro but the importance of some of these receptors in atherosclerosis has been confirmed in vivo in mice.
1) CD36
The most well studied receptor for Ox-PL is CD36. The Ox-PL involved in the binding to CD36 contains fragmentation products of arachidonate and linoleate that contain a terminal acid or aldehyde. This group of CD36-binding phospholipids is referred to as a group of Ox-PC binding to CD36 (Ox-PCCD36).28 Using model phospholipids Gao et al60 showed that a carboxylate tail at the terminal of the sn-2 position is most important for binding and that α,β unsaturation also plays a role as did chain length and the charge at the sn-3 position.60 Several lysines in CD36 were shown to be required for the interaction with Ox-PCCD36. CD36 also binds to oxidation products of docosahexaenoic acid that contains a terminal acid or aldehyde.61 Knockout of CD36 in mice modestly decreased the atherosclerosis.62
2) SRB1
Several of the Ox-PCCD36 lipids were also shown to bind to SRB1. In general, the binding to SRB1 showed comparable binding requirements to CD36 but SRB1 had a lower binding affinity for Ox-PL. Furthermore SRB1 requires longer chains at the sn-2 position for optimal binding.60 The Ox-PCCD36 binding site on SRB1 was identified and shown to also be a binding site for HDL. For this reason, treatment of hepatocytes with Ox-PCCD36 blocked SRB1-mediated uptake of cholesteryl esters.60
3) TLR2 and TLR4
There is evidence that TLR2 (Toll Receptor 2)30, 32, 49 and TLR450-52 may act as Ox-PL receptors. TLR2 was involved in the initiation of angiogenesis by the interaction of carboxyethylpyrrole (CEP)-containing proteins with TLR2.49 CEP-bound proteins and CEP itself were able to interact with and activate TLR2. In a separate study a role for TLR2 in the macrophage inflammatory response to Ox-PAPC was demonstrated.30 Treatment of mouse macrophages with Ox-PAPC caused an increase in expression of the inflammatory cytokines, and this was not observed in TLR2 null mice. This effect was stronger for Ox-PAPC lipids (like PEIPC) containing reducible groups in the sn-2 position and was inhibited by reduction of the lipid with sodium borohydride (NaBH4). Knockout of TLR2 in mice was shown to cause a general decrease in early atherosclerosis. However, the major effect was not mediated by bone marrow derived cells.63 It is possible that both roles for bone marrow-derived and non-bone marrow-derived effects are important at different stages of atherosclerosis. TLR4 has a role in Ox-PAPC activation of Hela cells52 and macrophages.50, 51 The binding of Ox-LDL to CD36 mediated increases in chemokines GRO1, MIP-2 (macrophage inhibitor protein 2) and RANTES (regulated upon activation, normal T-cell expressed, and secrete) in macrophages. This increase was inhibited when TLR4 or TLR6 were absent from the macrophages.51 These authors determined that a complex of CD36 with TLR4 and TLR6 was necessary for this activation and that Lyn (tyrosine protein kinase Lyn) activation in the complex was also required. The role of Ox-PL in the Ox-LDL activation was not determined. Dominant negative and antisense oligonucleotides to TLR4 blocked the effect of Ox-PAPC and POVPC on activation of the IL-8 promoter in Hela cells.52 Studies of the role of TLR4 have, however, been inconsistent in human aortic endothelial cells and may depend on the presence of co-receptors. Knockout of TLR4 in apoE null mice fed a chow diet caused a decrease in early atherosclerosis without a change in cholesterol levels.64 Some human studies suggest that a TLR4 polymorphism may be associated with atherosclerosis. It may be important in human studies to examine a combined risk of TLR4 and TLR6 polymorphisms.
4) EP2
The receptors described above all recognize multiple Ox-PL. A more specific receptor that has been identified is EP2, a prostaglandin and isoprostane receptor. The role of this receptor in Ox-PAPC action was examined because PEIPC contains an E2isoprostane group. Ox-PAPC and PEIPC were shown to bind to EP2 and this interaction regulates monocyte binding to endothelial cells.53 Prostaglandin E2 (PGE2) has been shown to have an effect on monocyte binding similar to PEIPC. However, EP2 was subsequently shown to regulate only a small subset of the PEIPC effects on HAEC. These studies have been done only in vitro and a role for EP2 should be examined in mouse models.
5) PAF receptor (PAFR)
The most extensively studied Ox-PL are acyl phospholipids that contain an ester-bonded fatty acid at the sn-1 position of the molecule. However, there are a comparable number of alkyl oxidized PL with ether bonds at the sn-1 position. Though the levels of these alkyl Ox-PL are much lower, they are highly potent activators of leukocyte interaction with endothelial cells and regulators of platelet function. The effects of these lipids have been shown to be regulated by the PAF receptor.54 There is also evidence that acyl Ox-PL may bind to the PAF receptor since the effects of Ox-PAPC and POVPC and several other acyl phospholipids can be inhibited by PAF receptor antagonists.54 Furthermore, WEB2086, a PAF receptor antagonist, decreases atherosclerosis in mice.65 The fact that a PAF receptor antagonist inhibits the development of atherosclerosis suggests that alkyl-linked Ox-PL as well as acyl-linked Ox-PL are important in the early stages of atherosclerosis.
6) TMEM30a
Several studies have suggested that phospholipid oxidation products can cross the plasma membrane. Studies using tagged Ox-PAPC showed its presence in several cell membrane compartments.66, 67 TMEM30a is a receptor that transports PC- and PE-containing phospholipids into cells. It is found in the plasma membrane and in membranes of several cell organelles. In vivo, PAF-AH (platelet activating factor acetyl hydrolase) was shown to play only a small role in the clearance of PAF and PAF-like alkyl Ox-PL from the blood, with transport into cells being more important in clearance. PAF and PAF-like lipids were mainly taken up into cells by TMEM30a.68 Knockdown of TMEM30a reduced the apoptotic effect of an endogenous alkyl phospholipid oxidation product. It remains to be determined if acyl Ox-PL can also be taken up by this receptor or by an alternate receptor. It would be important to confirm a role for this receptor in atherosclerosis in mice.
D. Non-Receptor Mechanism
1) Covalent binding to cell surface proteins
The importance of covalent or strong interactions with cell surface proteins as regulators of Ox-PL action has received increasing attention over the last several years. The study discussed above concerning the interaction of CEP with TLR2 provides evidence that such interactions occur in vivo and are important in the action of Ox-PL.49 In a number of diseases, including atherosclerosis, pyrroles have been shown to accumulate and may serve as disease markers.16
With respect to covalent binding of other Ox-PL to cell surface proteins, the binding of Ox-PAPE-N biotin (oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl-(N-biotinylethanolamine), which has similar activities to Ox-PAPC) to approximately 20 proteins in endothelial cells was reported several years ago.66 Recent studies have shown that Ox-PAPC can covalently interact with and activate two specific metalloproteinases, a dysintegrin and metalloproteinase 10 (ADAM10) and a dysintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4), which are present on the cell surface.17 This activation results in an increase in the transcription of pro-inflammatory genes including IL-8 and MCP-1. It would be important to look for covalent binding to ADAM10 and ADAMTS4 in vivo. A recent review has focused on protein modification by aldehydophospholipids and its functional consequences.69
2) Cholesterol depletion and membrane stiffening
Several groups have reported changes in membrane characteristics when Ox-PL, Ox-LDL or oxysterols are incorporated into the membrane.56, 57 A major finding in pig aortic endothelial cells is an increase in membrane disorder, increase in cell stiffness and a decrease in the cholesterol content of caveolar and non-caveolar lipid rafts after exposure of the cells to Ox-LDL.56 Although it is counter intuitive, three pieces of evidence suggest that cholesterol depletion has occurred in lipid rafts of these endothelial cells after treatment with Ox-LDL. First, the distribution of raft markers is changed. Second, Ox-LDL exposure causes physical changes to the membrane consistent with cholesterol loss. Third, the effects of Ox-LDL can be mimicked by cholesterol depletion with methyl-β-cyclodextrin (MBCD).56 One possible cause of this effect on membrane structure is an insertion of oxysterols into the membrane56. Another mechanism may relate to the observation that incorporation of Ox-PL into lipid monolayers results in monolayer expansion and subsequent loss of Ox-PL from the monolayer, thereby changing monolayer characteristics.57 In HAEC, Ox-PAPC causes caveolar cholesterol depletion and an increase in sterol regulatory element binding protein (SREBP) activation.58 Exposing HAEC to cholesterol reversed the effect of Ox-PAPC on IL-8 induction. There is also in vivo evidence for a decrease in membrane cholesterol in atherogenic conditions. First, in apoE null mice fed a high cholesterol diet, there is a decrease in endothelial caveolae on the cell surface.70 Second, SREBP is activated in areas of human atherosclerotic lesions where inflammatory cells are present.58 Taken together, these studies suggest that changes in cholesterol and in the fluidity of membranes where signaling molecules are present may lead to an increase in gene expression and other changes to endothelial cell function in response to Ox-PL. A study in human lesions examining the endothelial expression of several genes controlling cholesterol levels would clarify the role of cholesterol in regulating different stages of the disease.
E. Signaling
A number of signaling pathways have been shown to be activated by Ox-PL. Changes in second messenger occur as early as 2 minutes after treatment, and secondary signals may be sustained for many days. For some signaling events the entire process from the receptor to the downstream function has been identified. In other cases only part of the pathway has been determined. Since various parts of the pathway are potential drug targets, we have presented both complete and incomplete pathways. Some signaling studies have been done with Ox-LDL and may or may not apply to effects of oxidized phospholipids since Ox-LDL contains both sterol and phospholipid oxidation products. Therefore, we have only included Ox-LDL studies where the receptor or signaling pathway is also known to be activated by oxidized phospholipids.
1) Macrophages
Foam cell formation
CD36 is recognized to play a major role in foam cell formation.
Originally CD36 was thought to be a scavenger receptor and not a signaling receptor. It is now recognized that CD36 activation by Ox-PCCD36 lipids has important signaling functions that affect both foam cell formation and platelet activation. The ability of Ox-PCCD36 to stimulate foam cell formation was shown to relate to a stimulation of endosomal vesicle trafficking.71 This pathway involves the rapid activation of Lyn followed by the activation of VAV family of guanine nucleotide exchange factor (VAV) proteins that are known to be involved in receptor ligand endocytosis in other cell types. These VAV proteins constitutively interact with dynamin2. Upon activation of CD36, dynamin is phosphorylated and the dynamin/VAV complex moves from the plasma membrane to the perinuclear area. There is also an increase in Ca2+ entry into the cells that increases endocytosis and provides positive feedback in the process.
A secondary pathway regulated by C-Jun N-terminal kinase (JNK) phosphorylation also appears to be important since JNK null mice have decreased foam cell formation in vitro and in vivo. It will now be important to examine the importance of this pathway in a mouse model of atherosclerosis and determine if this pathway is activated in human lesions.
Increase in cytokine expression
TLR2 was found to be involved in the pro-inflammatory Ox-PAPC response in macrophages, including induction of cyclooxygenase 2 (COX2), IL-1β, MIP-2 and keratinocyte chemoattractant (KC).30 The response to high molecular weight species of Ox-PAPC, such as PEIPC, was blunted in bone marrow derived macrophage (BMDM) from TLR2-/- mice as compared to wild type mice. P38 and JNK are phosphorylated in wild type but not TLR2 knockout macrophages, suggesting that these proteins are involved in the pro-inflammatory response. The induction of the pro-inflammatory genes did not involve activation of NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells). Studies cited above63 suggest that macrophage TLR2 is not involved in atherosclerosis. However, there may be compensatory mechanisms in the TLR2 knockout mice. An examination of the pathways identified by Kadl et al. in macrophages from TLR2 WT mice might help to resolve the question of whether macrophage TLR2 is an important regulator of atherosclerosis.
Ox-PL activation of the PAF receptor with alkyl Ox-PL was shown to increase chemokine synthesis in both monocyte and neutrophils. Treatment with Ox-PL increased the levels of MIP-1 (macrophage inhibitor protein 1), MIP-2, and MCP-1. Levels of leukotriene B4 (LTB4), another monocyte chemotactic factor, were also increased.31, 72 The induction of MCP-1 synthesis was shown to require both NFκB activation and Ca2+/calcineurin signaling pathways. A dose response study comparing the effects of alkyl and acyl Ox-PL on chemokine synthesis by macrophages would suggest the relative roles of the PAF and TLR2 receptors.
One of the most important chemotactic factors for monocytes in atherosclerosis is MCP-1. MKP1 (mitogen activated protein kinase phosphatase 1) is highly and rapidly induced by Ox-PAPC and plays an important role in Ox-PAPC induction of MCP-1 in endothelial cells and macrophages.73 The exact mechanism controlling this regulation is not known. However, knockout of MKP1 in apoE null mice fed a chow diet for 24 weeks caused a major reduction in levels of MCP-1 in plasma and also a decrease in the size of atherosclerotic lesions.73 A microarray study on the effect of MKP1 might aid in the understanding of the regulatory effect on this molecule.
Apoptosis
Ox-PL stimulates macrophage apoptosis and play an important role in the uptake of apoptotic cells by macrophages. CD36 and TLR2 play important roles in the ability of Ox-PAPC to facilitate apoptosis in connection with the unfolded protein response (UPR).32 In cells with either CD36 or TLR2 knockout, the effect of Ox-PAPC on apoptosis was strongly inhibited. A TLR2/TLR6 heterodimer was necessary for this response. Ox-PAPC could be delivered either as free molecules or in combination with Lp(a), which carries Ox-PAPC in plasma. Ox-PAPC was not the only lipid that accelerated apoptosis. They observed that reactive oxygen species (ROS) were increased in Ox-PAPC-treated cells activation of NADPH oxidase 2 (NOX2) was necessary for ROS formation and the apoptotic response. The response did not develop in cells from NOX2 null animals. ERK/MAPK (extracellular-signal-regulated kinases/mitogen activated protein kinase) activation was necessary for Ox-PAPC-induced increases in ROS and apoptosis.
Macrophage apoptosis in advanced lesions of TLR2/TLR4 deficient mice was decreased. The relevance of this pathway in human lesions should be examined by staining human lesions for NOX2 and ERK activation in apoptotic macrophages. TLR4 was mainly responsible for apoptosis of macrophages undergoing efferocytosis due to Ox-PAPC treatment.74In HL-60 cells (a pre-myelocytic cell line) treated with hexadecylazelaoyl PC (HAz-PC) (an alkyl phospholipid as opposed to the acyl phospholipids used for the previous study) apoptotic death was shown to be increased by affecting mitochondrial integrity.75 This effect depended on uptake by TMEM30a. It will be important to determine whether TMEM30a is expressed in human macrophages undergoing apoptosis.
The uptake of apoptotic cells by macrophages has also been shown to be regulated by Ox-PL. Several groups have shown that during apoptosis macrophages produce Ox-PL.14, 27, 76 This Ox-PL is present on the surface of the apoptotic cells and contributes to their uptake by macrophages. PS-containing Ox-PL are more active than PC-containing phospholipids, however both are reactive. CD36 is strongly involved in the binding and uptake of apoptotic cells expressing Ox-PS/PCCD36.13, 14 The uptake of apoptotic cells by CD36 has the potential to be anti-atherogenic, while the uptake of lipid into foam cells is pro-atherogenic. The combination of these two functions may explain why CD36 knockout does not have a more robust effect in decreasing atherosclerosis. It would be interesting to use a CD36 inhibitory peptide to test effects of the receptor at different stages of atherosclerosis.
2) Platelet Activation and Thrombosis
Both the CD36 and PAF receptor were shown to play a role in platelet hypersensitivity to agents causing platelet aggregation. CD36 interaction with Ox-PCCD36 or Ox-LDL was shown to be involved in the hypersensitivity of hyperlipidemia.14 The signaling pathway by which platelets are activated by Ox-LDL interaction with CD36 has many similarities to the regulation of foam cell formation by Ox-LDL.77 CD36 engagement of Ox-LDL increased the activity of Fyn as shown by Fyn phosphorylation. This led to interaction of Fyn with VAV1 and phosphorylation of VAV1. Mice with deletion of VAV1 and VAV3 did not show platelet hypersensitivity. The exact mechanism by which VAV proteins affect platelets has not been identified. Previous studies have also shown that JNK activity is necessary for platelet hypersensitivity. Effects of a platelet specific knockout of VAV1 would clarify the issue of the direct involvement of platelet VAV in hypersensitivity.
The PAF receptor also contributes to early stages of platelet hypersensitivity. There is evidence that platelets can be activated by ether-containing oxidized phospholipids, one of which is HAz-PC.42 Treatment of platelets with HAz-PC leads to a priming of platelet activation to a sub-threshold level of thrombin, collagen and ADP. This leads to surface expression of P-selectin and an increase in calcium flux, which leads to platelet aggregation. Low levels of HAz-PC (10-7M) could induce this response, which was abolished at higher concentration. The effects of HAz-PC were reversed by PAFR antagonist WEB2086. A number of ether lipid oxidation products were effective and the effect did not correspond to chain length. The authors of this study speculate that, as more ligands binding CD36 accumulate in the vessel wall, CD36 becomes the dominant receptor mediating hypersensitivity.
Transcription of at least three proteins important in thrombosis is regulated by Ox-PAPC. Thrombomodulin down-regulation was shown to be regulated by a decrease in transcription mediated by decreased binding of retinoic acid receptor (RAR), retinoid X receptor (RXR), specificity protein 1 (Sp1) and specificity protein 3 (Sp3) to elements in the TM promoter.78 This decrease in binding was mostly due to decreased levels of these transcription factors in the nucleus. Ox-PAPC also strongly increases the levels of tissue factor mRNA.79 This occurs by activation of PKC, which then activates ERK within 10 minutes of Ox-PAPC treatment. ERK was shown to activate the transcription factor EGR1, which then binds to the TF promoter. NGFI-A binding protein 2 (NAB2) overexpression, which inhibits EGR1 activation, inhibited the effect of Ox-PAPC on tissue factor mRNA. 1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine (PGPC) but not POVPC mimicked this effect. Vascular endothelial growth factor receptor (VEGFR2) activation by Ox-PAPC also mediates ERK phosphorylation. Furthermore, siRNA to VEGFR2 inhibited the induction of tissue factor by Ox-PAPC.80 This suggests that VEGFR2 is the receptor mediating the pathway involved in EGR1 (early grown factor response protein 1) regulation of tissue factor.
A second pathway of Ox-PAPC regulation of tissue factor also involved Ox-PAPC activation of calcium entry into the cells, which then leads to nuclear factor of activated T cells (NFAT) translocation and binding of NFAT to the TF promoter.79Cyclosporin, which inhibits NFAT binding, decreased the effect of Ox-PAPC.
Immunohistochemical staining of human lesions for the activation of these pathways would provide evidence for their potential role in human atherosclerosis.
Ox-PL containing a terminal 9 carbon aldehyde or carboxylic acid, but not those containing a 5 carbon aldehyde or those derived from Ox-PAPC, inhibit the activity of isolated TFPI (tissue factor protein inhibitor, which inhibits the activity of tissue factor).81 The mechanism inhibiting the activity was direct binding to the active TFPI domains. Thus, procoagulant activity is promoted by Ox-PL regulation of multiple cell-associated proteins in the coagulation pathway.
3) Endothelial Cells
A large number of endothelial cell functions are regulated by a 4 hour treatment of HAEC with Ox-PAPC. Analysis of HAEC microarrays from 150 donors demonstrated a significant regulation of more than 1000 genes with at least 600 regulated in each individual donor. These genes can be divided into 11 major categories of endothelial cell function.21 It is clear that multiple pathways are involved in Ox-PAPC regulation of different groups of genes. In addition, Ox-PAPC affects important endothelial functions independent of transcription. Ox-PAPC rapidly activates monocyte binding to the endothelial cells and has a rapid effect on endothelial barrier properties independent of gene regulation. There is also regulation of endothelial function by pyrroles formed by Ox-PL. Thus, the endothelium is a major target of Ox-PL.
Transcription Independent Regulation
a) Monocyte Binding
Two major monocyte binding molecules on EC have been identified: vascular cell adhesion molecule-1 (VCAM-1) and the connecting segment 1 (CS-1) region of fibronectin, both of which bind monocyte integrin α4β1. VCAM-1 binding plays an important role in binding of monocytes to EC in mouse and rabbit atherosclerosis. However, VCAM-1 does not appear to play a major role in monocyte entry in large vessel endothelial cells of human lesions, although it is increased in small vessels of the adventitia in human lesions.82 In mice, a CS-1 blocking antibody inhibits monocyte endothelial interactions by approximately 30%, while a blocking antibody to VCAM-1 inhibits binding by 70%.83 The effect of Ox-PAPC on EC/monocyte interactions in HAEC involves increased deposition of an alternate splice form of fibronectin, which includes the CS-1 peptide.84 Interaction of Ox-PAPC and PEIPC with the EP2 receptor on endothelial cells has been shown to regulate monocyte binding to HAEC. This interaction causes an increase in cyclic adenosine mono-phosphate (cAMP), which increases R-Ras activation in part by first increasing and then strongly inhibiting H-Ras activation. The activation of R-Ras leads to activation of phosphoinositide 3 kinase (PI3K) which leads to activation of α5β1 integrin. Activation of this integrin on the EC causes the deposition of the CS-1 peptide on the cell surface. The CS-1 deposits in patches in the areas of the cell where monocyte binding is observed.
Staining of carotid endarterectomy samples confirmed the presence of CS-1 on large vessel endothelium in areas of monocyte entry and a lack of an increase in VCAM-1 in these areas.
b) Monolayer Barrier Regulation
The monolayer barrier (involving EC/EC interaction) is regulated by adherens and tight junctions. Low concentrations of Ox-PAPC (5-20μg/ml) increased the EC monolayer barrier function while higher concentrations (25-100μg/ml) reversibly decreased barrier function.24, 25 PEIPC (but not POVPC or PGPC) was able to mimic the Ox-PAPC effect to increase barrier function (JB, KB – unpublished data); whereas POVPC and PGPC exclusively decreased barrier function.2 Enhancement of barrier function involved activation of Rac1 and Cdc42, which then caused peripheral redistribution of focal adhesions, increased association of adherens junction complex containing VE-cadherin, α/β/γ catenin, and stimulated paxillin – β-catenin interactions.25 Low dose Ox-PAPC, via activation of Rap-1, enhances interactions between adherens junctions and tight junction proteins.85 These Ox-PAPC-induced associations were mediated by the Rap1-effector afadin, which promotes interactions between adherens junction protein p120-catenin and tight junction protein zona occludens 1 (ZO-1).85 Low concentrations of Ox-PAPC also induced Rac1- and p120-catenin-dependent peripheral accumulation and activation of Rho negative regulator, p190RhoGAP, which prevented endothelial monolayers from thrombin-induced barrier compromise via inhibition of Rho activity24, 86 (Figure 5A). Thus the increase in barrier function seen with low dose Ox-PAPC can be attributed to changes in several GTPase proteins including Rac-1, Cdc42 and Rap-1.
Figure 5.
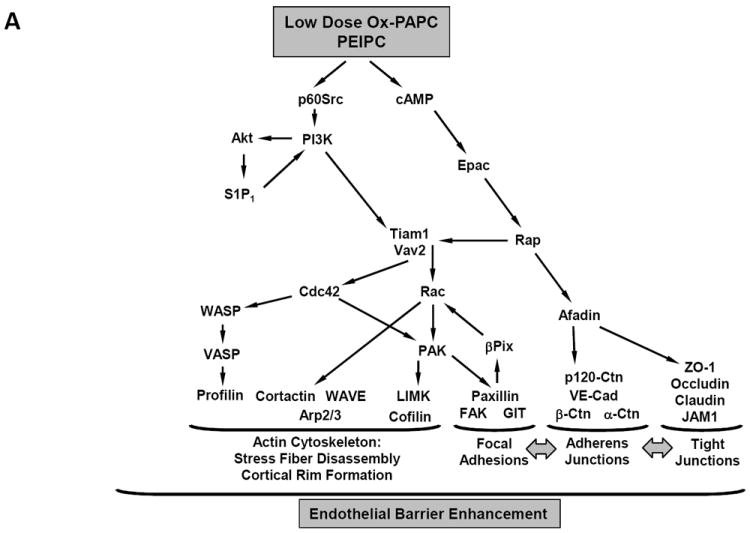
A. Barrier-protecitve mechanisms stimulated by low doses of OxPAPC
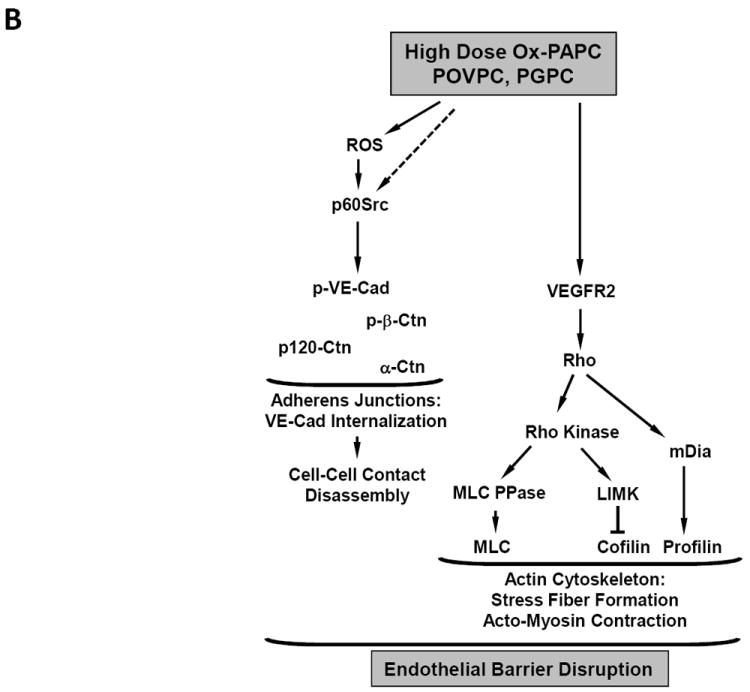
B. Barrier-disruptive mechanisms stimulated by high doses of OxPAPC
Signal transduction pathways leading to Ox-PAPC regulation of endothelial monolayer barrier function. (A) Effects of low concentrations that strengthen the barrier.(B) Effects of higher but non- toxic concentrations that breakdown the barrier.
Decreased barrier function in response to high dose Ox-PAPC also involves changes to both adherens and tight junctions. At early times after treatment with high dose Ox-PAPC, VE-cadherin, a key protein in maintaining barrier function, becomes highly phosphorylated in response to Src tyrosine kinase activation.25 ROS also contribute to this phosphorylation. VE-cadherin hyperphosphorylation leads to a dissociation of VE-cadherin interactions with β-catenin and paxillin, VE-cadherin disappearance from cell-cell contacts and internalization.25 These early changes could be reversed by ROS inhibitors. At later times, after high dose treatment with Ox-PAPC, there was also an increase in Rho activation, which is known to decrease barrier function. Rho activation increases myosin light chain (MLC) phosphorylation by inhibiting myosin phosphatase activity24 and induces F-actin rearrangement from the cell periphery to stress fibers. These events trigger actomyosin contraction and a formation of gaps between the cells. Although high Ox-PAPC doses did not cause immediate Rho activation, increased Rho activity was observed after 30 min of high Ox-PAPC treatment, via VEGFR2 activation.24 Activation of Rho signaling by vascular endothelial growth factor (VEGF) has previously been shown to cause breakdown of endothelial monolayer permeability.87 In addition to the weakening of adherens junctions, high dose Ox-PAPC led to breakdown of tight junctions and decreased occludin expression at mRNA and protein levels. High dose Ox-PAPC also increased occludin phosphorylation, which is known to decrease tight junction interactions.88 These changes in occludin could be reversed by inhibiting the formation of ROS induced by high dose Ox-PAPC (Figure 5B). A phospho-proteomic study has identified a large number of filament associated proteins phosphorylated in response to high dose Ox-PAPC.89
These effects of high and low dose Ox-PAPC on endothelial barrier function have been reported in the cultures of pulmonary and aortic EC24, 25, as well as in in vivo models of lung barrier dysfunction.50, 90 These data strongly suggest that the pathways described here may contribute to atherosclerotic changes in vascular endothelial barrier function and accumulation of lipoproteins. It will be important to examine the presence of these pathways in early human lesions and to correlate this with staining for oxidized lipids.
Transcription Dependent Regulation
a) Regulation of antioxidant enzymes by Ox-PAPC
Treatment of endothelial cells with Ox-PAPC rapidly leads to induction of a large number of genes protecting against ROS, including hemeoxygenase I (HO-1), genes regulating glutathione metabolism and thioredoxins.21 NRF2 (nuclear factor (erythroid derived) like 2) activation appears to play a major role in the Ox-PAPC regulation of a number of these genes and knockdown of NRF2 strongly decreases the effect of Ox-PAPC on these genes.21, 91 The mechanism of NRF2 activation by Ox-PAPC is not certain but there is evidence that lipid electrophiles, like 15-deoxy-Δ12,14-prostaglandin J2 (15dPGJ2), can directly modify Keap1 leading to NRF2 activation.92 A number of Ox-PL contain electrophilic groups that have the potential to modify Keap1. Another mechanism of NRF2 activation involves casein kinase 2 (CK2).93 Once activated NRF2 then binds to the anti-oxidant response element (ARE) in the promoter region of the target genes, increasing transcription. For hemeoxygenase 1(HO-1), the effect of NRF2 knockdown is variable from 30-70% decrease. Several pathways of HO-1 regulation have been described. In HAEC a major role for the plasma membrane electron transport (PMET) system in HO-1 induction by Ox-PAPC was described.94 This system is involved in moving electrons into and out of cells. Major proteins in the PMET complex are eNOX1 (ecto NADPH oxidase 1) and NQO1 (NAD(P)H dehydrogenase, quinone 1). With Ox-PAPC treatment the PMET system is activated and there is a transfer of electrons from an intracellular source (e.g. NADPH) to an outside extracellular acceptor such as oxygen. This electron loss from the cells decreases the overall levels of NADPH in the cell, leading to an increase in ROS, activation of NRF2, and an increase in HO-1. Knockdown of a PMET protein, eNOX1, inhibited Ox-PAPC activation of NRF2 and HO-1 gene expression.
Another pathway of HO-1 activation by Ox-PAPC involved activation of CREB downstream of protein kinases A and C, P38 and ERK.95 There was binding of CREB to an ARE element in the HO-1 promoter. Thus, induction of HO-1 and other redox proteins by Ox-PAPC is likely regulated by multiple pathways, and different pathways may be important in different cell types. Several studies have shown that the signal transduction pathway upregulating anti-oxidant genes is distinct from those regulating inflammatory or pro-coagulant molecules.1 Overexpression of several of these anti-oxidant genes decreases atherosclerosis in mice. Thus selective activation of the anti-oxidant pathways might provide a protective strategy in atherosclerosis.
b) UPR (Unfolded protein response)
A prominent response to Ox-PAPC in HAEC is the activation of the UPR response.20, 21 The UPR response has also been shown to be activated in both endothelial cells and macrophages of atherosclerotic lesions.20 In endothelial cells all three arms of the UPR are activated by a 4 hour treatment of HAEC with Ox-PAPC.96 The mRNA levels of activating transcription factor 3 (ATF3) and activating transcription factor 4 (ATF4) are increased as early as 1 hour after treatment. The UPR response, while protective at low levels can, at higher levels, lead to an increased inflammatory response and cell death, accelerating atherosclerosis. The mechanism of UPR activation by Ox-PAPC and HNE in endothelial cells has been the subject of several studies but a consensus has not yet emerged. In particular, the role of redox regulation of UPR is still unresolved. In human umbilical vein endothelial cells (HUVEC) the Ox-PAPC-dependent increase in ATF4 was dependent on NRF2 binding to an ARE in the ATF4 promoter.22 However, in HAEC the ability of Ox-PAPC to induce transcription of ATF4 was not affected by inhibiting NADPH oxidase 4 (NOX4), the major generator of ROS in response to Ox-PAPC.97 HNE induction of UPR also involves depletion of glutathione. However, glutathione depletion did not cause UPR activation, measured as X-box binding protein 1(XBP-1) activation.98 These authors suggested that direct binding of HNE to UPR proteins was likely responsible. One candidate for Ox-PAPC regulation of UPR came from studies of 150 endothelial donors, examining both untreated and treated HAEC.99 These studies revealed a genetic hotspot controlling the transcription of seven UPR-associated genes in treated cells and 33 genes involved directly or indirectly in the UPR response in untreated cells. The nearest gene to this hot spot was USP16 (ubiquitin specific peptidase 16). Knockdown of USP16 blocked the increase in these genes. Though previously unrecognized as a UPR regulator, this gene may be an important contributor to the UPR response in human patients. More studies will be necessary to resolve the important question of the mechanism of UPR regulation by Ox-PL.
c) Sterol Regulation
As described above Ox-PL induce lipid raft cholesterol depletion and SREBP activation.56, 58 This effect is not observed until approximately 1 hour after treatment but is sustained for at least 18 hours, and it likely plays a role in long term IL-8 induction. There is evidence that cholesterol depletion is mediated by ROS formation, both in response to VEGFR2 activation80 and in response to uncoupling of endothelial nitric oxide synthase (eNOS) that occurs upon eNOS release from caveolae.100 The eNOS uncoupling is a feed-forward mechanism that likely results in sustained SREBP activation. Studies on Ox-LDL suggest that cholesterol depletion can also result directly from incorporation of oxysterols into membranes.56Using other cell types and inducers, it was observed that activation of the UPR caused activation of SREBP.101 However, in HAEC, inhibition of several branches of the UPR did not affect the Ox-PAPC-induced increase in LDLR or the decrease in ABCA1.96 It will be important to determine the stages at which cholesterol depletion plays a role in altered endothelial function in atherosclerosis and whether at some stages there is actually an increase in endothelial cell cholesterol.
d) Regulation of inflammatory cytokines
One of the earliest gene responses to Ox-PAPC is an increase in cytokine synthesis. In contrast to inflammatory mediators lipopolysaccharide (LPS) and tumor necrosis factor (TNF), induction of cytokine synthesis is sustained for at least 12 hours in response to Ox-PAPC. Furthermore, induction of cytokines by Ox-PAPC does not involve activation of NFκB. Different pathways likely control the early and late cytokine responses to Ox-PAPC in endothelial cells.
EGFR (epidermal growth factor receptor)
One pathway regulating the early induction of cytokines involves the ability of Ox-PAPC to activate specific metalloproteinases causing the release of active receptor tyrosine kinase ligands. Activation of the metalloproteinases ADAM10 and ADAMTS4 was shown to lead to an increase in IL-8 synthesis in response to Ox-PAPC.17 Metalloproteinase activation leads to release of active HBEGF (heparin binding epidermal growth factor), which then binds, with the EGFR to induce IL-8 synthesis. A pathway downstream of the EGFR, involving Src, Jak and Stat activation of MCP-1 transcription has been previously identified.102 This same pathway was involved in Ox-PAPC induced IL-8 transcription.103 Thus the following pathway is proposed.
Using microarray analysis ADAM10 knockdown inhibits regulation of 30% of the Ox-PAPC response genes.17 One mechanism of this metalloproteinase activation may involve binding of Ox-PAPC to free thiols of cysteine residues. Free thiolsplay an important role in activation of some metalloproteinases. PEIPC, a bioactive component of Ox-PAPC, contains an electrophilic α,β unsaturated enone group fatty acid that readily forms Michael adducts with free thiols.104
VEGFR2
An important role for VEGFR2 activation in regulating several important pathways of Ox-PAPC action in HAEC was reported.80 Inhibition of VEGFR2 activation decreased induction of inflammatory, sterol synthesis and thrombotic genes by Ox-PAPC.
VEGFR2 activation requires Src activation, which may be a primary event or a necessary component of the activation complex. Downstream of VEGFR2, Rac1 is activated, and ultimately a NOX4 complex is formed that produces ROS.97 Inhibition of NOX4 prevents induction of cytokines and genes of sterol regulation.
UPR regulation of cytokine synthesis
It is important to recognize that some of the Ox-PAPC effects considered protective can also be pro-inflammatory. For example, specific UPR proteins stimulate the inflammatory response.96 Knockdown of UPR proteins strongly decreased basal expression and Ox-PAPC induction of interleukin 8 (IL-8), interleukin 6 (IL-6) and MCP-1.96 Knockdown of XBP-1 was most effective, causing a 90% decrease in both basal and Ox-PAPC induction of cytokines. Thus UPR knockdown does not target cytokine induction by Ox-PAPC specifically but would have an overall negative effect on cytokine levels. The mechanism of this regulation is still not certain. In another study, the UPR pathway was shown to regulate IL-8 induction by HNE.98 In this case inhibition of the UPR with overexpression of ATF6 decreased HNE induction of IL-8 but not basal levels. These studies suggest a general relationship between UPR and cytokines, but the signal transduction pathway controlling this regulation has not been identified.
Sterol Regulation of IL-8 induction
As described above, Ox-PAPC leads to activation of SREBP.58 SREBP activation was shown to lead to an increase in IL-8 synthesis. An SRE (sterol regulatory element) was identified in the IL-8 promoter. When this element was deleted, Ox-PAPC-induced IL-8 synthesis was strongly decreased. MBCD, which causes cholesterol depletion, induces IL-8, while cholesterol loading inhibits Ox-PAPC induction of IL-8.
In addition to the pathways discussed above, we obtained evidence that Ox-PAPC and PEIPC caused activation of PPAR alpha and Wy14643 (a PPAR alpha activator) increased IL-8 and MCP-1.105, 106 However, in HAEC, knockdown of PPAR alpha only marginally decreased IL-8 induction. Taken together, these studies suggest a number of pathways regulate the early and sustained induction of cytokines by Ox-PAPC in HAEC. The current data suggest that at early times receptor tyrosine kinases may play an important role. At later times UPR and SREBP may be most important. In order to effectively inhibit Ox-PAPC induction of cytokine expression in endothelial cells, it will likely be necessary to inhibit several pathways of cytokine regulation.
e) Angiogenesis
Angiogenesis is a relatively late effect of Ox-PAPC and is regulated by several pathways. A recent study has shown that pyrroles, such as CEP, are recognized by a Toll-like receptor 2/Toll-like receptor 1 (TLR2/TLR1) complex and can lead to angiogenesis.49 Many proteins containing CEP and CEP itself can activate TLR2/TLR1. Knockout of TLR2 but not CD36 or SRB1 inhibited the effect of CEP on angiogenesis. This pathway requires the presence of MYD88 (myeloid differentiation primary response gene (88)) and activation of NFκB. NFκB activation mediates Rac activation, followed by activation of integrin interactions with fibronectin. A similar pathway of angiogenesis occurs during development.107
A number of studies demonstrated that Ox-PAPC inhibits NFκB activation in response to bacterial products and does not activate NFκB.1, 2 However, CEP which is a product derived from Ox-PAPC stimulates NFκB activation, demonstrating the ability of specific lipid products to mediate opposite effects on TLR signaling pathways. A separate pathway of angiogenesis was described involving NRF2.22 In this study a 4 hour treatment of endothelial cells with Ox-PAPC increased the levels of vascular endothelial growth factor A (VEGFA), and overnight exposure led to an increase in angiogenesis. This increase in VEGFA was shown to be regulated by NRF2 activation of ATF4. An NRF2 binding element was identified in the ATF4 promoter, and an ATF4 binding site was observed in the VEGFA promoter.
Taken together, these two studies suggest a complex regulation of angiogenesis by several pathways. It would be valuable to examine levels of VEGFA vs. activated NFκB in angiogenic vessels of human lesions.
4) Smooth Muscle Cells
Phenotypic Switching and Smooth Muscle Replication
Less work has been done documenting the effects of Ox-PAPC on smooth muscle cells. However, there are major pro-atherogenic effects on the smooth muscle phenotype. These changes include loss of smooth muscle markers, an increase in matrix synthesis and an increase in replication. In atherosclerotic lesions, SMC in the intima lose marker proteins myosin heavy chain (MHC) and α-actin38 and increase matrix synthesis.39 Treatment of SMC with Ox-PAPC or POVPC caused a strong suppression of the synthesis of mRNA for major SMC proteins α-actin, MHC and myocardin. This suppression was shown to be regulated by activation of ELK-1108 and increased synthesis of KLF4 (transcription that requires SP-1).39 These two proteins formed a complex on the promoter that led to recruitment of HDACs inhibiting transcription.
Ox-PAPC, and two of its constituents, POVPC and PEIPC, also caused an increase in the synthesis of collagen VIII, fibronectin, laminin and versican. KLF4 transcript levels were strongly increased by Ox-PAPC, and knockdown of KLF4 strongly inhibited matrix synthesis in response to Ox-PAPC. Furthermore, increased binding of KLF4 to the promoter region of the collagen 8 gene was demonstrated by CHIP assay. Evidence was also presented that Sp1, a known regulator of KLF4, played a role in Ox-PAPC regulation of matrix proteins since knockdown of Sp1 inhibited KLF4-mediated mRNA increases in matrix proteins. KLF4 is a molecule previously shown to regulate phenotypic switching in smooth muscle cells and levels were increased in SMC of atherosclerotic lesions. In response to Ox-PAPC, there was also an an increase in the gene expression of MCP-1 and monocyte chemotactic protein 3 (MCP-3) in these SMC. Several pathways have been demonstrated to increase SMC proliferation in response to Ox-PAPC and its components. POVPC was demonstrated to activate galactosyl transferase-2 (GALT2) to produce lactoxylceramide (LacCer) 109. LacCer then activates Nox increasing superoxide and causing activation of P21 Ras which leads to an increase in c-fos and proliferating cell nuclear antigen (PCNA).
SMC phosphorylation of cnx43 has also been shown to increase SMC replication, and Ox-PAPC was shown to increase this phosphorylation.40, 110 Thus Ox-PL have a profound pro-atherogenic effect on SMC, causing phenotypic switching, cell replication, and increased production of inflammatory molecules.
Apoptosis
In human smooth muscle cells 10μM POVPC was shown to cause apoptosis as measured after 10 hours of treatment.41 This apoptotic effect was initiated by activation of acid sphinomyelinase, resulting in the conversion of sphingomyelin to ceramide. Ceramide activates the phosphorylation of JNK and P38 MAPKs, which have been shown to activate caspase 3 and apoptosis. SMC apoptosis is an important characteristic of human lesions. A better understanding of the pathways of apoptosis could be important in stabilizing the vessel wall.
Metabolism of Ox-PL
There is considerable evidence that formation of oxidized phospholipids in atherosclerotic lesions can be initiated enzymatically. This likely occurs in tissues rather than in the blood, since levels of Ox-PL in tissues are significantly higher than levels found in plasma.1, 2 Several enzymes have been shown to generate Ox-PL, including 12/15 LO and myeloperoxidase. Incubation of macrophages overexpressing 12/15 LO increased the formation of Ox-PL, and knockout of 12/15 LO in mice caused a 50% reduction in atherosclerosis.10 The importance of hematopoetic cells as the source of 12/15 LO was shown in bone marrow transplantion studies.111 An important role for myeloperoxidase in generating bioactive Ox-PL was shown by using isolated enzyme and comparing macrophages from normal and MPO-deficient patients.9 Furthermore MPO transgenic mice had increased atherosclerosis.112 The levels of MPO were increased in human lesions and levels of MPO in plasma were shown to predict the presence of CAD.9 While MPO-generated Ox-PL likely play an important role in the regulation of atherosclerosis, other products of MPO activity are likely also important.9 Clinical trials of MPO or 12/15 LO inhibitors should be considered. It would be useful to know which type of macrophages are producing MPO in atherosclerosis since it might be possible to specifically target these and avoid problems with infections that would come from targeting macrophages in general.
There are a number of enzymes that can degrade Ox-PL and have the potential to impact atherosclerosis. There are at least three enzymes in plasma that can hydrolyze the sn-2 position of Ox-PL: PAF-AH, certain subtypes of phospholipase A2 (PLA2) and LCAT.2 In cell culture hydrolysis of Ox-PAPC decreases transcriptional regulation of several genes.113 However, the enzyme used for some of these studies was different from hydrolases found in mouse or human plasma. These studies should be repeated with more appropriate enzymes and unesterified lipid oxidation products should be synthesized and tested. In spite of the ability of PAF-AH to hydrolyze Ox-PL there is evidence that this is not a major mechanism for the regulation of PAF or PAF-like Ox-PL levels in plasma, since clearance of PAF from plasma was unchanged in PAF-AH null mice and Ox-PL could competitively block this clearance.114 A major mechanism of removal of ether-containing Ox-PL from plasma is uptake by the phospholipid transfer protein TMEM30a discussed above.68, 114 Nonetheless, there is clear evidence that levels of PAF-AH can predict the development of CAD in humans. However, to date, PAF-AH inhibitor has not had a consistent positive effect on CAD in humans. While the major effect of LCAT is trans-esterification of cholesterol, it can also hydrolyze Ox-PL. The in vivo effects of LCAT on Ox-PL are controversial, with some studies demonstrating increased levels of Ox-PL in the absence of LCAT and others demonstrating an inhibition of oxidation by LCAT.2 Secretory phospholipase A2 (sPLA2) V, one of the most active phospholipases in the hydrolysis of lipoprotein phospholipids, promotes the inflammatory response in mice, probably by remodeling LDL to a form more rapidly taken up by scavenger receptors. The studies on PLA2 have recently been reviewed.115 In summary some of the PLA2 may serve as markers of CAD, but to date an increase in the activity of these molecules in humans has not been shown to negatively impact in CAD. Thus, more specific inhibitors are needed in order to make firm conclusions about the effects of sPLA2. A better understanding of the relative effects of PLA2 on effects of Ox-PL in vitro and on mouse atherosclerosis will help to clarify which of these molecules should be targeted in human trials.
In addition to hydrolysis of Ox-PL as a potential protective mechanism, oxidized fatty acids containing aldehydes and epoxides such as PEIPC and POVPC can be metabolized by aldose reductase (AR)116; while Ox-PL containing hydroperoxide groups such as 9-hydroperoxy eicosatetraenoic acid (9-HPETE) can be metabolized by glutathione peroxidases.117-119 Somewhat surprisingly, in cultured macrophages the metabolic product of AR (1-palmitoyl-2-(5)-hydrovaleryl-sn-glycero-3-phosphorylcholine (PHVPC)) was shown to increase the inflammatory response 10-100 fold as compared to only a 2-3 fold response to POVPC.120 However, atherosclerosis was higher in AR null/apoE null mice than in mice null for apoE alone.121 These authors carefully examined the relationship between levels of PAF-AH and levels of AR in regulating the macrophage response to POVPC.120 They determined that inhibitors of AR were more effective than inhibitors of PAF-AH in blocking the inflammatory response in macrophages. Thus, they conclude that in vivo the relative levels of PAF-AH and AR may be important in determining the effects of inhibitors of these two molecules on atherosclerosis. Taken together these studies point out important considerations in targeting enzymes that metabolize Ox-PL. Two other major enzymes involved in the metabolism of Ox-PL have glutathione peroxidase activity and are able to reduce phospholipid hydroperoxides to hydroxides: glutathione peroxidase 4 (GPX4) and peroxiredoxin 6 (PR6). PR6 has a PLA2 as well as a peroxidase activity.117 The ability of both of these enzymes to use Ox-PL as substrates has been verified.118 Overexpression of GPX4 in apoE null mice was shown to reduce lesion formation and to decrease the complexity of more advanced lesions.119 The ability of PR6 to reduce the size of atherosclerotic lesions has also been reported, however, the effect is very dependent on the strain tested. Overall, because of their effects on atherosclerosis in mice, the molecules that metabolize Ox-PL have promise as therapeutic targets.
Conclusions
The preponderance of studies demonstrates that Ox-PL is present in human and mouse atherosclerotic lesions and contribute to lesion formation. The amount of Ox-PL in lesions is sufficient to cause pro-atherogenic effects on cells in culture, and some of the pathways identified in vitro as regulated by Ox-PL are also activated in human or mouse lesions and effect atherosclerosis. Levels of Ox-PL and of MPO in blood of patients predict development of CAD. Furthermore injection of F(ab) fragments of antibodies to Ox-PL inhibit the development of atherosclerosis in mice.122 These studies suggest that Ox-PL can be both a marker and therapeutic target for CAD. Thus, the studies discussed here and in earlier reviews, identify a number of novel therapeutic targets (receptors and enzymes), signaling pathways, and Ox-PL-lowering approaches to confront the atherogenic effects of Ox-PL. They are briefly summarized below.
Based on studies in mice, one of the most effective strategies might be to alter levels of Ox-PL with antibodies to these lipids. These results suggest that Ox-PL vaccination would potentially protect against atherosclerosis. HDL has been shown to be able to bind Ox-PL but its cholesterol transport function is compromised. Another strategy is employing synthetic molecules that retain the Ox-PL binding function of HDL without altering the protective effects of HDL. D4F, an HDL mimetic peptide, is an example of this approach. Another possibility is to alter synthesis and/ or degradation of Ox-PL. Studies in mice and in humans suggest that inhibitors of MPO and 12/15 LO, both of which increase Ox-PL levels, may have a therapeutic potential. Another strategy is to increase enzymes that degrade Ox-PL. However, recent studies suggest that it will be important to determine the activity of the lipids after treatment with various PLA2 enzymes and reductases. In addition the relative importance of the individual enzymes in lowering levels of active Ox-PL and atherosclerosis need to be clarified.
In vitro and in vivo studies over the last 5 years have identified receptors and signaling pathways that could be targets to inhibit the pro-atherogenic effects of Ox-PL (Fig 6). These studies have made it clear that multiple receptors and signaling pathways can regulate one response such as angiogenesis. Therefore it may be necessary to target more than one receptor or signaling pathway. A striking finding of the recent studies is the importance of receptor complexes in many of the actions of Ox-PL. Several of these complexes contain TLRs and CD36. Studies in mice demonstrate that targeting of TLR2 and 4 reduces some aspects of atherosclerosis but some effects are limited to early stages. Because TLRs also protect against bacterial infection, it will be important to target inhibitors to atherosclerotic lesions. The importance of covalent binding of Ox-PL to proteins has emerged as an important aspect of their mechanism of action. It may be possible to inhibit covalent binding with compounds such as n-acetyl cysteine which can covalently bind Ox-PL and reduce many pro-atherogenic effects in vitro. In addition it is apparent that Ox-PL alter the lipid raft fraction and these changes in lipid rafts may contribute to the mechanisms of Ox-PL action. A better understanding of the mechanisms by which Ox-PL alter membrane composition will define important targets in this pathway. In general, in choosing receptors and pathways to target, it will be important to gain more understanding of the stages of atherosclerosis regulated by these receptors and pathways. Studies in mice at both early and late stages and studies of signaling pathways in human lesions will be very helpful in this process.
Figure 6.
Model of Ox-PL regulation of atherosclerosis:
(A) Early Lesions
(1) LDL enters the vessel wall and is oxidized by MPO and 12/15 LO to form oxidation products including Ox-PAPC. Low doses of Ox-PAPC decrease the permeability of the EC monolayer by forming adherens junctions. Higher levels of Ox-PAPC cause a strong increase in monolayer permeability due to junction breakdown and stress fiber formation, resulting in increased entry of LDL into the vessel wall. (4) Ox-PAPC binds to the E-type prostaglandin receptor (EP2) receptor, causing deposition of CS-1 Fibronectin on the apical surface which binds monocytes. (5) Ox-PAPC activates specific a dysintegrin and metalloproteinases (ADAMs) to cause release of active HBEGF and activation of epidermal growth factor receptor (EGFR), leading to IL-8 and MCP-1 synthesis. (6) Ox-PAPC also activates VEGFR2, leading to IL-8 and MCP-1 synthesis. (7) These chemokines facilitate entry of monocytes into the vessel wall. (8) Ox-PL acting on TLR2, TLR4/6/CD36 and PAFR cause some monocytes to differentiate into M1 macrophages producing chemokines. (9) Ox-PAPC causes differentiation of some macrophages into Mox, which have high levels of anti-oxidant enzymes and lower chemokine synthesis. (10) Ox-PAPC causes differentiation of some monocytes into dendritic cells with impaired presentation of lipid antigens. (11) Macrophages further oxidize LDL to form Ox-LDL. Ox-PCCD36 acting on CD36 in the presence of Ox-LDL causes foam cell formation.
(B) Advanced Lesions.
(1) In the presence of Ox-PAPC and PAF-like lipids, macrophages make: Interleukin 1 beta (IL-1β) and RANTES. These chemokines and a direct effect of Ox-PAPC on SMC cause migration and proliferation and matrix production of SMC. These SMC cover the foam cells that accumulate under the endothelium. The interaction of Ox-PAPC with CD36/TLR2 and with UPR activators and the interaction of PAF-like lipids with TMEM30a cause macrophage apoptosis. (4) Ox-PS/PCCD36 in the apopotic cell membrane bind to CD36 in macrophages, leading to macrophage uptake of the apoptotic cells. (5) Some apoptotic fragments stimulate EC to make IL-8, an angiogenic cytokine. (6) CEP activation of TLR2/TLR1 causing integrin activation and Ox-PAPC acting to increase VEGFA cause angiogenesis of adventitial vessels into the media and intima. (7) C-reactive protein (CRP) and Ox-PAPC interacting with CD36 stimulates macrophage production of metalloproteinase. This weakens the plaque and can lead to plaque rupture. (8) Ox-PAPC activation of VEGFR2 increases tissue factor synthesis in the endothelium. Ox-PAPC also causes and increases in Serpin B2 and a decrease in thrombomodulin. Ox-PCCD36 acting on CD36 and PAF-like lipids acting on PAFR cause increased aggregability of platelets. (Illustration Credit: Ben Smith).
In employing inhibitors to target the pro-atherogenic effects of Ox-PL it is important to recognize that Ox-PL can also have protective effects. For example, PEIPC at low concentrations protects endothelial barrier function. Ox-PAPC increases the levels of anti-oxidant enzymes, binds to complement factor H to inhibit inflammation and can under some circumstances promotes cell survival by inducing UPR and related pathways. However, some aspects of the UPR, such as effects on inflammation and apoptosis, can be pro-atherogenic making it important to gain a better understanding of the regulation of the major UPR responses in atherosclerosis. In general, at least some of the protective effects of Ox-PL are mediated by different signaling pathways from the atherogenic effects. It will be important to determine which Ox-PL lipid, which receptors and which signaling pathways promote atherogenesis and which are protective. With knowledge of the protective and the pro-atherogenic effects of Ox-PL in different vascular cell types, it should be possible to design experiments to test the effect of inhibition of specific pathways in specific cell types on the development of atherosclerosis in mice.
It also may be possible to identify individuals most likely to benefit from Ox-PL inhibition. Using endothelial cells from 150 heart transplant donors we have determined that there are major differences in the effects of Ox-PAPC in different individuals.21, 99 We have identified several genes that control aspects of this sensitivity. We also have data suggesting differences in sensitivity in macrophages from different individuals (AJL, unpublished observation). From these studies it may be possible to identify polymorphisms that effect susceptibility to Ox-PL and would indicate individuals who might most benefit from reduction in Ox-PL levels or signaling.
Acknowledgments
This work was supported by K99HL105577 for SL; HL87823, HL76259 and PO1HL58064 for KB; T32-HL69766 JRS; P01 HL30568 for AJL, JB and CR and HL64731 JB.
Abbreviations
- 12/15 LO
12/15-lipoxygenase
- 12S-HETE
12S hydroxyeicosatetraenoic acid
- 13-HODE-PC
1-palmitoyl-2-(13(S)-hydroxy-(9Z,11E)-octadeca-9,11-dienoyl)-sn-glycero-3-phosphocholine
- 15dPGJ2
15-deoxy-Δ12,14-prostaglandin J2
- 9-HPETE
9-hydroperoxy eicosatetraenoic acid
- GPX4
glutathione peroxidase 4
- ADAM10
a dysintegrin and metalloproteinase 10
- ADAMTS4
a dysintegrin and metalloproteinase with thrombospondin motifs 4
- ADP
adenosine diphosphate
- AIF
apoptosis inducing factor
- Apo-AI
apolipoprotein AI
- AR
aldose reductase
- ARE
antioxidant response element
- ATF3, ATF4
activating transcription factor 3, -4
- BMDM
bone marrow derived macrophage
- CAD
coronary artery disease
- cAMP
cyclic adenosine mono-phosphate
- CD11B
cluster of differentitation 11b antigen
- CD1a, CD1b, CD1C
cluster of differentiation 1 antigens
- CD36
cluster determinant 36
- CEP
2 (w-carboxyethyl)pyrrole
- CK2
casein kinase 2
- COX-2
cyclooxygenase 2
- CRP
C-reactive protein
- CS-1
connecting segment 1
- EGFR
epidermal growth factor receptor
- EGR1
early grown factor response protein 1
- eNOS
endothelial nitric oxide synthase
- EP2
E-type prostaglandin receptor
- Erk/MAPK
extracellular-signal-regulated kinases/mitogen activated protein kinase
- GRO1
GRO1 oncogene
- HAEC
human aortic endothelial cells
- HAz-PC
Hexadecylazelaoyl PC
- HBEGF
heparin binding epidermal growth factor
- HDL
high-density lipoprotein
- HNE
hydroxynonenal
- HO-1
hemeoxygenase 1
- HOHA-PC
1-palmitoyl-2-(4-hydroxy-7-oxohept-5-enoyl)-sn-glycero-3-phosphocholine
- HOOA-PC
1-palmitoyl-2-(5-hydroxy-8-oxooct-6-enoyl)-sn-glycero-3-phosphocholine
- HUVEC
human umbilical vein endothelial cells
- IL-1β
Interleukin 1 beta
- IL-6
interleukin 6
- IL-8
interleukin 8
- JNK
C-Jun N-terminal kinase
- KC
keratinocyte chemoattractant
- KLF4
Krueppel-like factor 4
- KOdiA-PC
1-palmitoyl-2-(7-carboxy-5-oxo-hept-6E-enoyl)-sn-glycero-3-phosphocholine
- LCAT
lecithin-cholesterol acyl transferase
- LDL
low-density lipoprotein
- LP(a)
lipoprotein(a)
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- Lyn
tyrosine protein kinase Lyn
- M1, M2 and M0
M1, M2 and M0 macrophages
- MBCD
methyl-β-cyclodextrin
- MCP-1, MCP-3
monocyte chemotactic protein 1, -3
- MHC
Myosin heavy chain
- MIP-1, MIP-2
macrophage inhibitor protein 1, -2
- MKP-1
MAP kinase phosphatase 1
- MLC
myosin light chain
- MPO
myeloperoxidase
- MYD88
myeloid differentiation primary response gene (88)
- NaBH4
sodium borohydride
- NFAT
nuclear factor of activated T cells
- NfκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NOX2, NOX4
NADPH oxidase 2, -4
- NQO1
NAD(P)H dehydrogenase, quinone 1
- NRF2
nuclear factor (erythroid derived) like 2
- Ox-LDL
oxidized low-density lipoprotein
- Ox-PAPC
oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
- OxPAPE-N-biotin
oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl-(N-biotinylethanolamine)
- Ox-PC
oxidized PC-containing phospholipids
- Ox-PCCD36
a group of Ox-PC binding to CD36
- Ox-PL
oxidized phospholipids
- Ox-PLPC
oxidized 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine
- Ox-PS
oxidized PS-containing phospholipids
- PAF
platelet activating factor
- PAF-AH
platelet activating factor acetyl hydrolase
- PAPC
1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
- PC
phosphatidylcholine
- PGPC
1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine
- PE
phosphatidylethanolamine
- PEIPC
1-palmitoyl-2-(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphocholine
- PGE2
prostaglandin E2
- PHVPC
1-palmitoyl-2-(5)-hydrovaleryl-sn-glycero-3-phosphorylcholine
- PLA2
phospholipase A2
- PLPC
1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine
- POVPC
1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- PR6
peroxyredoxin 6
- PS
phosphatidylserine
- RANTES
regulated upon activation, normal T-cell expressed, and secreted
- RAR
retinoic acid receptor
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- SMC
smooth muscle cells
- Sp1
specificity protein 1
- Sp3
specificity protein 3
- sPLA2
secretory phospholipase A2
- SRB1
scavenger receptor B1
- SRE
sterol regulatory element
- SREBP
sterol regulatory element binding protein
- TFPI
tissue factor protein inhibitor
- TLR2, 4
Toll-like receptor 2 or Toll-like receptor 4
- TM
thrombomodulin
- TMEM30a
transmembrane protein 30A
- TNF
tumor necrosis factor
- UPR
unfolded protein response
- Vav
Vav family of guanine nucleotide exchange factor
- VCAM-1
vascular cell adhesion molecule-1
- VEGFA
vascular endothelial growth factor A
- VEGFR2
vascular endothelial growth factor receptor
- XBP-1
X-box binding protein 1
Footnotes
Disclosures:
None
References
- 1.Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50(Suppl):S207–212. doi: 10.1194/jlr.R800074-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxidants & redox signaling. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn BH, McMahon M. Atherosclerosis and systemic lupus erythematosus: The role of altered lipids and of autoantibodies. Lupus. 2008;17:368–370. doi: 10.1177/0961203308089989. [DOI] [PubMed] [Google Scholar]
- 4.Tsimikas S, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, Sandhu MS, Miller ER, Benessiano J, Tedgui A, Witztum JL, Khaw KT, Boekholdt SM. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 5.Choi SH, Chae A, Miller E, Messig M, Ntanios F, DeMaria AN, Nissen SE, Witztum JL, Tsimikas S. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: Volume observations from the reversal (reversal of atherosclerosis with aggressive lipid lowering) study. J Am Coll Cardiol. 2008;52:24–32. doi: 10.1016/j.jacc.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 6.Fraley AE, Schwartz GG, Olsson AG, Kinlay S, Szarek M, Rifai N, Libby P, Ganz P, Witztum JL, Tsimikas S. Relationship of oxidized phospholipids and biomarkers of oxidized low-density lipoprotein with cardiovascular risk factors, inflammatory biomarkers, and effect of statin therapy in patients with acute coronary syndromes: Results from the miracl (myocardial ischemia reduction with aggressive cholesterol lowering) trial. J Am Coll Cardiol. 2009;53:2186–2196. doi: 10.1016/j.jacc.2009.02.041. [DOI] [PubMed] [Google Scholar]
- 7.Tsimikas S, Aikawa M, Miller FJ, Jr, Miller ER, Torzewski M, Lentz SR, Bergmark C, Heistad DD, Libby P, Witztum JL. Increased plasma oxidized phospholipid:Apolipoprotein b-100 ratio with concomitant depletion of oxidized phospholipids from atherosclerotic lesions after dietary lipid-lowering: A potential biomarker of early atherosclerosis regression. Arterioscler Thromb Vasc Biol. 2007;27:175–181. doi: 10.1161/01.ATV.0000251501.86410.03. [DOI] [PubMed] [Google Scholar]
- 8.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet cd36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nature medicine. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res. 2009;50(Suppl):S346–351. doi: 10.1194/jlr.R800086-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cyrus T, Pratico D, Zhao L, Witztum JL, Rader DJ, Rokach J, FitzGerald GA, Funk CD. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation. 2001;103:2277–2282. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- 11.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoa-i-mimetic peptides bind oxidized lipids with much higher affinity than human apoa-i. Journal of lipid research. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman P, Horkko S, Steinberg D, Witztum JL, Dennis EA. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of schiff base formation and aldol condensation. J Biol Chem. 2002;277:7010–7020. doi: 10.1074/jbc.M108860200. [DOI] [PubMed] [Google Scholar]
- 13.Boullier A, Friedman P, Harkewicz R, Hartvigsen K, Green SR, Almazan F, Dennis EA, Steinberg D, Witztum JL, Quehenberger O. Phosphocholine as a pattern recognition ligand for cd36. J Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-cd36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. The Journal of experimental medicine. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salomon RG. Forum review - levuglandins and isolevuglanins: Stealthy toxins of oxidative injury. Antioxidants & Redox Signaling. 2005;7:185–201. doi: 10.1089/ars.2005.7.185. [DOI] [PubMed] [Google Scholar]
- 16.Salomon RG, Hong L, Hollyfield JG. Discovery of carboxyethylpyrroles (ceps): Critical insights into amd, autism, cancer, and wound healing from basic research on the chemistry of oxidized phospholipids. Chemical research in toxicology. 2011;24:1803–1816. doi: 10.1021/tx200206v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Springstead JR, Parks B, Romanoski CE, Palvolgyi R, Ho T, Nguyen P, Lusis AJ, Berliner JA. Metalloproteinase processing of hbegf is a proximal event in the response of human aortic endothelial cells to oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2012 doi: 10.1161/ATVBAHA.111.241257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 19.Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamem.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, Pan C, Lee S, Vakili L, Yang WP, Kayne P, Mungrue IN, Araujo JA, Berliner JA, Lusis AJ. Network for activation of human endothelial cells by oxidized phospholipids: A critical role of heme oxygenase 1. Circulation research. 2011;109:e27–41. doi: 10.1161/CIRCRESAHA.111.241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR, Bochkov VN. Oxidized phospholipids regulate expression of atf4 and vegf in endothelial cells via nrf2-dependent mechanism: Novel point of convergence between electrophilic and unfolded protein stress pathways. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1007–1013. doi: 10.1161/ATVBAHA.110.204354. [DOI] [PubMed] [Google Scholar]
- 23.Doyle B, Caplice N. Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J Am Coll Cardiol. 2007;49:2073–2080. doi: 10.1016/j.jacc.2007.01.089. [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Lee S, Starosta V, Wu T, Ho T, Kim J, Berliner JA, Birukov KG. A role for vegfr2 activation in endothelial responses caused by barrier disruptive oxpapc concentrations. PLoS One. 2012;7:e30957. doi: 10.1371/journal.pone.0030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starosta V, Wu T, Zimman A, Pham D, Tian X, Oskolkova O, Bochkov V, Berliner JA, Birukova AA, Birukov KG. Differential regulation of endothelial cell permeability by high and low doses of oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine. Am J Respir Cell Mol Biol. 2012;46:331–341. doi: 10.1165/rcmb.2011-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong KW, Zhao W, Li N, Barajas B, Kleinman M, Sioutas C, Horvath S, Lusis AJ, Nel A, Araujo JA. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007;8:R149. doi: 10.1186/gb-2007-8-7-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor cd36. The Journal of biological chemistry. 2002;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- 29.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor cd36 and is enriched in atherosclerotic lesions. The Journal of biological chemistry. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 30.Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S, Grubbs N, Sharma R, Leitinger N. Oxidized phospholipid-induced inflammation is mediated by toll-like receptor 2. Free Radic Biol Med. 2011;51:1903–1909. doi: 10.1016/j.freeradbiomed.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silva AR, de Assis EF, Caiado LF, Marathe GK, Bozza MT, McIntyre TM, Zimmerman GA, Prescott SM, Bozza PT, Castro-Faria-Neto HC. Monocyte chemoattractant protein-1 and 5-lipoxygenase products recruit leukocytes in response to platelet-activating factor-like lipids in oxidized low-density lipoprotein. J Immunol. 2002;168:4112–4120. doi: 10.4049/jimmunol.168.8.4112. [DOI] [PubMed] [Google Scholar]
- 32.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger cd36-tlr2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, Witztum JL. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via nrf2. Circulation research. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shamshiev AT, Ampenberger F, Ernst B, Rohrer L, Marsland BJ, Kopf M. Dyslipidemia inhibits toll-like receptor-induced activation of cd8alpha-negative dendritic cells and protective th1 type immunity. J Exp Med. 2007;204:441–452. doi: 10.1084/jem.20061737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bluml S, Zupkovitz G, Kirchberger S, Seyerl M, Bochkov VN, Stuhlmeier K, Majdic O, Zlabinger GJ, Seiser C, Stockl J. Epigenetic regulation of dendritic cell differentiation and function by oxidized phospholipids. Blood. 2009;114:5481–5489. doi: 10.1182/blood-2008-11-191429. [DOI] [PubMed] [Google Scholar]
- 37.Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, Gutierrez MA, Navab M, Reddy ST, Witztum JL, Fogelman AM, Rea TH, Eisenberg D, Berliner J, Modlin RL. Host-derived oxidized phospholipids and hdl regulate innate immunity in human leprosy. J Clin Invest. 2008;118:2917–2928. doi: 10.1172/JCI34189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- 39.Cherepanova OA, Pidkovka NA, Sarmento OF, Yoshida T, Gan Q, Adiguzel E, Bendeck MP, Berliner J, Leitinger N, Owens GK. Oxidized phospholipids induce type viii collagen expression and vascular smooth muscle cell migration. Circ Res. 2009;104:609–618. doi: 10.1161/CIRCRESAHA.108.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnstone SR, Ross J, Rizzo MJ, Straub AC, Lampe PD, Leitinger N, Isakson BE. Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am J Pathol. 2009;175:916–924. doi: 10.2353/ajpath.2009.090160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loidl A, Sevcsik E, Riesenhuber G, Deigner HP, Hermetter A. Oxidized phospholipids in minimally modified low density lipoprotein induce apoptotic signaling via activation of acid sphingomyelinase in arterial smooth muscle cells. J Biol Chem. 2003;278:32921–32928. doi: 10.1074/jbc.M306088200. [DOI] [PubMed] [Google Scholar]
- 42.Chen R, Chen X, Salomon RG, McIntyre TM. Platelet activation by low concentrations of intact oxidized ldl particles involves the paf receptor. Arterioscler Thromb Vasc Biol. 2009;29:363–371. doi: 10.1161/ATVBAHA.108.178731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kuhn H, Hazen SL, Goodall AH, Hamali HA, Collins PW, O’Donnell VB. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. The Journal of biological chemistry. 2010;285:6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. Hdl and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nature reviews. Cardiology. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 45.Bowry VW, Stanley KK, Stocker R. High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc Natl Acad Sci U S A. 1992;89:10316–10320. doi: 10.1073/pnas.89.21.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szapacs ME, Kim HY, Porter NA, Liebler DC. Identification of proteins adducted by lipid peroxidation products in plasma and modifications of apolipoprotein a1 with a novel biotinylated phospholipid probe. J Proteome Res. 2008;7:4237–4246. doi: 10.1021/pr8001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oskolkova OV, Afonyushkin T, Preinerstorfer B, Bicker W, von Schlieffen E, Hainzl E, Demyanets S, Schabbauer G, Lindner W, Tselepis AD, Wojta J, Binder BR, Bochkov VN. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J Immunol. 2010;185:7706–7712. doi: 10.4049/jimmunol.0903594. [DOI] [PubMed] [Google Scholar]
- 48.Ashraf MZ, Kar NS, Chen X, Choi J, Salomon RG, Febbraio M, Podrez EA. Specific oxidized phospholipids inhibit scavenger receptor bi-mediated selective uptake of cholesteryl esters. J Biol Chem. 2008;283:10408–10414. doi: 10.1074/jbc.M710474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV. Oxidative stress induces angiogenesis by activating tlr2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. Cd36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walton KA, Hsieh X, Gharavi N, Wang S, Wang G, Yeh M, Cole AL, Berliner JA. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 53.Li R, Mouillesseaux KP, Montoya D, Cruz D, Gharavi N, Dun M, Koroniak L, Berliner JA. Identification of prostaglandin e2 receptor subtype 2 as a receptor activated by oxpapc. Circulation research. 2006;98:642–650. doi: 10.1161/01.RES.0000207394.39249.fc. [DOI] [PubMed] [Google Scholar]
- 54.Marathe GK, Zimmerman GA, Prescott SM, McIntyre TM. Activation of vascular cells by paf-like lipids in oxidized ldl. Vascul Pharmacol. 2002;38:193–200. doi: 10.1016/s1537-1891(02)00169-6. [DOI] [PubMed] [Google Scholar]
- 55.Smiley PL, Stremler KE, Prescott SM, Zimmerman GA, McIntyre TM. Oxidatively fragmented phosphatidylcholines activate human neutrophils through the receptor for platelet-activating factor. J Biol Chem. 1991;266:11104–11110. [PubMed] [Google Scholar]
- 56.Levitan I, Shentu TP. Impact of oxldl on cholesterol-rich membrane rafts. J Lipids. 2011;2011:730209. doi: 10.1155/2011/730209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabatini K, Mattila JP, Megli FM, Kinnunen PK. Characterization of two oxidatively modified phospholipids in mixed monolayers with dppc. Biophys J. 2006;90:4488–4499. doi: 10.1529/biophysj.105.080176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeh M, Cole AL, Choi J, Liu Y, Tulchinsky D, Qiao JH, Fishbein MC, Dooley AN, Hovnanian T, Mouilleseaux K, Vora DK, Yang WP, Gargalovic P, Kirchgessner T, Shyy JY, Berliner JA. Role for sterol regulatory element-binding protein in activation of endothelial cells by phospholipid oxidation products. Circ Res. 2004;95:780–788. doi: 10.1161/01.RES.0000146030.53089.18. [DOI] [PubMed] [Google Scholar]
- 59.Greenberg ME, Li XM, Gugiu BG, Gu X, Qin J, Salomon RG, Hazen SL. The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 60.Gao D, Ashraf MZ, Kar NS, Lin D, Sayre LM, Podrez EA. Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class b scavenger receptors cd36 and sr-bi. The Journal of biological chemistry. 2010;285:4447–4454. doi: 10.1074/jbc.M109.082800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun M, Finnemann SC, Febbraio M, Shan L, Annangudi SP, Podrez EA, Hoppe G, Darrow R, Organisciak DT, Salomon RG, Silverstein RL, Hazen SL. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for cd36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. The Journal of biological chemistry. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage cd36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2333–2338. doi: 10.1161/01.ATV.0000148007.06370.68. [DOI] [PubMed] [Google Scholar]
- 63.Mullick AE, Soldau K, Kiosses WB, Bell TA, 3rd, Tobias PS, Curtiss LK. Increased endothelial expression of toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. 2008;205:373–383. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein e. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subbanagounder G, Leitinger N, Shih PT, Faull KF, Berliner JA. Evidence that phospholipid oxidation products and/or platelet-activating factor play an important role in early atherogenesis : In vitro and in vivo inhibition by web 2086. Circ Res. 1999;85:311–318. doi: 10.1161/01.res.85.4.311. [DOI] [PubMed] [Google Scholar]
- 66.Gugiu BG, Mouillesseaux K, Duong V, Herzog T, Hekimian A, Koroniak L, Vondriska TM, Watson AD. Protein targets of oxidized phospholipids in endothelial cells. J Lipid Res. 2008;49:510–520. doi: 10.1194/jlr.M700264-JLR200. [DOI] [PubMed] [Google Scholar]
- 67.Moumtzi A, Trenker M, Flicker K, Zenzmaier E, Saf R, Hermetter A. Import and fate of fluorescent analogs of oxidized phospholipids in vascular smooth muscle cells. J Lipid Res. 2007;48:565–582. doi: 10.1194/jlr.M600394-JLR200. [DOI] [PubMed] [Google Scholar]
- 68.Chen R, Brady E, McIntyre TM. Human tmem30a promotes uptake of antitumor and bioactive choline phospholipids into mammalian cells. J Immunol. 2011;186:3215–3225. doi: 10.4049/jimmunol.1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stemmer U, Hermetter A. Protein modification by aldehydophospholipids and its functional consequences. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamem.2012.03.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kincer JF, Uittenbogaard A, Dressman J, Guerin TM, Febbraio M, Guo L, Smart EJ. Hypercholesterolemia promotes a cd36-dependent and endothelial nitric-oxide synthase-mediated vascular dysfunction. J Biol Chem. 2002;277:23525–23533. doi: 10.1074/jbc.M202465200. [DOI] [PubMed] [Google Scholar]
- 71.Rahaman SO, Zhou G, Silverstein RL. Vav protein guanine nucleotide exchange factor regulates cd36 protein-mediated macrophage foam cell formation via calcium and dynamin-dependent processes. J Biol Chem. 2011;286:36011–36019. doi: 10.1074/jbc.M111.265082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Venkatesha RT, Ahamed J, Nuesch C, Zaidi AK, Ali H. Platelet-activating factor-induced chemokine gene expression requires nf-kappab activation and ca2+/calcineurin signaling pathways. Inhibition by receptor phosphorylation and beta-arrestin recruitment. J Biol Chem. 2004;279:44606–44612. doi: 10.1074/jbc.M408035200. [DOI] [PubMed] [Google Scholar]
- 73.Imaizumi S, Grijalva V, Priceman S, Wu L, Su F, Farias-Eisner R, Hama S, Navab M, Fogelman AM, Reddy ST. Mitogen-activated protein kinase phosphatase-1 deficiency decreases atherosclerosis in apolipoprotein e null mice by reducing monocyte chemoattractant protein-1 levels. Mol Genet Metab. 2010;101:66–75. doi: 10.1016/j.ymgme.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR. Abca1 and abcg1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010;106:1861–1869. doi: 10.1161/CIRCRESAHA.110.217281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen R, Yang L, McIntyre TM. Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J Biol Chem. 2007;282:24842–24850. doi: 10.1074/jbc.M702865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kagan VE, Gleiss B, Tyurina YY, Tyurin VA, Elenstrom-Magnusson C, Liu SX, Serinkan FB, Arroyo A, Chandra J, Orrenius S, Fadeel B. A role for oxidative stress in apoptosis: Oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing fas-mediated apoptosis. J Immunol. 2002;169:487–499. doi: 10.4049/jimmunol.169.1.487. [DOI] [PubMed] [Google Scholar]
- 77.Chen K, Li W, Major J, Rahaman SO, Febbraio M, Silverstein RL. Vav guanine nucleotide exchange factors link hyperlipidemia and a prothrombotic state. Blood. 2011;117:5744–5750. doi: 10.1182/blood-2009-01-201970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ishii H, Tezuka T, Ishikawa H, Takada K, Oida K, Horie S. Oxidized phospholipids in oxidized low-density lipoprotein down-regulate thrombomodulin transcription in vascular endothelial cells through a decrease in the binding of rarbeta-rxralpha heterodimers and sp1 and sp3 to their binding sequences in the tm promoter. Blood. 2003;101:4765–4774. doi: 10.1182/blood-2002-08-2428. [DOI] [PubMed] [Google Scholar]
- 79.Bochkov VN, Mechtcheriakova D, Lucerna M, Huber J, Malli R, Graier WF, Hofer E, Binder BR, Leitinger N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of erk/egr-1 and ca(++)/nfat. Blood. 2002;99:199–206. doi: 10.1182/blood.v99.1.199. [DOI] [PubMed] [Google Scholar]
- 80.Zimman A, Mouillesseaux KP, Le T, Gharavi NM, Ryvkin A, Graeber TG, Chen TT, Watson AD, Berliner JA. Vascular endothelial growth factor receptor 2 plays a role in the activation of aortic endothelial cells by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2007;27:332–338. doi: 10.1161/01.ATV.0000252842.57585.df. [DOI] [PubMed] [Google Scholar]
- 81.Ohkura N, Hiraishi S, Itabe H, Hamuro T, Kamikubo Y, Takano T, Matsuda J, Horie S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid Redox Signal. 2004;6:705–712. doi: 10.1089/1523086041361686. [DOI] [PubMed] [Google Scholar]
- 82.O’Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, McCarty J, Ferguson M, Hudkins K, Benjamin CD, et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993;92:945–951. doi: 10.1172/JCI116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–159. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 84.Cole AL, Subbanagounder G, Mukhopadhyay S, Berliner JA, Vora DK. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a camp-dependent r-ras/pi3-kinase pathway. Arterioscler Thromb Vasc Biol. 2003;23:1384–1390. doi: 10.1161/01.ATV.0000081215.45714.71. [DOI] [PubMed] [Google Scholar]
- 85.Birukova AA, Zebda N, Fu P, Poroyko V, Cokic I, Birukov KG. Association between adherens junctions and tight junctions via rap1 promotes barrier protective effects of oxidized phospholipids. J Cell Physiol. 2010;226:2052–2062. doi: 10.1002/jcp.22543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birukova AA, Zebda N, Cokic I, Fu P, Wu T, Dubrovskyi O, Birukov KG. P190rhogap mediates protective effects of oxidized phospholipids in the models of ventilator-induced lung injury. Exp Cell Res. 2011;317:859–872. doi: 10.1016/j.yexcr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of rhoa/rho kinase signaling in vegf-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- 88.DeMaio L, Rouhanizadeh M, Reddy S, Sevanian A, Hwang J, Hsiai TK. Oxidized phospholipids mediate occludin expression and phosphorylation in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2006;290:H674–683. doi: 10.1152/ajpheart.00554.2005. [DOI] [PubMed] [Google Scholar]
- 89.Zimman A, Chen SS, Komisopoulou E, Titz B, Martinez-Pinna R, Kafi A, Berliner JA, Graeber TG. Activation of aortic endothelial cells by oxidized phospholipids: A phosphoproteomic analysis. J Proteome Res. 2010;9:2812–2824. doi: 10.1021/pr901194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nonas S, Miller I, Kawkitinarong K, Chatchavalvanich S, Gorshkova I, Bochkov VN, Leitinger N, Natarajan V, Garcia JG, Birukov KG. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury. Am J Respir Crit Care Med. 2006;173:1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li R, Chen W, Yanes R, Lee S, Berliner JA. Okl38 is an oxidative stress response gene stimulated by oxidized phospholipids. J Lipid Res. 2007;48:709–715. doi: 10.1194/jlr.M600501-JLR200. [DOI] [PubMed] [Google Scholar]
- 92.Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, Oskolkova O, Afonyushkin T, Koistinaho J, Yamamoto M, Bochkov VN, Yla-Herttuala S, Levonen AL. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008;103:e1–9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 93.Afonyushkin T, Oskolkova OV, Binder BR, Bochkov VN. Involvement of ck2 in activation of electrophilic genes in endothelial cells by oxidized phospholipids. J Lipid Res. 2010;52:98–103. doi: 10.1194/jlr.M009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee S, Li R, Kim B, Palvolgyi R, Ho T, Yang QZ, Xu J, Szeto WL, Honda H, Berliner JA. Ox-papc activation of pmet system increases expression of heme oxygenase-1 in human aortic endothelial cell. J Lipid Res. 2009;50:265–274. doi: 10.1194/jlr.M800317-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of camp-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 96.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 97.Lee S, Gharavi NM, Honda H, Chang I, Kim B, Jen N, Li R, Zimman A, Berliner JA. A role for nadph oxidase 4 in the activation of vascular endothelial cells by oxidized phospholipids. Free Radic Biol Med. 2009;47:145–151. doi: 10.1016/j.freeradbiomed.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vladykovskaya E, Sithu SD, Haberzettl P, Wickramasinghe NS, Merchant ML, Hill BG, McCracken J, Agarwal A, Dougherty S, Gordon SA, Schuschke DA, Barski OA, O’Toole T, D’Souza SE, Bhatnagar A, Srivastava S. The lipid peroxidation product, 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. J Biol Chem. 2012 doi: 10.1074/jbc.M111.320416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Romanoski CE, Lee S, Kim MJ, Ingram-Drake L, Plaisier CL, Yordanova R, Tilford C, Guan B, He A, Gargalovic PS, Kirchgessner TG, Berliner JA, Lusis AJ. Systems genetics analysis of gene-by-environment interactions in human cells. Am J Hum Genet. 2010;86:399–410. doi: 10.1016/j.ajhg.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gharavi NM, Baker NA, Mouillesseaux KP, Yeung W, Honda HM, Hsieh X, Yeh M, Smart EJ, Berliner JA. Role of endothelial nitric oxide synthase in the regulation of srebp activation by oxidized phospholipids. Circulation research. 2006;98:768–776. doi: 10.1161/01.RES.0000215343.89308.93. [DOI] [PubMed] [Google Scholar]
- 101.Colgan SM, Hashimi AA, Austin RC. Endoplasmic reticulum stress and lipid dysregulation. Expert Rev Mol Med. 2011;13:e4. doi: 10.1017/S1462399410001742. [DOI] [PubMed] [Google Scholar]
- 102.Singh NK, Wang D, Kundumani-Sridharan V, Van Quyen D, Niu J, Rao GN. 15-lipoxygenase-1-enhanced src-janus kinase 2-signal transducer and activator of transcription 3 stimulation and monocyte chemoattractant protein-1 expression require redox-sensitive activation of epidermal growth factor receptor in vascular wall remodeling. J Biol Chem. 2011;286:22478–22488. doi: 10.1074/jbc.M111.225060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gharavi NM, Alva JA, Mouillesseaux KP, Lai C, Yeh M, Yeung W, Johnson J, Szeto WL, Hong L, Fishbein M, Wei L, Pfeffer LM, Berliner JA. Role of the jak/stat pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem. 2007;282:31460–31468. doi: 10.1074/jbc.M704267200. [DOI] [PubMed] [Google Scholar]
- 104.Springstead JR, Gugiu BG, Lee S, Cha S, Watson AD, Berliner JA. Evidence for the importance of oxpapc interaction with cysteines in regulating endothelial cell function. J Lipid Res. 2012;53:1304–1315. doi: 10.1194/jlr.M025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee H, Shi W, Tontonoz P, Wang S, Subbanagounder G, Hedrick CC, Hama S, Borromeo C, Evans RM, Berliner JA, Nagy L. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ Res. 2000;87:516–521. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- 106.Subbanagounder G, Wong JW, Lee H, Faull KF, Miller E, Witztum JL, Berliner JA. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J Biol Chem. 2002;277:7271–7281. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- 107.Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–1838. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- 108.Yoshida T, Gan Q, Owens GK. Kruppel-like factor 4, elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol. 2008;295:C1175–1182. doi: 10.1152/ajpcell.00288.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chatterjee S, Berliner JA, Subbanagounder GG, Bhunia AK, Koh S. Identification of a biologically active component in minimally oxidized low density lipoprotein (mm-ldl) responsible for aortic smooth muscle cell proliferation. Glycoconj J. 2004;20:331–338. doi: 10.1023/B:GLYC.0000033629.54962.68. [DOI] [PubMed] [Google Scholar]
- 110.Johnstone SR, Kroncke BM, Straub AC, Best AK, Dunn CA, Mitchell LA, Peskova Y, Nakamoto RK, Koval M, Lo CW, Lampe PD, Columbus L, Isakson BE. Mapk phosphorylation of connexin 43 promotes binding of cyclin e and smooth muscle cell proliferation. Circ Res. 2012;111:201–211. doi: 10.1161/CIRCRESAHA.112.272302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Poeckel D, Zemski Berry KA, Murphy RC, Funk CD. Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in apoe knock-out mice. J Biol Chem. 2009;284:21077–21089. doi: 10.1074/jbc.M109.000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111:2798–2804. doi: 10.1161/CIRCULATIONAHA.104.516278. [DOI] [PubMed] [Google Scholar]
- 113.Subbanagounder G, Leitinger N, Schwenke DC, Wong JW, Lee H, Rizza C, Watson AD, Faull KF, Fogelman AM, Berliner JA. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000;20:2248–2254. doi: 10.1161/01.atv.20.10.2248. [DOI] [PubMed] [Google Scholar]
- 114.Liu J, Chen R, Marathe GK, Febbraio M, Zou W, McIntyre TM. Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis by lipoprotein-associated phospholipase a2/ paf acetylhydrolase. Circ Res. 2011;108:469–477. doi: 10.1161/CIRCRESAHA.110.228742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mallat Z, Lambeau G, Tedgui A. Lipoprotein-associated and secreted phospholipases a in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation. 2010;122:2183–2200. doi: 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- 116.Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, Bhatnagar A, Srivastava S. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J. 2007;405:95–105. doi: 10.1042/BJ20061743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase a2 activities. J Biol Chem. 2000;275:28421–28427. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- 118.Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 119.Guo Z, Ran Q, Roberts LJ, 2nd, Zhou L, Richardson A, Sharan C, Wu D, Yang H. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein e-deficient mice. Free Radic Biol Med. 2008;44:343–352. doi: 10.1016/j.freeradbiomed.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vladykovskaya E, Ozhegov E, Hoetker JD, Xie Z, Ahmed Y, Suttles J, Srivastava S, Bhatnagar A, Barski OA. Reductive metabolism increases the proinflammatory activity of aldehyde phospholipids. J Lipid Res. 2011;52:2209–2225. doi: 10.1194/jlr.M013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, Chung SS, Hunt G, Dawn B, Bhatnagar A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein e-null mice. Circ Res. 2009;105:793–802. doi: 10.1161/CIRCRESAHA.109.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tsimikas S, Miyanohara A, Hartvigsen K, Merki E, Shaw PX, Chou MY, Pattison J, Torzewski M, Sollors J, Friedmann T, Lai NC, Hammond HK, Getz GS, Reardon CA, Li AC, Banka CL, Witztum JL. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol. 2011;58:1715–1727. doi: 10.1016/j.jacc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. The Journal of biological chemistry. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 124.Greenberg ME, Li XM, Gugiu BG, Gu XD, Qin J, Salomon RG, Hazen SL. The lipid whisker model of the structure of oxidized cell membranes. Journal of Biological Chemistry. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 125.Subbanagounder G, Deng Y, Borromeo C, Dooley AN, Berliner JA, Salomon RG. Hydroxy alkenal phospholipids regulate inflammatory functions of endothelial cells. Vascular pharmacology. 2002;38:201–209. doi: 10.1016/s1537-1891(02)00170-2. [DOI] [PubMed] [Google Scholar]
- 126.Deng YH, Salomon RG. Total synthesis of gamma-hydroxy-alpha,beta-unsaturated aldehydic esters of cholesterol and 2-lysophosphatidylcholine. Journal of Organic Chemistry. 1998;63:7789–7794. [Google Scholar]
- 127.Choi J, Laird JM, Salomon RG. An efficient synthesis of gamma-hydroxy-alpha,beta-unsaturated aldehydic esters of 2-lysophosphatidylcholine. Bioorganic & medicinal chemistry. 2011;19:580–587. doi: 10.1016/j.bmc.2010.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gao S, Zhang R, Greenberg ME, Sun M, Chen X, Levison BS, Salomon RG, Hazen SL. Phospholipid hydroxyalkenals, a subset of recently discovered endogenous cd36 ligands, spontaneously generate novel furan-containing phospholipids lacking cd36 binding activity in vivo. The Journal of biological chemistry. 2006;281:31298–31308. doi: 10.1074/jbc.M604039200. [DOI] [PubMed] [Google Scholar]
- 129.Sun M, Deng Y, Batyreva E, Sha W, Salomon RG. Novel bioactive phospholipids: Practical total syntheses of products from the oxidation of arachidonic and linoleic esters of 2-lysophosphatidylcholine(1) The Journal of organic chemistry. 2002;67:3575–3584. doi: 10.1021/jo0105383. [DOI] [PubMed] [Google Scholar]
- 130.Legrand AB, Wang J, Sobo G, Blair IA, Brash AR, Oates JA. Incorporation of 12(s)-hydroxyeicosatetraenoic acid into the phosphatidylcholine signaling pathway. Biochimica et biophysica acta. 1996;1301:150–160. doi: 10.1016/0005-2760(96)00015-x. [DOI] [PubMed] [Google Scholar]
- 131.Zhang JY, Nobes BJ, Wang JM, Blair IA. Characterization of hydroxyeicosatetraenoic acids and hydroxyeicosatetraenoic acid phosphatidylcholines by liquid secondary-ion tandem mass-spectrometry. Biol Mass Spectrom. 1994;23:399–405. doi: 10.1002/bms.1200230704. [DOI] [PubMed] [Google Scholar]
- 132.Cho YH, Ziboh VA. Incorporation of 13-hydroxyoctadecadienoic acid (13-hode) into epidermal ceramides and phospholipids - phospholipase c-catalyzed release of novel 13-hode-containing diacylglycerol. Journal of lipid research. 1994;35:255–262. [PubMed] [Google Scholar]
- 133.Sun M, Salomon RG. Oxidative fragmentation of hydroxy octadecadienoates generates biologically active gamma-hydroxyalkenals. Journal of the American Chemical Society. 2004;126:5699–5708. doi: 10.1021/ja038756w. [DOI] [PubMed] [Google Scholar]
- 134.Watson AD, Subbanagounder G, Welsbie DS, Faull KF, Navab M, Jung ME, Fogelman AM, Berliner JA. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. The Journal of biological chemistry. 1999;274:24787–24798. doi: 10.1074/jbc.274.35.24787. [DOI] [PubMed] [Google Scholar]
- 135.Jung ME, Berliner JA, Koroniak L, Gugiu BG, Watson AD. Improved synthesis of the epoxy isoprostane phospholipid peipc and its reactivity with amines. Organic letters. 2008;10:4207–4209. doi: 10.1021/ol8014804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jung ME, Berliner JA, Angst D, Yue D, Koroniak L, Watson AD, Li R. Total synthesis of the epoxy isoprostane phospholipids peipc and pecpc. Organic letters. 2005;7:3933–3935. doi: 10.1021/ol051415y. [DOI] [PubMed] [Google Scholar]
- 137.Salomon RG, Subbanagounder G, Singh U, O’Neil J, Hoff HF. Oxidation of low-density lipoproteins produces levuglandin-protein adducts. Chemical research in toxicology. 1997;10:750–759. doi: 10.1021/tx970016b. [DOI] [PubMed] [Google Scholar]
- 138.Brose SA, Thuen BT, Golovko MY. Lc/ms/ms method for analysis of e-2 series prostaglandins and isoprostanes. Journal of lipid research. 2011;52:850–859. doi: 10.1194/jlr.D013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen R, Yang L, McIntyre TM. Cytotoxic phospholipid oxidation products - cell death from mitochondrial damage and the intrinsic caspase cascade. Journal of Biological Chemistry. 2007;282:24842–24850. doi: 10.1074/jbc.M702865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Milne GL, Porter NA. Separation and identification of phospholipid peroxidation products. Lipids. 2001;36:1265–1275. doi: 10.1007/s11745-001-0841-2. [DOI] [PubMed] [Google Scholar]