

Abstract

New arylthioindole derivatives having different cyclic substituents at position 2 of the indole were synthesized as anticancer agents. Several compounds inhibited tubulin polymerization at submicromolar concentration and inhibited cell growth at low nanomolar concentrations. Compounds 18 and 57 were superior to the previously synthesized 5. Compound 18 was exceptionally potent as an inhibitor of cell growth: it showed IC50 = 1.0 nM in MCF-7 cells, and it was uniformly active in the whole panel of cancer cells and superior to colchicine and combretastatin A-4. Compounds 18, 20, 55, and 57 were notably more potent than vinorelbine, vinblastine, and paclitaxel in the NCI/ADR-RES and Messa/Dx5 cell lines, which overexpress P-glycoprotein. Compounds 18 and 57 showed initial vascular disrupting effects in a tumor model of liver rhabdomyosarcomas at 15 mg/kg intravenous dosage. Derivative 18 showed water solubility and higher metabolic stability than 5 in human liver microsomes.
INTRODUCTION
Microtubules (MTs) are involved in many essential cellular functions, e.g., the maintenance of cell shape, cell motility, intracellular transport, and cell division. Cellular MTs undergo continuous polymerization and depolymerization transitions. Interference with this dynamic equilibrium, by either inhibiting tubulin polymerization or blocking MT disassembly, prevents proper MT function and ultimately leads to cell death. Because of their crucial role in the formation of the mitotic spindle during cell division, MTs are a highly attractive target for the development of new effective anticancer agents.1–5
Natural products such as colchicine (1),6,7 combretastatin A-4 (CSA4, 2)8 (Chart 1), and the Catharanthus alkaloids vincristine and vinblastine (VBL) inhibit MT assembly by preventing tubulin polymerization. On the other hand, taxoids and epothilones target a lumenal site on the β-subunit.9,10 These drugs enter the lumen through a binding site11 located at a pore on the MT surface formed by different tubulin heterodimers. At high concentrations, paclitaxel (PTX) stimulates MT polymerization and stabilizes microtubules, whereas at lower concentrations it inhibits MT dynamics with little effect on the proportion of tubulin in polymer.12 Despite substantial interest in the development of new MT-targeting drugs,13 several problems related to drug resistance and secondary toxicity remain unresolved. Therefore, new and improved tubulin inhibitors are needed.14
Chart 1. General Structure of ATI Derivatives 7–78 and Reference Compounds 1–6a.

aR1 = azolyl, azinyl, phenyl, or substituted phenyl, naphthyl, biphenyl, benzofused heterocyclyl, alkyl, cycloalkyl; R2 = H; X = S, C==O, CHOH, or CH2 (Tables 1 and 2).
Arylthioindoles (ATIs) are a class of potent inhibitors of tubulin polymerization and cancer cell growth developed by our research group.15–19 The ATIs inhibit tubulin polymerization by binding to the colchicine site, inhibiting the binding of [3H]colchicine to tubulin.15
In our previous work,19 we obtained new ATIs by replacing the 2-alkoxycarbonyl group with a bioisosteric five-membered heterocycle nucleus. The new ATIs (i.e., 3–6) inhibited tubulin polymerization, inhibited the growth of a panel of human transformed cell lines, and showed higher metabolic stability than the reference ester (see example in Chart 1).
These new ATIs showed significant therapeutic potential, as they induced mitotic arrest and apoptosis similarly to 2 and VBL and triggered caspase-3 expression in a significant fraction of cells in both p53-positive and p53-negative cell lines.20 Importantly, some ATIs were more effective than VRB, VBL, and PTX as growth inhibitors of the P-glycoprotein-overexpressing cell line NCI/ADR-RES. These compounds had a pharmacokinetic profile in the mouse characterized by low systemic clearance and excellent oral bioavailability. On the basis of these observations, the current study was designed to explore further chemical modifications of the five-membered heterocycle nucleus at position 2 of the indole. We describe the synthesis and biological evaluation of new ATI derivatives 7–78 with different biososteric rings at position 2, with a sulfur bridge between the ring systems or with other bioisosteric groups at position 3 of the indole (Table 1).
Table 1.
Inhibition of Tubulin Polymerization, Growth of MCF-7 Human Breast Carcinoma Cells, and Colchicine Binding by Compounds 7–78
 7-78 | |||||
|---|---|---|---|---|---|
| Compd | R1 | X | Tubulin Assemblya IC50 ± SD (μM) | MCF-7b IC50 ± SD (nM) | Colchicine Bindingc (% ± SD) |
| 7 |

|
C=O | 1.4 ± 0.2 | 10 ± 0 | 79 ± 3 |
| 8 |

|
C=O | 1.8 ± 0.2 | 35 ± 7 | 71 ± 0.7 |
| 9 |

|
C=O | 29 ± 1 | ndd | ndd |
| 10 |

|
CHOH | 1.8 ± 0.1 | 30 ± 0 | 70 ± 6 |
| 11 |

|
CHOH | 9.0 ± 1 | nd | nd |
| 12 |

|
CH2 | >40 | nd | nd |
| 13 |

|
CH2 | 3.0 ± 0.2 | 600 ± 0 | 56 ± 7 |
| 14 |

|
S | 0.92 ± 0.2 | 35 ± 20 | 81± 1 |
| 15 |

|
C=O | 1.7 ± 0.04 | 25 ± 7 | 68 ± 1 |
| 16 |

|
S | 1.2 ± 0.1 | 50 ± 10 | 78 ± 0.5 |
| 17 |

|
S | 4.6 ± 0.4 | 270 ± 60 | 29 ± 3 |
| 18 |

|
S | 1.3 ± 0.06 | 1.0 ± 0.5 | 92 ± 0.8 |
| 19 |

|
C=O | 3.5 ± 0.2 | 120 ± 0 | 42 ± 2 |
| 20 |
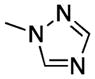
|
S | 1.2 ± 0.1 | 11 ± 4 | 83 ± 1 |
| 21 |

|
C=O | 1.9 ± 0.2 | 55 ± 7 | 78 ± 0.3 |
| 22 |

|
CH2 | 6.8 ± 1 | nd | nd |
| 23 |

|
C=O | 1.1 ± 0.007 | 40 ± 20 | 77 ± 0.8 |
| 24 |

|
CH2 | 6.5 ± 0.08 | nd | nd |
| 25 |

|
C=O | 2.1 ± 0.08 | 40 ± 0 | 61 ± 3 |
| 26 |

|
CH2 | 2.7 ± 0.08 | 340 ± 200 | 52 ± 4 |
| 27 |

|
S | 0.96 ± 0.1 | 80 ± 20 | 72 ± 6 |
| 28 |

|
S | 3.3 ± 0.6 | 160 ± 50 | 37 ± 0.8 |
| 29 |

|
C=O | 14 ± 0.8 | nd | nd |
| 30 |

|
CH2 | 9.4 ± 0.01 | nd | nd |
| 31 |

|
S | 4.5 ± 0.2 | 270 ± 60 | 51 ± 2 |
| 32 |

|
C=O | 3.4 ± 0.2 | 80 ± 0 | 44 ± 5 |
| 33 |

|
S | >40 | nd | nd |
| 34 |

|
S | 3.3 ± 0.1 | 52 ± 7 | nd |
| 35 |

|
C=O | 5.7 ± 0.06 | 87 ± 20 | nd |
| 36 |

|
CH2 | 3.7 ± 0.3 | 170 ± 60 | 50 ± 3 |
| 37 |

|
S | 0.94 ± 0.08 | 70 ± 40 | 61 ± 3 |
| 38 |

|
S | 1.8 ± 0.2 | 190 ± 20 | 50 ± 3 |
| 39 |

|
S | 1.7 ± 0.2 | 50 ± 0 | 74 ± 2 |
| 40 |

|
C=O | 2.1 ± 0.1 | 60 ± 10 | 55 ± 2 |
| 41 |

|
S | 1.7 ± 0.04 | 63 ± 4 | 75 ± 0.6 |
| 42 |

|
C=O | 7.6 ± 0.4 | nd | nd |
| 43 |

|
S | 1.8 ± 0.2 | 63 ± 4 | 74 ± 0.06 |
| 44 |

|
C=O | 9.4 ± 0.2 | nd | nd |
| 45 |

|
S | 3.3 ± 0.06 | 530 ± 60 | 25 ± 8 |
| 46 |

|
S | 3.6 ± 0.1 | 73 ± 20 | 32 ± 20 |
| 47 |

|
S | 0.94 ± 0.08 | 56 ± 7 | 47 ± 1 |
| 48 |

|
S | >40 | nd | nd |
| 49 |

|
S | 1.1 ± 0.08 | 140 ± 20 | 68 ± 3 |
| 50 |

|
S | >40 | nd | nd |
| 51 |

|
C=O | 4.1 ± 0.6 | 200 ± 40 | 24 ± 5 |
| 52 |

|
CH2 | >40 | nd | nd |
| 53 |

|
S | 1.5 ± 0.2 | 180 ± 80 | 48 ± 6 |
| 54 |

|
C=O | >20 | 330 ±100 | nd |
| 55 |

|
S | 1.3 ± 0.07 | 12 ± 7 | 88 ± 0.1 |
| 56 |

|
C=O | 6.3 ± 0.7 | 200 ± 100 | nd |
| 57 |

|
S | 0.95 ± 0.1 | 16 ± 10 | 91 ± 2 |
| 58 |

|
C=O | 6.0 ± 0.6 | 170 ± 60 | nd |
| 59 |

|
S | >40 | nd | nd |
| 60 |

|
C=O | >40 | nd | nd |
| 61 |

|
CH2 | >40 | nd | nd |
| 62 |

|
S | 1.0 ± 0.06 | 530 ± 50 | 51 ± 6 |
| 63 |

|
C=O | >40 | nd | nd |
| 64 |

|
CHOH | >40 | nd | nd |
| 65 |

|
CH2 | >40 | nd | nd |
| 66 |

|
S | 4.0 ± 0.1 | 900 ± 0 | 29 ± 6 |
| 67 |

|
S | 3.1 ± 0.3 | 450 ± 0 | 46 ± 7 |
| 68 |

|
S | 1.8 ± 0.1 | 150 ± 70 | 57 ± 3 |
| 69 |

|
S | 1.5 ± 0.1 | 110 ± 7 | 59 ± 0.7 |
| 70 |

|
S | 1.7 ± 0.1 | 580 ± 50 | 52 ± 5 |
| 71 |

|
S | >40 | nd | nd |
| 72 |
|
S | >40 | nd | nd |
| 73 |

|
S | 2.0 ± 0.08 | 150 ± 50 | 43 ± 1 |
| 74 |

|
C=O | 2.8 ± 0.2 | 110 ± 20 | 42 ± 5 |
| 75 |

|
S | 2.6 ± 0.08 | 200 ± 0 | 38 ± 5 |
| 76 |

|
C=O | 4.0 ± 0.08 | 170 ± 50 | 33 ± 5 |
| 77 |

|
S | >20 | 430 ± 200 | nd |
| 78 |

|
C=O | >20 | 1500 ± 700 | nd |
| 1 | - | - | 3.2 ± 0.4 | 5 ± 1 | - |
| 2 | - | - | 1.0 ± 0.1 | 13 ± 3 | 98 ± 0.6 |
| 3e |

|
S | 1.1 ± 0.05 | 18 ± 6 | 90 ± 4 |
| 4e |

|
S | 1.2 ± 0.2 | 20 ± 0 | 85 ± 1 |
| 5e |

|
S | 0.74 ± 0.05 | 39 ± 10 | 86 ± 0.7 |
| 6e |

|
C=O | 1.0 ± 0.1 | 36 ± 6 | 75 ± 3 |
Inhibition of tubulin polymerization. Tubulin was 10 μM during polymerization.
Inhibition of growth of MCF-7 human breast carcinoma cells.
Inhibition of [3H]colchicine binding. Tubulin was at 1 μM. Both [3H]colchicine and inhibitor were at 5 μM.
Compounds that inhibited tubulin assembly with IC50 ≤ 5 μM were tested in the cellular and colchicine binding assays.
Lit.19 Plots of inhibition of tubulin polymerization and MCF-7 cell growth by compounds 7–78 are shown in Supporting Information.
CHEMISTRY
ATI derivatives 17, 18, 20, 27, 33, 34, 38, 39, 41, 43, 45–47, 53, 55, 57, 73, 75, and 77 were synthesized by microwave reaction of an appropriate indole 79–87 with bis(3,4,5-tri-methoxyphenyl)disulfide in the presence of sodium hydride in a closed vessel at 110 °C while irradiating at 150 W for 2 min (Scheme 1). Compounds 14, 16, 37, 48, 59, 62, 72 were obtained by heating at 60 °C in DMF the corresponding indoles 98–104 with bis(3,4,5-trimethoxyphenyl)disulfide in the presence of sodium hydride. Acids 105 and 106 were similarly transformed into the corresponding sulfides 107 and 108, respectively. Acid 107 was then converted into ATI 28 by treatment with bromoethylamine hydrobromide in the presence of benzotriazol-1-yloxytris(dimethylamino)phosphonium hexa-fluorophosphate (BOP reagent) and triethylamine. Iodo derivative 108 was transformed into ATI 66 or 67 by heating at 110 °C with an appropriate boronic acid pinacol ester in the presence of 1,1′-bis(diphenylphosphino)ferrocene]dichloro-palladium(II) complex with dichloromethane (Pd(dppf)-Cl2·CH2Cl2) and potassium phosphate in anhydrous DMF. Organostannane reactions of 108 with 2-(triethylstannyl)-1H-indole-1-carboxylic acid in the presence of bis(triphenylphosphine)palladium(II) dichloride (PdCl2(PPh3)2) in boiling ethanol for 48 h or 2-(tributylstannyl)oxazole, tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3), and tricyclohexyl-phosphine P(Cy)3 in anhydrous DMF at 150 °C and 150 W afforded 68 or 31, respectively. The synthesis of 69 was performed by heating 108 at 100 °C in aqueous 1,4-dioxane in the presence of 2-benzofuranylboronic acid, palladium(II) acetate, and potassium phosphate. ATI 70 was synthesized by reaction of 108 with benzo[b]thien-2-ylboronic acid in the presence of tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) and potassium carbonate in tetrahydrofuran at 60 °C. Compound 71 was obtained from 108 and benzo[b]thien-3-ylboronic acid in the presence of Pd2(dba)3, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos), and potassium phosphate in n-butanol at 100 °C.
Scheme 1a.

aReagents and reaction conditions: (a) (i) indoles 79–97 (R1 = imidazol-2-yl (79), imidazol-1-yl (80),21 1,2,4-triazol-1-yl (81), thiazol-2-yl (82), 4,5-dihydro-1H-imidazol-2-yl (83), phenyl (84), 4-chlorophenyl (85), 2-fluorophenyl (86), 3-fluorophenyl (87), 4-fluorophenyl (88), 2-methoxyphenyl (89), 3-methoxyphenyl (90), 4-methoxyphenyl (91), pyridin-2-yl (92), pyridin-3-yl (93), pyridin-4-yl (94), cyclobutyl (95), cyclopentyl (96), cyclohexyl (97)), NaH, anhydrous DMF, 25 °C, 10 min; (ii) closed vessel, 110 °C, 150 W, 2 min, yield 3–77%; (b) (i) indoles 98–104 (R1 = pyrazol-4-yl (98), pyrazol-3-yl (99),22 4-tolyl (100), 3-isopropoxy-4-methoxyphenyl (101), naphth-1-yl (102), naphth-2-yl (103), 1,1′-biphenyl-4-yl (104), COOH (105), I (106)23), NaH, anhydrous DMF, 0 °C, 15 min, Ar stream; (ii) 60 °C, 12 h, Ar stream, 35–72%; (c) Et3N, BOP reagent, anhydrous DMF, 25 °C, 2 h, 30%; (d) 6-quinolineboronic acid pinacol ester (for 66) or 1-Boc-indole-5-boronic acid pinacol ester (for 67), Pd(dppf)Cl2·CH2Cl2, K3PO4, anhydrous DMF, Ar stream, 110 °C, 12 h, yield 26–70%; (e) 2-(triethylstannyl)-1H-indole-1-carboxylic acid24 PdCl2(PPh3)2, ethanol, reflux, 78 °C, 48 h, Ar stream, yield 20%; (f) 2-(tributylstannyl)oxazole, Pd2(dba)3, P(Cy)3, anhydrous DMF, closed vessel, 150 °C, 150 W, 10 min, yield 35%; (g) 2-benzofuranylboronic acid, Pd(OCOMe)2, K3PO4, 1,4-dioxane/H2O, 100 °C, 24 h, Ar stream, yield 88%; (h) benzo[b]thien-2-ylboronic acid, Pd(PPh3)4, Na2CO3, THF, 60 °C, 24 h, Ar stream, yield 10%; (i) benzo[b]thien-3-ylboronic acid, Pd2(dba)3, SPhos, K3PO4, n-BuOH, Ar stream, 100 °C, 12 h, yield 36%.
Ketones 7, 15, 19, 54, 56, 58, and 60 were prepared by reaction of indoles 80, 92–94, 98, 102, and 109 with methyl-magnesium bromide in the presence of anhydrous zinc chloride at 25 °C and subsequent treatment with 3,4,5-trimethoxybenzoyl chloride and tin(IV) chloride (Scheme 2). Ketones 9, 21, 23, 25, 32, 35, 40, 42, 44, 51, 63, 74, 76, and 78 were obtained by reaction of the appropriate indoles 86–88, 91, 94–96, 103, and 110–114 with 3,4,5-trimethoxybenzoyl chloride in the presence of anhydrous aluminum chloride. Reduction of ketones 9, 21, 23, 25, 35, 51, 60, and 63 with borane–tetrahydrofuran complex solution in acetonitrile/methanol at 50 °C for 1 h afforded the corresponding methylene compounds 12, 22, 24, 26, 36, 52, 61, and 65. Sodium hydroxide hydrolysis of 9, 11 (obtained by sodium borohydride reduction of 9), or 12 in boiling methanol provided ketone 8, 10, or 23, respectively.
Scheme 2a.

aReagents and reaction conditions: (a) (i) indoles 80, 92–94, 98, 102, 109 (R1 = imidazol-1-yl (80),21 pyridin-2-yl (92), pyridin-3-yl (93), pyridin-4-yl (94), pyrazol-4-yl (98), naphth-1-yl (102), pyrrol-2-yl (109),25 MeMgBr, anhydrous ZnCl2, anhydrous CH2Cl2, 25 °C, 1 h, Ar stream; (ii) 3,4,-5-trimethoxybenzoyl chloride, 25 °C, 1 h, Ar stream; (iii) SnCl4, 25 °C, 12 h, Ar stream, yield 3–67%; (b) indoles 86–88, 91, 94–96, 103, 110–114 (R1 = phenyl (84), 2-fluorophenyl (86), 3-fluorophenyl (87), 4-fluorophenyl (88), 4-methoxyphenyl (91), cyclobutyl (94), cyclopentyl (95), cyclohexyl (96), naphth-2-yl (103), 1-(phenylsulfonyl)-1H-pyrrol-3-yl (110),19 furan-2-yl (111),19 furan-3-yl (112),19 thiophen-3-yl (113),19 oxazol-2-yl (114)), 3,4,5-trimethoxybenzoyl chloride, anhydrous AlCl3, 1,2-dichloroethane, closed vessel, 110 °C, 150 W, 2 min, yield 5–95%; (c): BH3/THF, MeCN/MeOH, 50 °C, 1 h, Ar stream, yield 4–99%; (d) 2 M NaOH, MeOH, reflux, 3 h, yield 20–98%; (e) NaBH4, THF/H2O, 80 °C, 2 h, yield 44–97%.
2-(1H-Pyrazol-4-yl)-1H-indole (98) was synthesized by microwave reaction of 106 with 1-Boc-pyrazole-4-boronic acid pinacol ester in the presence of PdCl2(PPh3)2, 1 M Na2CO3, and DMF in a closed vessel at 160 °C while irradiating at 250 W for 10 min (Scheme 3). Solvent-free reaction at 150 °C of 106 with 1,2,4-triazole furnished 2-(1H-1,2,4-triazol-1-yl)-1H-indole (81). 2-(1H-Indol-2-yl)oxazole (114) was obtained as 31 by microwave reaction of 106 with 2-(tributylstannyl)oxazole in the presence of Pd2(dba)3 and P(Cy)3 in anhydrous DMF at 150 °C and 150 W. 2-(4,5-Dihydro-1H-imidazol-2-yl)-1H-indole (83) was prepared by treatment of 1H-indole-2-carbaldehyde (115)26 with ethylenediamine and subsequent iodine oxidation in the presence of potassium carbonate. The 1-phenylethanones were converted into 2-bromo-1-phenylethanones 116–121 with bromine in dichloromethane and then transformed into the corresponding 2-phenylindoles 86–90, 101, and 102 by heating at 170 °C with aniline in N,N-dimethylaniline. Microwave reaction of 2-bromo-1-(thiazol-2-yl)ethanone27 (116) with aniline in a closed vessel at 150 °C and 100 W for 1 min afforded 88. o-Toluidides 123–127 were transformed into the corresponding indoles 100, 104, and 95–97 with butyllithium in anhydrous tetrahydrofuran, while the mixture was warmed from –40 to 25 °C. Phenylhydrazono derivatives 128–133 underwent Fischer cyclization in polyphosphoric acid at 120 °C to give indoles 85, 91–94, and 103.
Scheme 3a.

aR2 = 2-F-Ph (86), 3-F-Ph (87), 4-F-Ph (88), 2-MeO-Ph (89), 3-MeO-Ph (90), 3-i-PrO-4-MeO-Ph (101), 1-naphthyl (102); R3 = cyclobutyl (95), cyclopentyl (96), cyclohexyl (97), 4-Me-Ph (100), 1,1′-biphenyl-4-yl (104); R4 = 4-Cl-Ph (85), 4-MeO-Ph (91), pyridin-2-yl (92), pyridin-3-yl (93) pyridin-4-yl (94), 2-naphthyl (103). Reagents and reaction conditions: (a) PdCl2(PPh3)2, 1 M Na2CO3, DMF, closed vessel, 160 °C, 250 W, 10 min, yield 26%; (b) 150 °C, 2 h, yield 42%; (c) Pd2(dba)3, P(Cy)3, anhydrous DMF, closed vessel, 150 °C, 150 W, 10 min, yield 26%; (d) (i) t-BuOH, Ar stream, 30 min; (ii) I2, K2CO3, 70 °C, 3 h, yield 43%; (e) (diacetoxyiodo)benzene, K2CO3, DMSO, 25 °C, 12 h; (f) anhydrous DMF, closed vessel, 150 °C, 100 W, 1 min, PowerMAX, yield 52%; (g) Br2, anhydrous CH2Cl2, 25 °C, 2 h, yield 21–90%; (h) R2 = 2-F (116), 3-F (117), 4-F (118), 2-MeO (119),28 3-MeO (120),28 3-i-PrO-4-MeO (121), 1-naphthyl (122), N,N-dimethylaniline, 170 °C, 15 min, yield 12–42%; (i) R3 = 4-tolyl (123), 1,1′-biphenyl-4-yl (124), cyclobutyl (125), cyclopentyl (126), cyclohexyl (127), anhydrous THF, 25 °C, 12 h, yield 40–90%; (j) (i) n-BuLi, anhydrous THF, –40 °C, 1 h, Ar stream; (ii) 0 °C, 1 h; (iii) 25 °C, 12 h, yield 22–59%; (k) R4 = 4-Cl-Ph (128), 4-MeO-Ph (129), pyridin-2-yl (130),29 pyridin-3-yl (131),29 pyridin-4-yl (132),29 2-naphthyl (133),29 CH3COONa, EtOH, open vessel, 250 W, cooling-while-heating, 80 °C, 5 min, yield 92–95%; (l) polyphosphoric acid, 120 °C, 1 h, yield 37–80%.
RESULTS AND DISCUSSION
Inhibition of Tubulin Polymerization
The effects of ATIs 7–78 on tubulin polymerization in vitro are shown in Table 1. On the basis of our previous results,18,19 we synthesized these compounds to obtain structure–activity relationship (SAR) information regarding position 2 of the indole moiety and the sulfur bridging group. Several new ATIs (7, 8, 10, 15, 16, 18, 20, 21, 23, 38, 39, 41, 43, 49, 53, 55, 68–70, and 73) inhibited tubulin polymerization with IC50 of 1.0–2.0 μM, and six compounds (14, 27, 37, 47, 57, and 62) yielded IC50 ≤ 1.0 μM, compared with 1.0 μM obtained for CSA4 (2) and 3.2 μM for colchicine (1).
For SAR studies regarding the sulfur bridging group, we synthesized 3-aroylindoles and several corresponding alcohol and methylene derivatives as potential bioisosteres of previously reported ATIs.19 As tubulin polymerization inhibitors, ketones 7 and 8 were less potent than their sulfur counterparts 3 and 4, respectively, as well as less active than the similar compounds 5 and 6. Similar results were were also obtained with ketones 21, 23, and 25 in comparison with the corresponding arylthioin-doles.19 Reduction of the ketone 8 to alcohol 10 resulted in an equipotent inhibitor of tubulin polymerization. Replacement of the sulfur bridging atom with a methylene group invariably caused a drop, sometimes dramatic, in inhibitory activity (cf. 4 with 13, 34 with 36, 28 with 30, 47 with 52, and 62 with 65).
Replacement of the pyrrol-3-yl group of 4 with a pyrazol-4-yl moiety provided ATI compound 14 (IC50 = 0.92 μM), which was more potent than the parent compound as an inhibitor of tubulin polymerization. This observation prompted the synthesis of new ATIs bearing a pyrazol-3-yl (16), imidazol-2-yl (17), imidazol-1-yl (18), 1,2,4-triazol-1-yl (20), thiazol-2-yl (27), or oxazol-2-yl (31) moiety at position 2 of the indole. With the exception of 17 and 31, these compounds potently inhibited tubulin polymerization with IC50 ranging from 0.96 μM (27) to 1.3 μM (18). Again, the ketones were less potent than the parent sulfur compounds (compare 14 with 15, and 18 with 19).
Introduction of a methyl, methoxy, chlorine, or fluorine moiety at the para or meta position of a phenyl ring at indole position 2 also provided potent tubulin polymerization inhibitors (i.e., 37, 38, 41, 43, and 47), compared with the unsubstituted 34. Interestingly, ATI 49, bearing the 3-hydroxy-4-methoxyphenyl substitution pattern (the same as 2) at indole position 2, inhibited tubulin assembly with IC50 = 1.1 μM, compared with 1.0 μM for 2. Introduction of a pyridin-2-yl (53, IC50 = 1.5 μM), pyridin-3-yl (55, IC50 = 1.3 μM), or pyridin-4-yl (57, IC50 = 0.95 μM) group resulted in potent inhibitors of tubulin assembly. The 2-naphthyl derivative 62 inhibited potently the tubulin polymerization (IC50 = 1.0 μM), but the 1-naphthyl derivative 59 was inactive. Compounds 68–70, bearing a benzofused heterocycl-2-yl substituent at position 2 of the indole, inhibited tubulin assembly with IC50 in the range of 1.5–1.8 μM.
Molecular modeling studies were consistent with the conclusion that the binding modes of these new ATIs in the colchicine site on tubulin were similar to those of previously reported ATIs19 (Supporting Information).
The new ATIs were also examined for potential inhibition of the binding of [3H]colchicine to tubulin (Table 1). Compounds 7, 8, 10, 14, 16, 18, 20, 21, 23, 27, 39, 41, 43, 55, and 57 inhibited the binding reaction by at least 70%. Among them, 18 (92%), 55 (88%), and 57 (91%) were the strongest inhibitors of this binding reaction, compared with 2 (98% inhibition).
Cell Growth Inhibition
ATIs 7, 8, 10, 14–16, 18, 20, 21, 23, 25, 34, 39, 40, 47, 55, and 57 inhibited the growth of human MCF-7 nonmetastatic breast cancer epithelial cells with IC50 ≤ 60 nM (Table 1). Five compounds (7, 18, 20, 55, and 57) had IC50 ≤ 16 nM, with 18 (IC50 = 1.0 nM) being the most potent. As an inhibitor of MCF-7 cell growth, 18 was 5 and 13 times more potent than 1 and 2, respectively. Moreover, this ATI was 18–39 times more effective than the reference ATI compounds 3–6.
Many of the new agents were potent inhibitors of tubulin polymerization; however, compounds bearing the heterocycle nucleus at the indole position 2 were more effective as inhibitors of the growth of MCF-7 cells. ATI 49, having the same substitution pattern as 2, was only a relatively weak inhibitor of MCF-7 cell growth (IC50 = 140 μM). ATI derivatives 18 (IC50 = 1.0 nM) and 20 (IC50 = 11 nM), bearing an imidazol-1-yl or 1,2,4-triazol-1-yl nucleus, respectively, at indole position 2, were powerful inhibitors of the MCF-7 cell line. In contrast to 18, compounds 14 (IC50 = 35 nM), 16 (IC50 = 50 nM), 17 (IC50 = 270 nM), 27 (IC50 = 80 nM), and 32 (IC50 = 270 nM), each having two heteroatoms in the 2-heterocyclyl moiety, were less active inhibitors of the growth of this cell line.
Compounds 7, 8, 10, 14, 15, 18, 20, 23, 25, 34, 47, 55, and 57 were evaluated for growth inhibition of HeLa, HT29 (human colon adenocarcinoma), A549 (human lung carcinoma), HCT 116, and HCT 15 (human colon carcinoma cell lines) in comparison with 1, 2, vinblastine (VBL), and paclitaxel (PTX) (Table 2). In terms of average IC50 for the five cell lines, the control compounds ranged from 17 to 40 nM (ignoring the high value obtained with 2 in the A549 cells). The average value for 18, 21 nM, was in the same range, and six other compounds (7, 8, 14, 20, 55, and 57) had average IC50 values less than 100 nM. All these compounds had IC50 vlues of 35 nM or less in the screening assay in the MCF7 cells.
Table 2.
Growth Inhibition of the HeLa, HT29, A549, HCT 116, and HCT 15 Cell Lines by Compounds 7, 8, 10, 14, 15, 18, 20, 23, 25, 34, 47, 55, and 57a
| compd | IC50 ± SD (nM)
|
averageb | ||||
|---|---|---|---|---|---|---|
| HeLa | HT-29 | A549 | HCT 116 | HCT 15 | ||
| 7 | 94 ± 4 | 182 ± 1 | 50 ± 6 | 89 ± 37 | 38 ± 22 | 91 |
| 8 | 120 ± 10 | 80 ± 16 | 68 ± 4 | 62 ± 5 | 52 ± 2 | 76 |
| 10 | 134 ± 60 | 88 ± 3 | 94 ± 4 | 177 ± 41 | 75 ± 14 | 114 |
| 14 | 188 ± 20 | 94 ± 30 | 80 ± 6 | 67 ± 2 | 62 ± 0.4 | 98 |
| 15 | 97 ± 3 | 282 ± 5 | 112 ± 20 | 154 ± 34 | 175 ± 2 | 164 |
| 18 | 30 ± 7 | 30 ± 4 | 20 ± 7 | 16 ± 6 | 11 ± 3 | 21 |
| 20 | 180 ± 15 | 90 ± 25 | 75 ± 5 | 68 ± 2 | 50 ± 0.9 | 93 |
| 23 | 154 ± 80 | 184 ± 70 | 82 ± 30 | 90 ± 43 | 53 ± 4 | 113 |
| 25 | 350 ± 9 | 282 ± 6 | 90 ± 5 | 533 ± 95 | 119 ± 18 | 275 |
| 34 | 288 ± 80 | 105 ± 10 | 137 ± 3 | 186 ± 1 | 160 ± 24 | 175 |
| 47 | 482 ± 60 | 188 ± 10 | 285 ± 2 | 593 ± 18 | 394 ± 91 | 388 |
| 55 | 48 ± 4 | 45 ± 6 | 28 ± 1 | 183 ± 5 | 84 ± 20 | 78 |
| 57 | 47 ± 1 | 78 ± 32 | 75 ± 2 | 59 ± 5 | 47 ± 13 | 61 |
| 1 | 28 ± 9 | 18 ± 4 | 20 ± 8 | 20 ± 2 | 86 ± 10 | 34 |
| 2 | 20 ± 5 | 130 ± 12 | >10000 | 5 ± 0.4 | 3 ± 9 | >2032 |
| VBL | 10 ± 0.6 | 30 ± 0.8 | 20 ± 2 | 3 ± 2 | 24 ± 10 | 17 |
| PTX | 5 ± 1 | 8 ± 1.5 | 7 ± 2 | 4 ± 0.4 | 90 ± 17 | 23 |
Inhibition of growth of the indicated cell lines.
Average activity of each compound was established for comparative purposes.
MDR Cell Lines
Compounds 18, 20, 55, and 57 were compared with vinorelbine (VRB), VBL, PTX 1 and 2 in the ovarian carcinoma cell lines OVCAR-8 and its cognate P-glycoprotein (Pgp) overexpressing line NCI/ADR-RES and with the human uterine sarcoma cell line Messa and its cognate line MDR line Messa/Dx5 (Table 3). Except for 2, the standard agents were relatively inactive in both MDR lines. The four ATIs 18, 20, 55, and 57 closely ressembled 2 in having full activity in both MDR lines.
Table 3.
Growth Inhibition of the OVCAR-8 and NCI/ADR-RES and Messa and Messa/Dx Cell Lines by Compounds 18, 20, 55, and 57 and Reference Compounds VRB, VBL, PTX, 1, and 2a
| compd | IC50 ± SD (nM)
|
Messa/Dx5b | ||
|---|---|---|---|---|
| OVCAR-8 | NCI/ADR-RES | Messab | ||
| 18 | 16 ± 6 | 20 ± 10 | 7 ± 0.3 | 13 ± 3 |
| 20 | 28 ± 10 | 15 ± 7 | 45 ± 4 | 39 ± 6 |
| 55 | 17 ± 6 | 23 ± 10 | 60 ± 7 | 71 ± 2 |
| 57 | 7.0 ± 3 | 14 ± 7 | 49 ± 2 | 53 ± 14 |
| 1 | ndc | ndc | 11 ± 6 | 329 ± 166 |
| 2 | 1.3 ± 0.6 | 1.3 ± 0.6 | 2.7 ± 2 | 2.6 ± 1 |
| VRB | 300 ± 0 | 5000 ± 1000 | ndc | ndc |
| VBL | 15 ± 7 | 200 ± 0 | 3 ± 2 | 144 ± 61 |
| PTX | 5.0 ± 2 | 3300 ± 1000 | 4 ± 1 | 1764 ± 477 |
Inhibition of growth of the indicated cell lines.
Growth inhibition data of compounds 7, 8, 10, 14, 15, 23, 25, 34, and 47 are shown in Supporting Information
No data.
Effects on Cell Cycle Progression
The most potent compound 18, as well as 57, was tested in dose–response experiments in comparison with 2 and VBL for effects on cell cycle distribution. HeLa cell cultures were treated with increasing concentrations of each compound, and 0.1% DMSO, the compound solvent, was used as a control. Representative images (DNA distribution by flow cytometry and wide-field microscopic images) of unfixed cultures taken after 24 h of treatment are shown in Figure 1A. Compound 57 at 100 nM induced a significant increase in the proportion of cells in the G2/M phase and, presumably, in mitosis, based on their rounded morphology in the microscopic image. After treatment with 10 nM 18, a significant proportion of the cells had assumed the rounded shape characteristic of mitotic arrest, and at 50 nM virtually the entire population was rounded and arrested in G2/M, based on DNA content.
Figure 1.

(A) Analysis of HeLa cell cultures treated with 18 or 57 for 24 h by wide-field microscopy (left panels) and flow cytometry (right panels). The left panels in each group show representative fields from unfixed cultures (10× objective): cells arrested in mitosis detach from the culture dish surface, leaving ample empty spaces, and are recognizable from their rounded-up refractive morphology. Cycling cultures are adherent with only rare rounded mitotic cells. Flow cytometry profiles of the cultures are shown on the right. The histrograms represent the ditribution of cells according to their DNA content calculated from the emitted PI fluorescence. Fluorescence intensity values are plotted on the x axis and cell numbers are plotted on the y axis. The 2C peak (in gray) identifies G1 cells. The 4C peak (in blue) identifies G2/M cells. Cells with intermediate DNA content are in S phase. (B) Mean frequency and SD of the frequency of cells with a 4C content (G2 + M) under the indicated conditions. From three to five assays were carried out for every treatment, and data from 20 000 cells per assay were acquired.
To confirm the initial microscopic observations, treated cultures were incubated with propidium iodide and subjected to quantitative flow cytometric analysis of the cell cycle phase distribution. Typical cell cycle profiles of PI-stained cultures after 24 h of treatment are shown in Figure 1A, and average values calculated from three to five independent assays per compound are shown in Figure 1B. Both compounds 18 and 57 arrested cell cycle progression in the G2/M phases (4C DNA content) when used at 100 nM. Plots of the concentration of the tested ATI against the fraction of G2/M-arrested cells in treated cultures (Figure 1B) indicated that 18 was a potent inhibitor of cell cycle progression already at 20 nM and induced a significant proportion of cells (46%) to arrest with a 4C DNA content, compared with 10.5% G2/M cells in control cultures. At higher concentrations, 18 progressively increased cell cycle arrest: at 50 nM, over 60% cells in treated cultures were in G2/M phase (Figure 1B), similar to the values obtained with both VBL and CSA-4 (2). The accumulation of cells with a replicated genome demonstrated that 18, like the control drugs, prevented or impaired mitotic cell division.
Compound 57 had somewhat milder effects on cell cycle progression (Figure 1B), compared with 18. Only about 48% of cells accumulated in the G2/M region with 57 at 50 nM; lower doses were virtually ineffective, with the proportion of G2/M cells essentially identical to the values observed in untretated controls. Only when the concentration was raised to 100 nM was the majority (65%) of cells arrested in G2/M.
Inhibition of Microtubule Assembly and Induction of Mitotic Arrest
We analyzed cell cultures in dose–response experiments using fluorescence microscopy to gain information on the effects of 18 and 57 on cellular MTs. After treatment with increasing concentrations of 18 or 57 for 24 h, we stained treated cells for α-tubulin, cyclin B1, and DAPI in order to discriminate between arrest in G2 interphase (i.e., interphase cells expressing cyclin B1, Figure 2A, left panel) or in prometaphase (Figure 2A, central panel). This is relevant because the molecular features of mitotic cell death are distinct, at least in part, from the classical apoptotic pathway induced in interphase.30 In some experiments, we also stained lamin B1 to visualize the integrity of the nuclear membrane and assess the stringency of mitotic arrest induced by ATI compounds. Indeed, cells with a defective mitotic apparatus do not necessarily arrest stably in prometaphase, but depending on the extent of MT damage, they may finally exit mitosis (mitotic slippage) with abnormal chromosome segregation and/or failed cell division giving rise to multi-nucleated cells (Figure 2A, right panel).
Figure 2.

(A) Examples of immunofluorescenctly stained cells. In all panels, the DNA is blue (stained with DAPI), α-tubulin is green, and cyclin B1 is red, as indicated. Left: A G2 interphase cell identified by cyclin B1 expression. Middle: A cell arrested in prometaphase with depolymerized microtubules (shown with and without merging with cyclin B1 staining). Right: A multinucleated cell derived from mitotic slippage. (B) Distribution of cells in dose–response experiments with 18 and 57. The histograms represent the mean frequency, and bars represent the SD of interphases (I), mitoses (M), and multinculeated cells (MN) from three independent experiments.
In dose–response experiments (Figure 2B, bottom panel), we found that 57 at 10 and 20 nM did not affect mitotic progression (only 10% of all cells displayed mitotic properites, similar to the baseline frequency observed in control cells treated with DMSO). Although these concentrations induced many abnormal mitotic spindles (aberrantly shaped, multipolar, and/or with sparse and rare MTs), cells were distributed in all mitotic stages, indicating that these abnormalities did not evoke a stringent spindle checkpoint, and cells progressed to divide. At 50 nM, 57 induced some mitotic arrest: 47% of all cells were arrested in prometaphase with reduced numbers of MTs. This arrest was not absolutely stringent, since 16% of cells in the culture were aberrant, multinucleated, postmitotic species. 100 nM 57 arrested almost 65% of cells stably in prometaphase with completely absent MTs and condensed chromosomes. Virtually no mitotic slippage was observed with this concentration. At 200 nM, the mitotic index decreased while the fraction of G2 interphases correspondingly increased, indicating such a strong effect on interphase MTs that cells were prevented from entering mitosis altogether.31 Indeed, a similar decrease in mitotic arrest, with a corresponding increase in G2 cells, is observed at high concentrations of 2 or VBL.
Compound 18 showed the highest effectiveness, in terms of inducing mitotic arrest at 50 nM in HeLa cell cultures (Figure 2B, top panel). At this concentration, 18 induced effective mitotic arrest in prometaphase with totally absent MTs and no significant mitotic slippage. ATI 18 blocked mitotic progression even at lower concentration (20 nM), yet the arrest was not fully sustained with this concentration, and some cells slipped through mitosis with a defective mitotic apparatus, generating multi-nucleated cells (18% of all cells). At 100 nM, 18 prevented M entry, suggesting an excessively strong effect on interphase MTs. Together, these data indicate that 18 is a strong inhibtior of MT polymerization and induced effective mitotic arrest at 50 nM, similar to 2 or VBL, whereas 57 induced a durable mitotic block when used at 100 nM.
Induction of Mitotic Cell Death
We examined whether the new ATIs 18 and 57, in addition to arresting mitosis, also triggered cell death. HeLa cell cultures were treated for 24 h as described above, and they were then incubated with annexin V in the absence of permeabilization in order to detect apoptotic cells with a damaged plasma membrane. They were then analyzed by flow cytometry (Figure 3A). Significant cell death was detected in cultures exposed to 18 (50 and 100 nM) or 57 (100 nM) concentrations at which the cell populations displayed significant mitotic arrest (Figures 1 and 2). Biparametric analysis of annexin V reactivity versus genomic DNA content revealed that necrosis was negligible (Figure 3B). These data therefore show that both 18 and 57 induce mitotic cell death.
Figure 3.

(A) Frequency of apoptotic cells in cultures treated with the indicated concentrations of 18 or 57. The histograms represent mean frequencies, and bars represent the SDs of annexin V reactive cells from three independent experiments. (B) Biparametric analysis of cell death in dose–response experiments with 18. Early apoptotic, late apotptotic, and necrotic cells are distinguished by their differential reactivity to annexin V but not PI, to both annexin V and PI, and to PI but not annexin V, respectively. Results from three independent experiments are shown for each concentration. No condition induced significant necrosis compared with the baseline level seen in DMSO-treated controls (data from 20 000 cells acquired per sample).
Loss of Mitochondrial Potential
We evaluated the loss of mitochondrial potential (ΔΨm), a hallmark for early induction of cellular apoptosis,32 to elucidate further whether compounds 18 and 57 would induce apoptosis or necrotic death. The loss of ΔΨm was indicated by a decrease in red 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzimidazoylcarbocyanine iodide (JC-1) aggregates, accompanied by a concomitant accumulation in green JC-1 monomers. The collapse in ΔΨm, as measured as reduction in the red/green fluorescence ratio, was assessed after cell treatment with 18 or 57. As shown in Figure 4, after a 24 h treatment, 57 was able to induce a more significant reduction in ΔΨm than was 18. The standard compound carbonyl cyanide m-chlorophenylhydrazone (CCCP) caused about 50% potential dissipation at 30 μM.
Figure 4.

Effects of a 24 h treatment with 100 nM 57 or 18 or 30 μM CCCP on mitochondrial transmembrane potential as assessed by JC-1 staining. Graphs indicate the ΔΨdissipation expressed as red/green (R/G) fluorescence ratio. Each value has been normalized versus the R/G ratio of the vehicle treated control to which an arbitrary value of 100% has been assigned. Data were the mean of two independent experiments performed in triplicate ((**) p < 0.01 and (***) p < 0.001, one-way ANOVA, Bonferroni’s corrected t test for post hoc pairwise comparisons).
ROS Generation
Mitochondria are an important intra-cellular source of reactive oxygen species (ROS).33 We measured the ability of compounds 18 and 57 to generate ROS in U87MG cells, using hydrogen peroxide specific probe 6-carboxy-2′, 7′-lichlorodihydrofiuorescein diacetate (DCFH2-DA).
The IC50 values of compounds 18 and 57 in U87MG (human glioblastoma) cell growth/survival after a 24 h treatment were determined. Compounds 18 and 57 showed dose-dependent inhibitory effects on cell survival, with compound 18 having IC50 = 85 ± 8 nM and 57 having IC50 = 103 ± 9 nM. Compound-treated cells showed an increase in the percentage of cell death compared with control cells, reaching statistical significance at 100 nM.
According to the observed loss of mitochondrial potential, both compounds 18 and 57 were able to increase ROS levels. The effect was not detectable immediately, but it reached statistical significance after a 1 h treatment (fluorescence intensity with respect to control: 18, 216%; 57, 254%; both greater than the value of 208% obtained with the usual standard, hydrogen peroxide (Figure 5)).
Figure 5.

Effects of treatment with 100 nM 57 or 18 and 400 mM H2O2 on ROS formation in U87MG cells after exposures of 1, 5, 15, and 60 min. The results are expressed as % of mean fluorescence intensity relative to the control and normalized for cell number. Data were the mean of two independent experiments performed in triplicate ((*) p < 0.05, (**) p < 0.01, and (***) p < 0.001, one-way ANOVA, Bonferroni’s corrected t test for post hoc pairwise comparisons).
In Vivo Vascular Disrupting Effects
In preclinical development of new anticancer drugs such as vascular disrupting agents (VDAs), in vivo validation proved to be an indispensible step in the bench-to-bedside chain, where animal models with tumor growth in visceral organs could better simulate clinical scenarios in comparison with the mostly applied subcutaneous implants.34 Magnetic resonance imaging (MRI) has greatly facilitated in vivo noninvasive assessment of tumoricidal effects on deeply seeded lesions in the animal models.35 Consequently, we were able to observe that at the same intravenous dosage of 15 mg/kg, both compounds 18 and 57 exerted initial vascular disrupting effects in the applied tumor model of liver rhabdomyosarcomas. The observed tumor vascular shutdown effect seems stronger and lasts longer with 18 (B′ and C′) compared to 57 (B and C). The reason for such different effects will be further elucidated (Figure 6).
Figure 6.

Contrast enhanced MRI was used in rats with bifocal liver growths of rhabdomyosarcoma (smaller and larger arrows denote smaller and larger lesions, respectively). At baseline prior to treatment, the liver tumors appear as homogeneous hyperintense spheroidal nodules, suggesting their hypervascularity (A, A′). At 1 h after intravenous injection of either 57 (B) or 18 (B′), the tumors were partially enhanced because of their early vascular disrupting effects, which appeared stronger with 18 (B′) than 57 (B). At 24 h after treatment, the tumors treated with 57 were enhanced completely (C), suggesting only transient early vascular disruption effect and the absence of any consequent tumor necrosis. However, tumors treated with 18 showed rim (arrowheads) and partial central enhancement (C′), suggesting the presence of both tumor necrosis and viable residues.
Metabolic Stability
Compounds 18, 20, 55, and 57 were examined in a microsomal stability assay in comparison with 7-ethoxycoumarin and propranolol as control compounds, using both human and mouse liver microsomes, to estimate compound stability to phase I oxidative metabolism (Table 4). Compound 18 showed the highest metabolic stability with 48.6% and 10.3% remaining after 30 min in human and mouse liver microsomes, respectively. Compounds 20, 55, and 57 showed medium metabolic stability with human liver microsomes and low metabolic stability with mouse liver microsomes (relative stabilities are defined in a Table 4 footnote).
Table 4.
Metabolic Stability with Human and Mouse Liver Microsomesa and Aqueous Solubility of Compounds 18, 20, 55, and 57
| compd | % remaining at 30 minb
|
solubility (μM)b,d | |
|---|---|---|---|
| human liver microsomes | mouse liver microsomes | ||
| 18 | 48.6 ± 1.9 | 10.3 ± 0.4 | 64.5 ± 0.7 |
| 20 | 12.0 ± 1.1 | 0.6 ± 0.1 | 4.0 ± 0.1 |
| 55 | 19.3 ± 0.9 | 2.8 ± 0.3 | 20.5 ± 0.7 |
| 57 | 17.7 ± 0.5 | 7.9 ± 0.2 | 5.0 ± 0.1 |
| 7-ethoxycoumarinc | 6.6 ± 0.2 | 0.07 ± 0.02 | |
| propranololc | 54.1 ± 0.4 | 20.6 ± 0.5 | |
Metabolic stability: >50, good; 10–50, medium; <10, low.
Results are expressed as mean ± SD, n = 2.
The standard compounds 7-ethoxycoumarin and propranolol showed metabolic stability in agreement with the literature and internal validation data.36
pH 7.4, high throughput screening solubility assay.
Aqueous Solubility
The solubility in aqueous pH 7.4 buffer of compounds 18, 20, 55, and 57 was measured in a high throughput screening solubility assay. The solubility of compound 18 was 64.5 μM, while under the same conditions compounds 20 (4 μM), 55 (20.5 μM), and 57 (8 μM) showed lower solubility (Table 4). The higher solubility of compound 18 could explain its greater activity as a cell growth inhibitor (see Tables 1 and 2), although these compounds were comparable inhibitors of tubulin assembly in the biochemical assays.
Caco-2 Cell Permeability
The intestinal permeability of compounds 18, 20, 55, and 57 was evaluated in the human Caco-2 model in comparison with caffeine (high permeability) and cimetidine (low permeability, Pgp substrate). The apparent permeability (Papp) of 18, 20, 55, or 57 from the A (apical) to B (basolateral) side together with B to A was measured in order to predict the absorption from the lumen of the gut and potential efflux phenomena. Compound 20 showed the highest permeability in both A → B (Papp = 132.5 nm/s) and B → A (Papp = 133.2 nm/s) directions. Compounds 18, 55, and 57 showed medium–high permeability (Table 5). None of the compounds tested showed efflux phenomena (i.e., ratio of Papp(B→A) to Papp(A→B) of <2).
Table 5.
Caco-2 Cell Permeability and CYP450 Isoform Inhibition of Compounds 18, 20, 55, and 57
| compd |
Papp (nm/s)a
|
CYP450 isoform (% inhibition at 1 μM)
|
|||||
|---|---|---|---|---|---|---|---|
| PA→B | PB→A | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |
| 18 | 55.3 ± 11.1 | 60.0 ± 5.6 | 22.9 ± 0.03 | 82.5 ± 0.01 | 95.9 ± 0.05 | 10.6 ± 0.16 | 94.5 ± 0.55 |
| 20 | 132.5 ± 19.7 | 133.2 ± 4.2 | <5 | 83.9 ± 0.21 | 93.1 ± 0.18 | <5 | 51.4 ± 0.18 |
| 55 | 53.1 ± 6.0 | 65.8 ± 2.5 | 23.4 ± 0.31 | 84.9 ± 0.70 | 101.8 ± 1.10 | 6.6 ± 0.64 | 76.4 ± 1.42 |
| 57 | 44.5 ± 4.0 | 54.2 ± 5.3 | 14.5 ± 1.66 | 100.2 ± 0.09 | 100.4 ± 1.36 | 43.1 ± 3.21 | 58.0 ± 1.26 |
Papp (nm/s): >50, high; 10–50, medium; <10, low; caffeine PA–B reference control, 206 ± 35 nm/s; cimetidine PA–B and PB–A reference control, 1.1 ± 0.1 and 21.3 ± 3.4 nm/s, respectively.
CYP450 Inhibition
To assess further the safety of compounds 18, 20, 55, and 57, we carried out a CYP inhibition assay. CYP inhibition can cause drug–drug interactions by increasing concentrations to other drugs to toxic levels. The inhibition of the enzymatic activity of human cloned P450 isoforms CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 was measured using specific substrates for each isoform that produced a fluorescent metabolite upon CYP metabolism. Compounds 18, 20, 55, and 57 at 1 μM were weak inhibitors of the isoforms CYP1A2 and CYP2D6. For these isoforms, inhibition was <50%, ranging from <5% (20 in CYP1A2 and CYP2D6 isoforms) to 43% (57 in CYP2D6 isoform). The isoform CYP3A4 was inhibited by 20, 55, and 77 by 51%, 76%, and 57%, respectively; CYP2C19 and CYP2C9 were inhibited to a greater extent by 18, 20, 55, and 57.
CONCLUSION
We synthesized 71 new arylthioindole/aroylindole compounds as potential anticancer agents having different (hetero)cyclic substituents at indole position 2. Several new ATIs inhibited tubulin polymerization with IC50 ranging from <1.0 to 2.0 μM, and six compounds yielded submicromolar IC50. These compounds inhibited the growth of human MCF-7 cells, and five of them inhibited the growth of these cells with low nanomolar IC50. ATI 18 strongly inhibited MCF-7 cell growth (IC50 = 1.0 nM) and was uniformly active in a larger panel of cancer cells. Compound 18 was superior to reference compounds 1 and 2 and ATIs 3–6.19 ATIs 18, 20, 55, and 57 were more potent than 1, 2, VRB, VBL, and PTX in two MDR cell lines, the Pgp overexpressing NCI-ADR-RES line and the Messa/Dx5 line. Compounds 18 and 57 arrested cell cycle progression at the G2/M phase. Compound 18 was a potent inhibitor of cell cycle progression at 20 nM, strongly inhibited MT polymerization, and induced effective mitotic arrest and cell death, while 57 showed somewhat weaker effects. Compounds 18 and 57 elicited dose- and time-dependent inhibition of U87MG cell growth and induced mitochondrial membrane potential collapse, a hallmark of early apoptosis. The compounds also evoked significant ROS generation in GBM cells. At 15 mg/kg intravenous dosage, both compounds 18 and 57 exerted initial vascular disrupting effects in a tumor model of liver rhabdomyosarcomas. In a microsomal stability assay, compound 18 showed the highest metabolic stability with both human and mouse liver microsomes of all the ATIs examined. The higher solubility in aqueous pH 7.4 buffer of compound 18 (64.5 μM) could explain its greater inhibitory effects on cell growth (MCF-7 cells, IC50 = 1.0 nM) among compounds with comparable inhibitory effects on tubulin assembly. In conclusion, we succeeded in improving the ATI class of cancer agents through the synthesis of new compounds. Compounds 18 and 57 proved to be superior to 5, the reference ATI of the previous series,19 in the whole cancer cell panel, including two MDR cell lines. Derivative 18 showed higher metabolic stability than 5 in human liver microsomes (48.6% remaining), showed the highest water solubility, and displayed medium-high Caco-2 cell permeability. The present results highlight the therapeutic potential of the ATI class as anticancer agents and prompt their further development.
EXPERIMENTAL SECTION
Microwave-assisted reactions were performed on a CEM Discover SP single mode reactor, controlling the instrument settings with PC-running CEM Synergy 1.49 software. Closed vessel experiments were carried out in capped microwave-dedicated vials (10 mL) with cylindrical stirring bar (length 8 mm, diameter 3 mm). Open vessel experiments were carried out in 100 mL round-bottom flasks equipped with a Dimroth reflux condenser and a cylindrical stirring bar (length 20 mm, diameter 6 mm). Stirring, temperature, irradiation power, maximum pressure (Pmax), PowerMAX (simultaneous cooling-while-heating), ActiVent (simultaneous venting-while-heating), and ramp and hold times were set as indicated. Temperature of the reaction was monitored by a built-in infrared sensor. After completion of the reaction, the mixture was cooled to 25 °C via air-jet cooling. Melting points (mp) were determined on a Stuart Scientific SMP1 apparatus and are uncorrected. Infrared spectra (IR) were run on a Perkin-Elmer SpectrumOne FT-ATR spectrophotometer. Band position and absorption ranges are given in cm−1. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker 400 MHz FT spectrometer in the indicated solvent and corresponding fid files processed by MestreLab Research S.L. MestreReNova 6.2.1-769 software. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane. Column chromatography was performed on columns packed with alumina from Merck (70–230 mesh) or silica gel from Macherey-Nagel (70–230 mesh). Aluminum oxide thin layer chromatography (TLC) cards from Fluka (aluminum oxide precoated aluminum cards with fluorescent indicator visualizable at 254 nm) and silica gel TLC cards from Macherey-Nagel (silica gel precoated aluminum cards with fluorescent indicator visualizable at 254 nm) were used for TLC. Developed plates were visualized by a Spectroline ENF 260C/FE UV apparatus. Organic solutions were dried over anhydrous Na2SO4. Evaporation of the solvents was carried out on a Buchi rotavapor R-210 equipped with a Buchi V-850 vacuum controller and a Buchi V-700 or V-710 vacuum pump. All reagents and solvents are commercially available and were used as purchased, without further purification. Elemental analyses of the compounds were found within ±0.4% of the theoretical values. The purity of tested compounds was >95%.
2-(1H-Imidazol-1-yl)-1H-indole (80),21 2-(1H-pyrazol-3-yl)-1H-indole (99),22 2-iodo-1H-indole (106),23 2-(1H-pyrrol-2-yl)-1H-indole (109),25 2-(1-(phenylsulfonyl)-1H-pyrrol-3-yl)-1H-indole (110),19 2-(furan-2-yl)-1H-indole (111),19 2-(furan-3-yl)-1H-indole (112),19 1H-indole-2-carbaldehyde (115),26 2-(1-(2-phenylhydrazono)ethyl)-pyridine (130),29 3-(1-(2-phenylhydrazono)ethyl)pyridine (131),29 4-(1-(2-phenylhydrazono)ethyl)pyridine (132),29 and 1-(1-(naphthalen-2-yl)ethylidene)-2-phenylhydrazine (133)29 were synthesized according to the indicated literature. 2-Phenyl-1H-indole (84), 1H-indole-2-carboxylic acid (105), 2-bromo-1-(2-methoxyphenyl)ethanone (119), and 2-bromo-1-(3-methoxyphenyl)ethanone (120) were purchased from Sigma-Aldrich.
Arylthio-1H-indoles. General Procedure for the Preparation of Compounds 17, 18, 20, 27, 33, 34, 38, 39, 41, 43, 45–47, 53, 55, 57, 73, 75, and 77. Example: 2-(1H-Imidazol-2-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (17)
2-(1H-Imidazol-2-yl)-1H-indole (79) (0.044 g, 0.00024 mol) was added to a suspension of NaH (0.014 g, 0.00036 mol, 60% dispersion in mineral oil) in anhydrous DMF (2 mL). After 10 min, bis(3,4,5-trimethoxyphenyl)disulfide (0.1 g, 0.000 264 mol) was added, and the reaction mixture was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C while stirring. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min. The reaction mixture was quenched on crushed ice and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 7:3 as eluent) to furnish 17 (0.02 g, 22%), mp 135–140 °C (from ethanol). 1H NMR (CDCl3): δ 3.62 (s, 6H), 3.76 (s, 3H), 6.38 (s, 2H), 7.14–7.26 (m, 4H), 7.36–7.38 (m, 1H), 7.63 (d, J = 7.9 Hz, 1H), 11.01 (broad s, disappeared on treatment with D2O, 1H), 11.58 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2923, 3345 cm−1. Anal. (C20H19N3O3S (381.45)) C, H, N, S.
2-(1H-Imidazol-1-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (18)
18 was synthesized similarly to 17, starting from 80.21 Yield 77%, mp 193–197 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.57 (s, 6H), 3.58 (s, 3H), 6.31 (s, 2H), 7.17–7.21 (m, 2H), 7.26–7.30 (m, 1H), 7.49–7.55 (m, 2H), 7.68–7.69 (m, 1H), 8.14–7.15 (m, 1H), 12.23 ppm (broad s, disappeared on treatment with D2O, 1H). 13C NMR (DMSO-d6): δ 56.33, 60.48, 91.97, 103.95, 112.62, 119.17, 120.49, 121.66, 123.68, 129.27, 129.77, 132.91, 133.81, 136.2, 136.23, 137.78, 153.79 ppm. IR: ν 2930 cm−1. Anal. (C20H19N3O3S (381.45)) C, H, N, S.
2-(1H-1,2,4-Triazol-1-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (20)
20 was synthesized similarly to 17, starting from 81. Yield 34%, mp 140–145 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.58 (s, 9H), 6.37 (s, 2H), 7.21 (t, J = 7.7 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.52–7.58 (m, 2H), 8.41 (s, 1H), 9.12 (s, 1H), 12.80 ppm (broad s, disappeared on treatment with D2O, 1H). 13C NMR (DMSO-d6): δ 56.32, 60.47, 93.24, 104.45, 113.01, 119.50, 121.78, 124.14, 128.78, 132.15, 134.11, 135.32, 136.38, 145.67, 153.04, 153.75 ppm. IR: ν 3354 cm−1. Anal. (C19H18N4O3S (382.44)) C, H, N, S.
2-(3-((3,4,5-Trimethoxyphenyl)thio)-1H-indol-2-yl)thiazole (27)
27 was synthesized similarly to 17, starting from 82. Yield 10%, mp 160–165 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.77 (s, 3H), 6.41 (s, 2H), 7.19–7.23 (m, 1H), 7.31–7.35 (m, 1H), 7.43–7.45 (m, 2H), 7.72 (dd, J = 0.8 and 7.9 Hz, 1H), 7.90 (d, J = 3.2 Hz, 1H), 9.87 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3339 cm−1. Anal. (C20H18N2O3S2 (379.47)) C, H, N, S.
2-(4,5-Dihydro-1H-imidazol-2-yl)-3-((3,4,5-trimethoxyphenyl)-thio)-1H-indole (33)
33 was synthesized similarly to 17, starting from 83. Yield 8%, mp 150–155 °C (from ethanol). 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.76 (s, 3H), 3.92 (m, 4H), 6.33 (s, 2H), 7.15 (t, J = 7.1 Hz, 1H), 7.32 (t, J = 7.0 Hz, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 8.1 Hz, 1H), 8.31 (broad s, disappeared on treatment with D2O, 1H), 12.10 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2852, 2921 cm−1. Anal. (C20H21N3O3S (383.46)) C, H, N, S.
2-Phenyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (34)
34 was synthesized similarly to 17, starting from 84. Yield 3%, mp 158–160 °C (from ethanol). 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.78 (s, 3H), 6.37 (s, 2H), 7.18–7.22 (m, 1H), 7.27–7.30 (m, 2H), 7.41–7.50 (m, 3H), 7.69 (d, J = 7.9 Hz, 1H), 7.80–7.82 (m, 2H), 8.56 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3329 cm−1. Anal. (C23H21NO3S (391.48)) C, H, N, S.
2-(4-Chlorophenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (38)
38 was synthesized similarly to 17, starting from 85. Yield 11%, mp 190–195 °C (from ethanol). 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.78 (s, 3H), 6.34 (s, 2H), 7.19–7.23 (m, 1H), 7.29–7.31 (m, 1H), 7.43–7.46 (m, 3H), 7.68–7.70 (m, 1H), 7.72–7.75 (m, 2H), 8.59 ppm. (broad s, disappeared on treatment with D2O, 1H). IR: ν 3335 cm−1. Anal. (C23H20ClNO3S (425.93)) C, H, N, Cl, S.
2-(2-Fluorophenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (39)
39 was synthesized similarly to 17, starting from 86. Yield 39% mp 175–180 °C (from toluene). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.56 (s, 3H), 6.26 (s, 2H), 7.13 (t, J = 7.6 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 7.33–7.42 (m, 2H), 7.49–7.54 (m, 3H), 7.65 (t, J = 7.5 Hz, 1H), 12.07 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3342 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, N, F, S.
2-(3-Fluorophenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (41)
41 was synthesized similarly to 17, starting from 87. Yield 65%, mp 180–185 °C (from toluene). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.64 (s, 6H), 6.79 (s, 2H), 7.07–7.35 (m, 5H), 7.45–7.48 (m, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 7.8 Hz, 1H), 12.27 (broad s, disappeared on treatment with D2O, 1H). IR: ν 1571, 3187 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, N, F, S.
2-(4-Fluorophenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (43)
43 was synthesized similarly to 17, starting from 88. Yield 37%, mp 170–175 °C (from toluene). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.57 (s, 3H), 6.30 (s, 2H), 7.12 (t, J = 7.0 Hz, 1H), 7.23 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 8.9 Hz, 2H), 7.49 (t, J = 7.2 Hz, 2H), 7.90–7.93 (m, 2H), 12.09 (broad s, disappeared on treatment with D2O, 1H). IR: ν 3318 cm−1. Anal. (C23H20FNO3S (409.47)) C, H, N, F, S.
2-(2-Methoxyphenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (45)
45 was synthesized similarly to 17, starting from 89. Yield 5%, mp 160–165 °C (from ethanol). 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.77 (s, 3H), 3.92 (s, 3H), 6.35 (s, 2H), 7.04–7.08 (m, 2H), 7.15–7.19 (m, 1H), 7.25–7.29 (m, 1H), 7.36–7.40 (m, 1H), 7.45–7.47 (m, 1H), 7.67–7.69 (m, 1H), 7.97–7.99 (m, 1H), 9.40 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3338 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
2-(3-Methoxyphenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (46)
46 was synthesized similarly to 17, starting from 90. Yield 4%, mp 192–194 °C (from ethanol). 1H NMR (CDCl3): δ 3.63 (s, 6H), 3.75 (s, 3H), 3.76 (s, 3H), 6.36 (s, 2H), 6.93–6.96 (m, 1H), 7.18–7.21 (m, 1H), 7.25–7.40 (m, 4H), 7.44–7.46 (m, 1H), 7.69–7.71 (m, 1H), 8.57 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3333 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
2-(4-Methoxyphenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (47)
47 was synthesized similarly to 17, starting from 91. Yield 8%, mp 165–170 °C (from ethanol). 1H NMR (CDCl3): δ 3.63 (s, 6H), 3.78 (s, 3H), 3.85 (s, 3H), 6.37 (s, 2H), 6.97–6.99 (m, 2H), 7.17–7.23 (m, 2H), 7.41–7.43 (m, 1H), 7.66–7.74 (m, 3H), 8.68 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3324 cm−1. Anal. (C24H23NO4S (421.51)) C, H, N, S.
2-(Pyridin-2-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (53)
53 was synthesized similarly to 17, starting from 92. Yield 26% as an oil. 1H NMR (CDCl3): δ 3.63 (s, 6H), 3.76 (s, 3H), 6.39 (s, 2H), 7.18 (t, J = 7.1 Hz, 1H), 7.24–7.27 (m, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.70–7.77 (m, 2H), 8.63–8.64 (m, 1H), 8.73 (d, J = 8.1 Hz, 1H), 10.23 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2828, 2933, 3003, 3056 cm−1. Anal. (C22H20N2O3S (392.47)) C, H, N, S.
2-(Pyridin-3-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (55)
55 was synthesized similarly to 17, starting from 93. Yield 20%, mp 188–191 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.76 (s, 3H), 6.33 (s, 2H), 7.22 (m, 1H), 7.31 (m, 1H), 7.37–7.41 (m 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 8.16–8.17 (m, 1H), 8.62 (m, 1H), 9.02 (m, 1H), 9.26 ppm (broad s, disappeared on treatment with D2O, 1H). 13C NMR (DMSO-d6): δ 56.28, 60.48, 98.8, 103.33, 112.63, 119.31, 121.21, 123.66, 124.12, 127.75, 130.72, 133.85, 135.99, 136.82, 139.7, 149.19, 149.68, 153.77 ppm. IR: ν 2923, 3674 cm−1. Anal. (C22H20N2O3S (392.47)) C, H, N.
2-(Pyridin-4-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (57)
57 was synthesized similarly to 17, starting from 94. Yield 60% as a yellow solid, mp 215–218 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.57 (s, 3H), 6.32 (s, 2H), 7.14–7.18 (m, 1H), 7.27–7.31 (m, 1H), 7.53–7.57 (m, 2H), 7.92 (dd, J = 1.7 and 4.5 Hz, 2H), 8.71 (dd, J = 1.6 and 4.5 Hz, 2H), 12.36 ppm (broad s, disappeared on treatment with D2O, 1H). 13C NMR (DMSO-d6): δ 56.27, 60.47, 100.49, 103.60, 112.82, 119.63, 121.38, 122.5, 124.25, 130.89, 133.39, 135.99, 136.92, 138.76, 139.02, 150.52, 153.8 ppm. IR: ν 3320 cm−1. Anal. (C22H20N2O3S (392.47)) C, H, N.
2-Cyclobutyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (73)
73 was synthesized similarly to 17, starting from 95. Yield 56%, mp 149–153 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.85–1.92 (m, 1H), 1.98–2.10 (m, 1H), 2.23–2.28 (m, 2H), 2.28–2.30 (m, 2H), 2.57 (s, 9H), 3.94–4.03 (m, 1H), 6.27 (s, 2H), 7.01–7.05 (m, 1H), 7.11–7.15 (m, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 11.74 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3361 cm−1. Anal. (C21H23NO3S (369.48)) C, H, N, S.
2-Cyclopentyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (75)
75 was synthesized similarly to 17, starting from 96. Yield 56%, mp 178–183 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.66–1.68 (m, 2H), 1.17–1.85 (m, 4H), 1.95–1.98 (m, 2H), 3.52–3.54 (m, 1H), 3.56 (s, 6H), 3.57 (s, 3H), 6.28 (s, 2H), 7.0–7.04 (m, 1H), 7.09–7.13 (m, 1H), 7.36–7.41 (m, 2H), 11.51 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3331 cm−1. Anal. (C22H25NO3S (383.50)) C, H, N, S.
2-Cyclohexyl-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (77)
77 was synthesized similarly to 17, starting from 97. Yield 48%, mp 214–219 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.24–1.42 (m, 3H), 1.66–1.83 (m, 7H), 3.09–3.17 (m, 1H), 3.56 (s, 6H), 3.57 (s, 3H), 6.28 (s, 2H), 7.0–7.02 (m, 1H), 7.09–7.13 (m, 1H), 7.35–7.41 (m, 2H), 11.52 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3327 cm−1. Anal. (C23H27NO3S (397.53)) C, H, N, S.
General Procedure for the Preparation of Compounds 14, 16, 37, 48, 59, 62, and 72. Example: 2-(1H-Pyrazol-4-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (14)
2-(1H-Pyrazol-4-yl)-1H-indole (98) (0.08 g, 0.000 44 mol) was added to an ice-cooled suspension of NaH (0.058 g, 0.001 45 mol, 60% dispersion in mineral oil) in anhydrous DMF (3 mL) under an Ar stream. After 15 min, bis(3,4,5-trimethoxyphenyl)disulfide (0.21 g, 0.000 53 mol) was added, and the reaction mixture was heated at 60 °C for 12 h under an Ar stream. After cooling, the reaction mixture was quenched on crushed ice and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 7:3) to furnish 14 (0.1 g, 59%), mp 178–182 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.53 (s, 6H), 3.57 (s, 3H), 6.33 (s, 2H), 7.06–7.10 (m, 1H), 7.15–7.19 (m, 1H), 7.44–7.46 (m, 2H), 8.23 (m, 2H), 11.88 (broad s, disappeared on treatment with D2O, 1H), 13.17 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3158, 3291 cm−1. Anal. (C20H19N3O3S (381.45)) C, H, N, S.
2-(1H-Pyrazol-3-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (16)
16 was synthesized similarly to 14, starting from 99.22 Yield 35%, mp 194–197 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.51 (s, 6H), 3.55 (s, 3H), 6.35 (s, 2H), 6.97–6.98 (m, 1H), 7.07 (t, J = 7.6 Hz, 1H), 7.17 (t, J = 7.9 Hz, 1H), 7.46–7.49 (m, 2H), 7.83 (s, 1H), 11.97 (broad s, disappeared on treatment with D2O, 1H), 13.23 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2936, 3348 cm−1. Anal. (C20H19N3O3S (381.45)) C, H, N, S.
2-(p-Tolyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (37)
37 was synthesized similarly to 14, starting from 100. Yield 72%, mp 168–171 °C (from ethanol). 1H NMR (DMSO-d6): δ 2.36 (s, 3H), 3.52 (s, 6H), 3.56 (s, 3H), 6.29 (s, 2H), 7.09–7.13 (m, 1H), 7.19–7.23 (m, 1H), 7.32–7.34 (m, 2H), 7.47–7.51 (m, 2H), 7.78–7.80 (m, 2H), 12.02 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3344 cm−1. Anal. (C24H23NO3S (405.51)) C, H, N, S.
2-(3-Isopropoxy-4-methoxyphenyl)-3-((3,4,5-trimethoxyphenyl)-thio)-1H-indole (48)
48 was synthesized similarly to 14, starting from 101. Yield 53%, mp 110–113 °C (from ethanol). 1H NMR (CDCl3): δ 1.29 (d, J = 6.1 Hz, 6H), 3.65 (s, 6H), 3.77 (s, 3H), 3.90 (s, 3H), 4.35 (m, 1H), 6.36 (s, 2H), 6.97 (d, J = 8.4 Hz, 1H), 7.19–7.29 (m, 3H), 7.41 (m, 1H), 7.44–7.46 (m, 1H), 7.70–7.72 (m, 1H), 8.56 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3337 cm−1. Anal. (C27H29NO5S (479.59)) C, H, N, S.
2-(Naphthalen-1-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (59)
59 was synthesized similarly to 14, starting from 102. Yield 44%, mp 227–230 °C (from ethanol). 1H NMR (CDCl3): δ 3.56 (s, 6H), 3.71 (s, 3H), 6.30 (s, 2H), 7.22–7.26 (m, 1H), 7.29–7.33 (m, 1H), 7.39–7.55 (m, 4H), 7.61 (dd, J = 1.2 and 7.1 Hz, 1H), 7.74–7.76 (m, 1H), 7.82–7.84 (m, 1H), 7.90–7.95 (m, 2H), 8.59 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3229 cm−1. Anal. (C27H23NO3S (441.54)) C, H, N, S.
2-(Naphthalen-2-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (62)
62 was synthesized similarly to 14, starting from 103. Yield 44%, mp 157–160 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.79 (s, 3H), 6.42 (s, 2H), 7.22–7.34 (m, 2H), 7.50–7.56 (m, 3H), 7.74–7.75 (m, 1H), 7.88–7.90 (m, 2H), 7.93–8.00 (m, 2H), 8.25 (m, 1H), 8.71 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3318 cm−1. Anal. (C27H23NO3S (441.54)) C, H, N, S.
2-((1,1′-Biphenyl)-4-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (72)
72 was synthesized similarly to 14, starting from 104. Yield 52%, mp 210–215 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.57 (s, 3H), 6.33 (s, 2H), 7.11–7.15 (m, 1H), 7.23–7.25 (m, 1H), 7.38–7.42 (m, 1H), 7.48–7.54 (m, 4H), 7.75–7.77 (m, 2H), 7.84–7.86 (m, 2H), 8.01–8.03 (m, 2H), 12.14 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3329 cm−1. Anal. (C29H25NO3S (467.58)) C, H, N, S.
2-(3-((3,4,5-Trimethoxyphenyl)thio)-1H-indol-2-yl)-4,5-dihy-droxazole (28)
2-Bromoethylamine hydrobromide (0.63 g, 0.0031 mol) was added to a solution of 3-((3,4,5-trimethoxyphenyl)thio)-1H-indole-2-carboxylic acid (107) (1.0 g, 0.0028 mol), triethylamine (0.85 g, 1.2 mL, 0.0084 mol), and BOP reagent (1.24 g, 0.0028 mol) in anhydrous DMF (15 mL). The reaction mixture was stirred at 25 °C for 2 h and then diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 7:3 as eluent) to furnish 28 (0.31 g, 30%), mp 190–195 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.55 (s, 6H), 3.56 (s, 3H), 4.00 (t, J = 9.7 Hz, 2H), 4.43 (t, J = 9.5 Hz, 2H), 6.42 (s, 2H), 7.07–7.11 (m, 1H), 7.23–7.27 (m, 1H), 7.43–7.49 (m, 2H), 12.21 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3198 cm−1. Anal. (C20H20N2O4S (384.45)) C, H, N, S.
2-(3-((3,4,5-Trimethoxyphenyl)thio)-1H-indol-2-yl)oxazole (31)
A mixture of 2-iodo-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (108) (0.1 g, 0.000 23 mol), 2-(tributylstannyl)oxazole (0.25 g, 0.1 mL, 0.000 69 mol), P(Cy)3 (0.0064 g, 0.000 023 mol) in anhydrous DMF (2 mL) was degassed for 10 min. Pd2(dba)3 (0.01 g, 0.000 011 mol) was added, and the reaction mixture was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 150 W was used, the temperature being ramped from 25 to 150 °C while stirring. Once 150 °C was reached, taking about 2 min, the reaction mixture was held at this temperature for 10 min. The reaction was quenched on a mixture of a saturated aqueous solution of potassium carbonate (20 mL) and ethyl acetate while stirring for 30 min. The organic layer was separated and washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:3 as eluent) to furnish 31 (0.03, 35%), mp 80–83 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.56 (s, 6H), 3.57 (s, 3H), 6.38 (s, 2H), 7.06–7.10 (m, 1H), 7.16–7.20 (m, 1H), 7.40–7.52 (m, 3H), 7.76–7.77 (m, 1H), 11.66 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3340 cm−1. Anal. (C20H18N2O4S (382.43)) C, H, N, S.
6-(3-((3,4,5-Trimethoxyphenyl)thio)-1H-indol-2-yl)quinoline (66)
A mixture of 108 (0.1 g, 0.000 23 mol), 6-quinolineboronic acid pinacol ester (0.076 g, 0.0003 mol), and potassium phosphate (0.14 g, 0.000 68 mol) in anhydrous DMF (10 mL) was degassed for 30 min. Pd(dppf)Cl2.CH2Cl2 (0.0054 g, 0.000 006 6 mol, complex with dichloromethane (1:1), Pd 13%) was added under an Ar stream, and the reaction mixture was heated at 110 °C for 12 h. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 8:2 as eluent) to furnish 66 as a slurry (0.07 g, 70%). 1H NMR (DMSO-d6): δ 3.52 (s, 6H), 3.55 (s, 3H), 6.35 (s, 2H), 7.14–7.16 (m, 1H), 7.25–7.28 (m, 1H), 7.54–7.61 (m, 3H), 8.15 (d, J = 8.8 Hz, 1H), 8.32 (dd, J = 2.0 and 8.8 Hz, 1H), 8.42 (d, J = 7.3 Hz, 1H), 8.47 (d, J = 1.8 Hz, 1H), 8.95 (dd, J = 1.7 and 4.2 Hz, 1H), 12.29 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2922, 3305 cm−1. Anal. (C26H22N2O3S (442.53)) C, H, N, S.
3-((3,4,5-Trimethoxyphenyl)thio)-1H,1′H-2,5′-bisindole (67)
67 was synthesized similarly to 66, starting from 108 and 1-Boc-indole-5-boronic acid pinacol ester. Yield 26% as a slurry. 1H NMR (CDCl3): δ 3.64 (s, 6H), 3.79 (s, 3H), 6.41 (s, 2H), 6.61–6.62 (m, 1H), 7.21–7.28 (m, 3H), 7.45–7.47 (m, 2H), 7.64–7.71 (m, 2H), 8.05–8.06 (m, 1H), 8.32 (broad s, disappeared on treatment with D2O, 1H), 8.65 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2963, 2918, 3350 cm−1. Anal. (C25H22N2O3S (430.52)) C, H, N, S.
3-((3,4,5-Trimethoxyphenyl)thio)-1H,1′H-2,2′-bisindole (68)
A mixture of 108 (0.15 g, 0.000 34 mol), 2-(triethylstannyl)-1H-indole-1-carboxylic acid24 (0.25 g, 0.000 683 mol), and ethanol (5 mL) was degassed for 30 min. PdCl2(PPh3)2 (0.021 g, 0.000 031 mol) was added under an Ar stream, and the reaction mixture was heated at 78 °C for 48 h. After cooling, the mixture was filtered through a pad of Celite, then diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:2 as eluent) to furnish 68 (0.03 g, 20%), mp 220–225 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.52 (s, 6H), 3.55 (s, 3H), 6.39 (s, 2H), 7.02–7.06 (m, 1H), 7.13–7.18 (m, 3H), 7.22–7.26 (m, 1H), 7.49–7.55 (m, 3H), 7.59–7.61 (m, 1H), 11.37 (broad s, disappeared on treatment with D2O, 1H), 12.02 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3326, 3388 cm−1. Anal. (C25H22N2O3S (430.52)) C, H, N, S.
2-(Benzofuran-2-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (69)
A mixture of 108 (0.1 g, 0.000 23 mol), 2-benzofuranylboronic acid (0.29 g, 0.0018 mol), and potassium carbonate (0.28 g, 0.02 mol) in 1,4-dioxane (12 mL) containing water (2 mL) was degassed for 30 min. Pd(OCOMe)2 (0.052 g, 0.002 34 mol) was added under an Ar stream, and the reaction mixture was heated at 100 °C for 24 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:2 as eluent) to furnish 69 (0.11 g, 88%), mp 190–195 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.54 (s, 6H), 3.56 (s, 3H), 6.42 (s, 2H), 7.14–7.18 (m, 1H), 7.27–7.33 (m, 2H), 7.37–7.42 (m, 1H), 7.54–7.57 (m, 2H), 7.64–7.67 (m, 2H), 7.75–7.76 (m, 1H), 12.49 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3338 cm−1. Anal. (C25H21NO4S (431.50)) C, H, N, S.
2-(Benzo[b]thiophen-2-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (70)
A mixture of 108 (0.1 g, 0.000 23 mol), benzo[b]thien-2-ylboronic acid (0.04 g, 0.000 225 mol), and 1 M potassium carbonate (0.6 mL) in THF (5 mL) was degassed for 30 min. Pd(PPh3)4 (0.01 g, 0.009 mmol) was added under an Ar stream, and the reaction mixture was heated at 60 °C for 24 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, dichloromethane/ethyl acetate = 99:1 as eluent) to furnish 70 (0.01 g, 10%), mp 220–225 °C (from ethanol). 1H NMR (CDCl3): δ 3.66 (s, 6H), 3.78 (s, 3H), 6.44 (s, 2H), 7.21–7.23 (m, 1H), 7.28–7.39 (m, 3H), 7.46 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.80–7.86 (m, 3H), 8.83 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3303 cm−1. Anal. (C25H21NO3S2 (447.57)) C, H, N, S.
2-(Benzo[b]thiophen-3-yl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (71)
A mixture of 108 (0.1 g, 0.000 23 mol), SPhos (0.0075 g, 0.0182 mmol), Pd2(dba)3 (0.0042 g, 0.004 54 mmol), and potassium carbonate (0.0964 g, 0.000 454 mol) was degassed for 30 min. A solution of benzo[b]thien-3-ylboronic acid (0.061 g, 0.000 34 mol) in n-BuOH (2 mL) was added under an Ar stream, and the reaction mixture was heated at 100 °C for 12 h. After cooling, the mixture was diluted with water, made acidic with 1 M HCl (pH ≈ 3–4), and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:2 as eluent) to furnish 71 (0.037 g, 36%), mp 170–174 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.48 (s, 6H), 3.53 (s, 3H), 6.25 (s, 2H), 7.16–7.18 (m, 1H), 7.24–7.28 (m, 1H), 7.45–7.47 (m, 2H), 7.53–7.56 (m, 2H), 7.87–7.89 (m, 1H), 8.18 (s, 1H), 8.09–8.12 (m, 1H), 12.17 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3237 cm−1. Anal. (C25H21NO3S2 (447.57)) C, H, N, S.
2-Methoxy-5-(3-((3,4,5-trimethoxyphenyl)thio)-1H-indol-2-yl)phenol (49)
Anhydrous aluminum chloride (0.14 g, 0.001 mol) was added to a solution of 48 (0.17 g, 0.000 35 mol) in anhydrous dichloromethane (10 mL). The reaction mixture was stirred at 25 °C for 1.5 h. After dilution with a saturated aqueous solution of ammonium chloride, the mixture was extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 49 (0.05 g, 35%), mp 110–113 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.77 (s, 3H), 3.95 (s, 3H), 5.69 (s, 1H), 6.36 (s, 2H), 6.94 (d, J = 8.3 Hz, 1H), 7.16–7.20 (m, 1H), 7.24–7.28 (m, 1H), 7.34 (dd, J = 2.2 and 8.3 Hz, 1H), 7.38 (d, J = 2.2 Hz, 1H), 7.42–7.44 (m, 1H), 7.66–7.68 (m, 1H), 8.49 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2933, 3329 cm−1. Anal. (C24H23NO5S (437.51)) C, H, N, S.
2-(3,4-Dimethoxyphenyl)-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (50)
Iodomethane (0.033 g, 0.01 mL, 0.000 23 mol) was added to a mixture of 49 (0.1 g, 0.000 23 mol) and anhydrous potassium carbonate (0.032 g, 0.000 23 mol) in anhydrous DMF (5 mL). The reaction mixture was stirred at 25 °C for 2 h, then diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 50 (0.02 g, 20%), mp 117–118 °C (from ethanol). 1H NMR (CDCl3): δ 3.65 (s, 6H), 3.77 (s, 3H), 3.78 (s, 3H), 3.94 (s, 3H), 6.38 (s, 2H), 6.97 (d, J = 8.3 Hz, 1H), 7.19–7.23 (m, 1H), 7.26–7.31 (m, 2H), 7.40 (d, J = 2.1 Hz, 1H), 7.45–7.47 (m, 1H), 7.70–7.72 (m, 1H), 8.57 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3344 cm−1. Anal. (C25H25NO5S (451.53)) C, H, N, S.
Aroyl-1H-indoles. General Procedure for the Preparation of Compounds 7, 15, 19, 54, 56, 58, and 60. Example: (2-(1H-Pyrrol-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (7)
Methylmagnesium bromide (1.4 mL, 0.0041 mol, 3.0 M in Et2O) was added dropwise over 5 min into a mixture of 2-(1H-pyrrol-2-yl)-1H-indole25 (109) (0.5 g, 0.003 mol) and anhydrous ZnCl2 (0.82 g, 0.006 mol) in anhydrous dichloromethane (20 mL) at 25 °C under an Ar stream. After 1 h, a solution of 3,4,5-trimethoxybenzoyl chloride (0.76 g, 0.0033 mol) in the same solvent (16 mL) was added dropwise over 5 min at 25 °C under an Ar stream. After an additional 1 h, SnCl4 (3 mL, 0.003 mol, 1.0 M in dichloromethane) was added dropwise, and the reaction mixture was stirred at 25 °C for 12 h under an Ar stream. The reaction was quenched on crushed ice and extracted with dichloro-methane The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 7 (0.03 g, 3%), mp 59–62 °C (from toluene). 1H NMR (DMSO-d6): δ 3.70 (s, 6H), 3.75 (s, 3H), 6.24–6.26 (m, 1H), 6.84–6.85 (m, 1H), 6.96–7.07 (m, 5H), 7.14–7.18 (m, 1H), 7.43–7.45 (m, 1H), 11.94 (broad s, disappeared on treatment with D2O, 1H), 12.15 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1623, 3344 cm−1. Anal. (C22H20N2O4 (376.41)) C, H, N.
(2-(1H-Pyrazol-4-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (15)
15 was synthesized similarly to 7, starting from 98. Yield 21%, mp 112–117 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.65 (s, 6H), 3.71 (s, 3H), 6.95 (s, 2H), 7.06–7.10 (m, 1H), 7.16–7.20 (m, 1H), 7.44–7.51 (m, 2H), 7.86 (m, 2H), 12.02 (broad s, disappeared on treatment with D2O, 1H), 13.04 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1634, 2962, 3243 cm−1. Anal. (C21H19N3O4 (377.39)) C, H, N.
(2-(1H-Imidazol-1-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (19)
19 was synthesized similarly to 7 starting from 80.21 Yield 26%, mp 210–215 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.66 (s, 3H), 3.69 (s, 6H), 6.79 (s, 2H), 6.88–6.89 (m, 1H), 7.24–7.32 (m, 2H), 7.39–7.40 (m, 1H), 7.50–7.52 (m, 1H), 7.79–7.80 (m, 1H), 7.97–7.99 (m, 1H), 12.66 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1618, 3354 cm−1. Anal. (C21H19N3O4 (377.39)) C, H, N.
(2-(Pyridin-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (54)
54 was synthesized similarly to 7, starting from 92. Yield 67%, mp 65–70 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.61 (s, 6H), 3.64 (s, 3H), 6.89 (s, 2H), 7.19–7.21 (m, 1H), 7.26–7.28 (m, 2H), 7.39–7.41 (m, 1H), 7.56–7.58 (m, 1H), 7.63–7.65 (m, 1H), 7.79–7.81 (m, 1H), 8.55–8.57 (m, 1H), 12.36 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1625, 3241 cm−1. Anal. (C23H20N2O4 (388.42)) C, H, N.
(2-(Pyridin-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (56)
56 was synthesized similarly to 7, starting from 93. Yield 45%, mp 130–135 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.60 (s, 3H), 3.61 (s, 6H), 6.82 (s, 2H), 7.21–7.31 (m, 3H), 7.53–7.55 (m, 1H), 7.75–7.78 (m, 1H), 7.96 (d, J = 7.7 Hz, 1H), 8.44 (dd, J = 1.6 and 4.8 Hz, 1H), 8.56–8.57 (m, 1H), 12.38 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1597, 3094 cm−1. Anal. (C23H20N2O4 (388.42)) C, H, N.
(2-(Pyridin-4-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (58)
58 was synthesized similarly to 7, starting from 94. Yield 10%, mp 50–55 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.62 (s, 3H), 3.63 (s, 6H), 6.86 (s, 2H), 7.21–7.25 (m, 1H), 7.29–7.33 (m, 1H), 7.35–7.37 (m, 2H), 7.55–7.57 (m, 1H), 7.87–7.90 (m, 1H), 8.46–8.47 (m, 2H), 12.43 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1602, 3304 cm−1. Anal. (C23H20N2O4 (388.42)) C, H, N.
(2-(Naphthalen-1-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (60)
60 was synthesized similarly to 7, starting from 102. Yield 50%, mp 177–179 °C (from ethanol). 1H NMR (CDCl3): δ 3.40 (s, 6H), 3.64 (s, 3H), 6.61 (s, 2H), 7.26–7.30 (m, 1H), 7.34–7.36 (m, 1H), 7.37–7.41 (m, 2H), 7.43–7.53 (m, 3H), 7.73 (d, J = 8.2 Hz, 1H), 7.78–7.83 (m, 1H), 7.94–8.00 (m, 1H), 8.31–8.36 (m, 1H), 8.81 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1605, 3158 cm−1. Anal. (C28H23NO4 (437.49)) C, H, N.
General Procedure for the Preparation of Compounds 9, 21, 23, 25, 32, 35, 40, 42, 44, 51, 63, 74, 76, and 78. Example: (2-(1-(Phenylsulfonyl)-1H-pyrrol-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (9)
A mixture of 2-(1-(phenyl-sulfonyl)-1H-pyrrol-3-yl)-1H-indole19 (110) (0.26 g, 0.000 82 mol), 3,4,5-trimethoxybenzoyl chloride (0.23 g, 0.000 98 mol), and anhydrous aluminum chloride (0.13 g, 0.000 98 mol) in anhydrous 1,2-dichloro-ethane (2 mL) was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C while stirring. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min. The reaction mixture was quenched on 1 M HCl/crushed ice and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 9 (0.27 g, 65%), mp 124–126 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.57 (s, 6H), 3.67 (s, 3H), 6.44 (s, 1H), 6.92 (s, 2H), 7.07 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 7.35 (s, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.66 (t, J = 7.8 Hz, 2H), 7.76–7.78 (m, 2H), 7.96 (d, J = 7.4 Hz, 2H), 12.04 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1603, 3303 cm−1. Anal. (C28H24N2O6S (516.56)) C, H, N, S.
(2-(Furan-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (21)
21 was synthesized similarly to 9, starting from 111.19 Yield 5%, mp 115–117 °C (from ethanol). 1H NMR (CDCl3): δ 3.80 (s, 6H), 3.95 (s, 3H), 6.50–6.51 (m, 1H), 7.10–7.15 (m, 3H), 7.22–7.26 (m, 2H), 7.41–7.46 (m, 2H), 7.52–7.53 (m, 1H), 9.11 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1643, 3358 cm−1. Anal. (C22H19NO5 (377.99)) C, H, N.
(2-(Furan-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (23)
23 was synthesized similarly to 9, starting from 112.19 Yield 32%, mp 115–117 °C (from ethanol). 1H NMR (CDCl3): δ 3.78 (s, 6H), 3.93 (s, 3H), 6.60–6.61 (m, 1H), 7.13 (s, 2H), 7.15–7.19 (m, 1H), 7.25–7.27 (m, 1H), 7.43–7.46 (m, 2H), 7.58–7.60 (m, 1H), 8.05–8.06 (m, 1H), 8.70 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1598, 3345 cm −1. Anal. (C22H19NO5 (377.99)) C, H, N.
(2-(Thiophen-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (25)
25 was synthesized similarly to 9, starting from 113.19 Yield 25%, mp 118–120 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 6H), 3.87 (s, 3H), 7.04 (s, 2H), 7.11–7.12 (m, 1H), 7.23–7.28 (m, 2H), 7.29–7.33 (m, 1H), 7.44–7.47 (m, 1H), 7.50–7.51 (m, 1H), 7.91–7.93 (m, 1H), 8.60 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1606, 3337 cm−1. Anal. (C22H19NO4S (393.46)) C, H, N, S.
(2-(Oxazol-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (32)
32 was synthesized similarly to 9, starting from 114. Yield 28%, mp 208–210 °C (from ethanol). 1H NMR (CDCl3): δ 3.79 (s, 6H), 3.94 (s, 3H), 7.20–7.26 (m, 4H), 7.35–7.39 (m, 1H), 7.49–7.56 (m, 2H), 7.73–7.75 (m, 1H), 9.69 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1608, 3137 cm−1. Anal. (C21H18N2O5 (378.38)) C, H, N.
(2-Phenyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (35)
35 was synthesized similarly to 9, starting from 84. Yield 95%, mp 207–212 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.60 (s, 6H), 6.82 (s, 2H), 7.18–7.28 (m, 5H), 7.36–7.38 (m, 2H), 7.50–7.53 (m, 1H), 7.92–7.94 (m, 1H), 12.20 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1590, 3213 cm−1. Anal. (C24H21NO4 (387.43)) C, H, N.
(2-(2-Fluorophenyl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (40)
40 was synthesized similarly to 9, starting from 86. Yield 65%, mp 180–185 °C (from toluene). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.64 (s, 6H), 6.79 (s, 2H), 7.07–7.35 (m, 5H), 7.45–7.48 (m, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 7.8 Hz, 1H), 12.27 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1591, 3187 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, N, F.
(2-(3-Fluorophenyl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (42)
42 was synthesized similarly to 9, starting from 87. Yield 52%, mp 190–195 °C (from toluene). 1H NMR (DMSO-d6): δ 3.60 (s, 3H), 3.62 (s, 6H), 6.78 (s, 2H), 7.11 (t, J = 8.8 Hz, 2H), 7.20 (t, J = 7.6 Hz, 1H), 7.27 (t, J = 7.2 Hz, 1H), 7.39–7.42 (m, 2H), 7.51 (d, J = 7.9 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 12.21 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1594, 3221 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(2-(4-Fluorophenyl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (44)
44 was synthesized similarly to 9, starting from 88. Yield 64%, mp 210–215 °C (from toluene). 1H NMR (DMSO-d6): δ 3.60 (s, 3H), 3.62 (s, 6H), 6.78 (s, 2H), 7.10 (t, J = 8.9 Hz, 2H), 7.20 (t, J = 7.3 Hz, 1H), 7.27 (t, J = 7.1 Hz, 2H), 7.38–7.42 (m, 2H), 7.51 (d, J = 8.2 Hz, 1H), 12.22 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1592, 3218 cm−1. Anal. (C24H20FNO4 (405.42)) C, H, F, N.
(2-(4-Methoxyphenyl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (51)
51 was synthesized similarly to 9, starting from 91. Yield 9%, mp 170–175 °C (from ethanol). 1H NMR (CDCl3): δ 3.72 (s, 6H), 3.78 (s, 3H), 3.84 (s, 3H), 6.76–6.79 (m, 2H), 7.00 (s, 2H), 7.27–7.34 (m, 4H), 7.46–7.48 (m, 1H), 8.03–8.05 (m, 1H), 8.70 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1614, 3144 cm−1. Anal. (C25H23NO5 (417.45)) C, H, N.
(2-(Naphthalen-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (63)
63 was synthesized similarly to 9, starting from 103. Yield 33%, mp 182–186 °C (from ethanol). 1H NMR (CDCl3): δ 3.56 (s, 3H), 3.63 (s, 6H), 6.98 (s, 2H), 7.29–7.37 (m, 2H), 7.43 (dd, J = 1.8 and 8.5 Hz, 1H), 7.47–7.52 (m, 3H), 7.68–7.78 (m, 3H), 7.87 (m, 1H), 8.10–8.11 (m, 1H), 8.80 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1609, 3170 cm−1. Anal. (C28H23NO4 (437.49)) C, H, N.
(2-Cyclobutyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (74)
74 was synthesized similarly to 9, starting from 94. Yield 75%, mp 60–63 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.84–1.94 (m, 2H), 2.14–2.22 (m, 2H), 2.29–2.39 (m, 2H), 3.76 (s, 6H), 3.77 (s, 3H), 3.79–3.81 (m, 1H), 6.94 (s, 2H), 7.02–7.06 (m, 1H), 7.12–7.16 (m, 1H), 7.40–7.42 (m, 1H), 7.44–7.47 (m, 1H), 11.95 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1600, 3217 cm−1. Anal. (C22H23NO4 (365.42)) C, H, N.
(2-Cyclopentyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (76)
76 was synthesized similarly to 9, starting from 95. Yield 68%, mp 110–115 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.57–1.62 (m, 2H), 1.75–1.85 (m, 4H), 1.95–1.99 (m, 2H), 3.37–3.45 (m, 1H), 3.76 (s, 6H), 3.78 (s, 3H), 6.97 (s, 2H), 7.0–7.04 (m, 1H), 7.10–7.14 (m, 1H), 7.29–7.31 (m, 1H), 7.41–7.43 (m, 1H), 11.77 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1603, 3261 cm−1. Anal. (C23H25NO4 (379.45)) C, H, N.
(2-Cyclohexyl-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (78)
78 was synthesized similarly to 9, starting from 96. Yield 68%, mp 140–145 °C (from ethanol). 1H NMR (DMSO-d6): δ 1.08–1.26 (m, 3H), 1.59–1.70 (m, 3H), 1.77–1.83 (m, 4H), 2.89–2.95 (m, 1H), 3.75 (s, 6H), 3.76 (s, 3H), 6.95 (s, 2H), 7.01–7.05 (m, 1H), 7.10–7.14 (m, 1H), 7.38–7.42 (m, 2H), 11.80 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1602, 3285 cm−1. Anal. (C24H27NO4 (393.48)) C, H, N.
((2-(1H-Pyrrol-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (8)
A mixture of 9 (0.1 g, 0.000 19 mol) and 2 M NaOH (0.6 mL) in methanol (2 mL) was heated at reflux for 3 h. After cooling, the reaction mixture was made acidic with 2 M HCl (pH ≈ 3–4) and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 8 (0.7 g, 98%), mp 114–115 °C (from ethanol). 1H NMR (CDCl3): δ 3.77 (s, 6H), 3.90 (s, 3H), 6.42–6.44 (m, 1H), 6.77–6.79 (m, 1H), 7.10 (s, 2H), 7.14–7.25 (m, 3H), 7.40–7.42 (m, 1H), 7.67–7.71 (m, 1H), 8.38 (broad s, disappeared on treatment with D2O, 1H), 8.53 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1614, 3241, 3333 cm−1. Anal. (C22H20N2O4 (376.41)) C, H, N.
(2-(4,5-Dihydrooxazol-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxy-phenyl)methanone (29)
29 was synthesized similarly to 28, starting from 3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylic acid. Yield 47%, mp 220–225 °C (from ethanol). 1H NMR (CDCl3): δ 3.84 (s, 6H), 3.94 (s, 3H), 3.99 (m, 2H), 4.19 (m, 2H), 7.19 (s, 2H), 7.21–7.23 (m, 1H), 7.34–7.37 (m, 1H), 7.45–7.47 (m, 1H), 7.70–7.72 (m, 1H), 10.24 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3182 cm−1. Anal. (C21H20N2O5 (380.39)) C, H, N.
3-(3,4,5-Trimethoxybenzoyl)-1H-indole-2-carboxylic Acid
A mixture of methyl 3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate18 (0.42 g, 0.000 65 mol) and 3 N NaOH (2 mL) was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C, while stirring. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min, then made acidic with 3 N HCl (pH ≈ 3–4) and extracted with ethyl acetate. The organic layer was washed with brine, dried, filtered, and evaporated to give 3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylic acid (0.23 g, 98%). 1H NMR (CDCl3): δ 3.84 (s, 6H), 1.09 (s, 3H), 7.11 (s, 2H), 7.16–7.20 (m, 1H), 7.23–7.25 (m, 1H), 7.39–7.43 (m, 1H), 7.67 (m, 1H), 10.88 (broad s, disappeared on treatment with D2O, 1H), 12.95 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1696, 2837, 2933, 3265 cm−1. Anal. (C19H17NO6 (355.34)) C, H, N.
Arylmethyl-1H-indoles. General Procedure for the Preparation of Compounds 12, 22, 24, 26, 36, 52, 61, and 65. Example: 2-(1-(Phenylsulfonyl)-1H-pyrrol-3-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (12)
Borane–tetrahydrofuran complex, 1 M solution in THF (0.85 mL, 0.000 85 mol), was added to an ice-cooled solution of 9 (0.1 g, 0.019 mol) in acetonitrile (1.2 mL) containing methanol (0.017 mL) under an Ar stream. The reaction mixture was heated at 50 °C for 1 h. After cooling, the reaction mixture was quenched on crushed ice and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 12 (0.08 g, 84%), mp 176–180 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.55 (s, 6H), 3.59 (s, 3H), 4.15 (s, 2H), 6.46 (s, 2H), 6.81–6.82 (m, 1H), 6.97–6.99 (m, 1H), 7.06–7.08 (m, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.48–7.50 (m, 2H), 7.62–7.66 (m, 3H), 7.75–7.76 (m, 1H), 7.94–7.96 (m, 2H), 11.16 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3293 cm−1. Anal. (C28H26N2O5S (502.58)) C, H, N, S.
2-(Furan-2-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (22)
22 was synthesized similarly to 12, starting from 21. Yield 9%, mp 78–80 °C (from ethanol). 1H NMR (CDCl3): δ 3.76 (s, 6H), 3.82 (s, 3H), 4.30 (s, 2H), 6.51–6.55 (m, 4H), 7.09–7.13 (m, 1H), 7.21–7.24 (m, 1H), 7.40–7.43 (m, 1H), 7.52–7.54 (m, 2H), 8.49 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3347 cm−1. Anal. (C22H21NO4 (363.41)) C, H, N.
2-(Furan-3-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (24)
24 was synthesized similarly to 12, starting from 23. Yield 13%, mp 102–105 °C (from ethanol). 1H NMR (CDCl3): δ 3.73 (s, 6H), 3.80 (s, 3H), 4.18 (s, 2H), 6.44 (s, 2H), 6.65–6.66 (m, 1H), 7.07–7.11 (m, 1H), 7.17–7.19 (m, 1H), 7.37–7.39 (m, 1H), 7.47–7.49 (m, 1H), 7.52–7.53 (m, 1H), 7.63–7.64 (m, 1H), 8.07 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3320 cm−1. Anal. (C22H21NO4 (363.41)) C, H, N.
2-(Thiophen-3-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (26)
26 was synthesized similarly to 12, starting from 25. Yield 61%, mp 165–168 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 6H), 3.83 (s, 3H), 4.25 (s, 2H), 6.48 (s, 2H), 7.10–7.14 (m, 1H), 7.21–7.25 (m, 1H), 7.34–7.35 (m, 1H), 7.40–7.46 (m, 3H), 7.51–7.53 (m, 1H), 8.22 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3353 cm−1. Anal. (C22H21NO3S (379.47)) C, H, N, S.
2-Phenyl-3-(3,4,5-trimethoxybenzyl)-1H-indole (36)
36 was synthesized similarly to 12, starting from 35. Yield 26%, mp 187–192 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.60 (s, 6H), 4.17 (s, 2H), 6.46 (s, 2H), 6.97–7.01 (m, 1H), 7.09–7.13 (m, 1H), 7.37–7.41 (m, 2H), 7.47–7.53 (m, 3H), 7.64–7.66 (m, 2H), 11.28 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3351 cm−1. Anal. (C24H23NO3 (373.44)) C, H, N.
2-(4-Methoxyphenyl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (52)
52 was synthesized similarly to 12, starting from 51. Yield 4% as an oil. 1H NMR (CDCl3): δ 3.73 (s, 6H), 3.82 (s, 3H), 3.86 (s, 3H), 4.19 (s, 2H), 6.46 (s, 2H), 6.98–7.00 (m, 2H), 7.07–7.11 (m, 1H), 7.18–7.22 (m, 1H), 7.39–7.41 (m, 1H), 7.46–7.49 (m, 3H), 8.09 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3356 cm−1. Anal. (C25H25NO4 (403.47)) C, H, N.
2-(Naphthalen-1-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (61)
61 was synthesized similarly to 12, starting from 60. Yield 99%, mp 142–147 °C (from ethanol). 1H NMR (CDCl3): δ 3.60 (s, 6H), 3.74 (s, 3H), 4.03 (s, 2H), 6.26 (s, 2H), 7.18–7.20 (m, 1H), 7.25–7.29 (m, 1H), 7.43–7.47 (m, 2H), 7.52–7.58 (m, 3H), 7.63–7.65 (m, 1H), 7.84–7.86 (m, 1H), 7.94–7.96 (m, 2H), 8.19 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3317 cm−1. Anal. (C28H25NO3 (423.50)) C, H, N.
2-(Naphthalen-2-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (65)
65 was synthesized similarly to 12, starting from 63. Yield 73%, mp 138–140 °C (from ethanol). 1H NMR (CDCl3): δ 3.73 (s, 6H), 3.83 (s, 3H), 4.29 (s, 2H), 6.52 (s, 2H), 7.13–7.17 (m, 1H), 7.24–7.28 (m, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.51–7.58 (m, 3H), 7.70 (dd, J = 1.8 and 8.5 Hz, 1H), 7.82–7.94 (m, 3H), 7.99 (m, 1H), 8.30 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3359 cm−1. Anal. (C28H25NO3 (423.50)) C, H, N.
2-(1H-Pyrrol-3-yl)-3-(3,4,5-trimethoxybenzyl)-1H-indole (13)
13 was synthesized similarly to 8, from 12. Yield 20%, mp 195–197 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.59 (s, 3H), 3.61 (s, 6H), 4.14 (s, 2H), 6.48–6.49 (m, 1H), 6.53 (s, 2H), 6.86–6.92 (m, 2H), 6.97–7.01 (m, 1H), 7.12–7.13 (m, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 10.88 (broad s, disappeared on treatment with D2O, 1H), 11.03 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3242 cm−1. Anal. (C22H22N2O3 (362.42)) C, H, N.
2-(3-(3,4,5-Trimethoxybenzyl)-1H-indol-2-yl)-4,5-dihydroxazole (30)
30 was synthesized similarly to 28, starting from 3-(3,4,5-tri-methoxybenzyl)-1H-indole-2-carboxylic acid. Yield 80%, mp 160–165 °C (from ethanol). 1H NMR (CDCl3): δ 3.77 (s, 6H), 3.80 (s, 3H), 4.09 (t, J = 9.5 Hz, 2H), 4.44–4.52 (m, 4H), 6.55 (s, 2H), 7.07–7.11 (m, 1H), 7.25–7.29 (m, 1H), 7.36 (dd, J = 0.75 and 8.3 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 9.10 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3257 cm−1. Anal. (C21H22N2O4 (366.41)) C, H, N.
3-(3,4,5-Trimethoxybenzyl)-1H-indole-2-carboxylic Acid
The compound was obtained from methyl 3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate.18 Yield 96%, mp 240–245 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.56 (s, 3H), 3.66 (s, 6H), 4.37 (s, 2H), 6.65 (s, 2H), 6.99–7.03 (m, 1H), 7.18–7.23 (m, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 11.48 (broad s, disappeared on treatment with D2O, 1H), 13.05 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1663, 2840, 3354 cm−1. Anal. (C19H19NO5 (341.36)) C, H, N.
Arylmethanol-1H-indoles. (2-(1H-Pyrrol-3-yl)-1H-indol-3-yl)-(3,4,5-trimethoxyphenyl)methanol (10)
10 was synthesized similarly to 8, starting from 11. Yield 98%, mp 189–191 °C (from ethanol). 1H NMR (CDCl3): δ 3.57 (s, 6H), 3.86 (s, 3H), 5.13 (m, 1H), 6.00 (s, disappeared on treatment with D2O, 1H), 6.38–6.39 (m, 2H), 6.54 (s, 1H), 6.62–6.64 (m, 1H), 6.75–6.78 (m, 3H), 6.85–6.88 (m, 1H), 7.00–7.03 (m, 1H), 8.01 (broad s, disappeared on treatment with D2O, 1H), 8.16 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2852, 2922, 3382 cm−1. Anal. (C22H22N2O4 (378.42)) C, H, N.
General Procedure for the Preparation of Compounds 11 and 64. Example: (2-(1-(Phenylsulfonyl)-1H-pyrrol-3-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanol (11)
To a solution of 9 (0.5 g, 0.0013 mol) in THF (2.6 mL) containing 0.08 mL of water was carefully added sodium borohydride (0.049 g, 0.0013 mol). The reaction mixture was heated at 80 °C for 2 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, acetone/n-hexane = 1:1 as eluent) to furnish 11 (0.54 g, 97%), mp 90–94 °C (from ethanol). 1H NMR (DMSO-d6): 3.58 (s, 6H), 3.60 (s, 3H), 5.71–5.73 (m, 1H), 6.05 (s, disappeared on treatment with D2O, 1H), 6.63 (s, 2H), 6.83–6.86 (m, 2H), 7.01 (t, J = 6.6 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.47–7.50 (m, 1H), 7.63–7.67 (m, 2H), 7.74–7.77 (m, 2H), 7.98 (d, J = 7.1 Hz, 2H), 11.14 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2938, 3341 cm−1. Anal. (C28H26N2O6S (518.58)) C, H, N, S.
(2-(Naphthalen-2-yl)-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanol (64)
64 was synthesized similarly to 11, starting from 63. Yield 44%, mp 145–150 °C (from ethanol). 1H NMR (CDCl3): δ 2.29 (d, J = 4.2 Hz, 1H), 3.76 (s, 6H), 3.84 (s, 3H), 6.29 (d, J = 4.2 Hz, 1H), 6.79 (s, 2H), 7.07–7.11 (m, 1H), 7.22–7.26 (m, 1H), 7.43–7.46 (m, 1H), 7.55–7.60 (m, 3H), 7.72 (dd, J = 1.8 and 8.5 Hz, 1H), 7.87–7.91 (m, 2H), 7.96 (d, J = 8.4 Hz, 1H), 8.06 (m, 1H), 8.35 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3286, 3295, 3293 cm−1. Anal. (C28H25NO4 (439.50)) C, H, N.
Synthetic Intermediates. 2-(1H-Imidazol-2-yl)-1H-indole (79). (Diacetoxyiodo)benzene (0.52 g, 0.0016 mol) was added to a mixture of 2-(4,5-dihydro-1H-imidazol-2-yl)-1H-indole (83) (0.27 g, 0.0015 mol) and potassium carbonate (0.22 g, 0.0016 mol) in DMSO (15 mL). The reaction mixture was stirred at 25 °C for 12 h under an Ar stream, then diluted with a saturated aqueous solution of sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 79 (0.08 g, 29%), mp 130–133 °C (from ethanol). 1H NMR (DMSO-d6): δ 7.12 (t, J = 7.5 Hz, 1H), 7.20 (t, J = 7.4 Hz, 1H), 7.23–7.25 (m, 3H), 7.32 (d, J = 7.8 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 11.90 (broad s, disappeared on treatment with D2O, 1H), 12.19 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2920, 3060, 3120, 3380 cm−1.
2-(1H-1,2,4-Triazol-1-yl)-1H-indole (81)
A mixture of 2-iodo-1H-indole23 (106) (0.5 g, 0.0021 mol) and triazole (0.14 g, 0.021 mol) was heated at 150 °C for 2 h. After cooling, the mixture was purified by column chromatography (silica gel, ethyl acetate as eluent) to give 81 (0.16 g, 42%) as a powder. 1H NMR (DMSO-d6): δ 6.76 (s, 1H), 7.08 (t, J = 7.2 Hz, 1H), 7.17 (t, J = 7.1 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 8.32 (s, 1H), 9.22 (broad s, disappeared on treatment with D2O, 1H), 12.19 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3112 cm−1.
2-(1H-Indol-2-yl)thiazole (82)
A mixture 2-bromo-1-(thiazol-2-yl)ethanone (122) (0.5 g, 0.0024 mol) and aniline (0.67 g, 0.6 mL, 0.0072 mol) in anhydrous DMF (2 mL) was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 100 W was used, the temperature being ramped from 25 to 150 °C, while stirring. Once 150 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 1 min, while cooling, then diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 2:7 as eluent) to furnish 82 (0.25 g, 52%), mp 112–115 °C (from ethanol).
2-(4,5-Dihydro-1H-imidazol-2-yl)-1H-indole (83)
Ethylenediamine (0.23 g, 0.25 mL, 0.0038 mol) was added to a solution of 1H-indole-2-carbaldehyde26 (115) (0.5 g, 0.0034 mol) in tert-butanol (34 mL). The reaction mixture was stirred at 25 °C for 30 min under an Ar stream. Iodine (1.08 g, 0.00425 mol) and potassium carbonate (1.41 g, 0.01 mol) were added, and the reaction mixture was heated at 70 °C for 3 h. After cooling, the mixture was diluted with a saturated aqueous solution of sodium sulfite and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, chloroform/ethanol = 8:2 as eluent) to furnish 83 (0.27 g, 43%). 1H NMR (D MSO-d6): δ 3.70 (s, 4H), 7.13 (t, J = 7.6 Hz, 1H), 7.25 (t, J = 6.9 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 8.68 (broad s, disappeared on treatment with D2O, 1H), 11.79 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3387, 2855 cm−1.
General Procedure for the Preparation of Compounds 85, 91–94, and 103. Example: 2-(4-Chlorophenyl)-1H-indole (85)
1-(1-(4-Chlorophenyl)ethylidene)-2-phenylhydrazine (128) (2.82 g, 0.011 mol) was added in portions to polyphosphoric acid (28 g) at 120 °C. The reaction mixture was stirred at the same temperature for 1 h. After cooling, it was quenched on crushed ice and treated with 3 N NaOH with stirring, then extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 as eluent) to furnish 85 (2.1 g, 80%), mp 210–212 °C (from ethanol). Lit.37 mp 206.3–209.0 °C.
2-(4-Methoxyphenyl)-1H-indole (91)
91 was synthesized similarly to 85, starting from 1-(1-(4-methoxyphenyl)ethylidene)-2-phenylhydrazine (129). Yield 37%, mp 222–224 °C (from toluene). Lit.38 mp 224–227 °C.
2-(Pyridin-2-yl)-1H-indole (92)
92 was synthesized similarly to 85, starting from 2-(1-(2-phenylhydrazono)ethyl)pyridine29 (130). Yield 73%, mp 154–156 °C (from ethanol). Lit.39 mp 156.0–156.5 °C.
2-(Pyridin-3-yl)-1H-indole (93)
93 was synthesized similarly to 85, starting from 3-(1-(2-phenylhydrazono)ethyl)pyridine29 (131). Yield 73%, mp 168–170 °C (from ethanol). Lit.40 mp 169–170 °C.
2-(Pyridin-4-yl)-1H-indole (94)
94 was synthesized similarly to 85, starting from 4-(1-(2-phenylhydrazono)ethyl)pyridine29 (132). Yield 68%, mp 237–239 °C (from ethanol). Lit.41 mp 238–239 °C.
2-(Naphthalen-2-yl)-1H-indole (103)
103 was synthesized similarly to 85, starting from 1-(1-(naphthalen-2-yl)ethylidene)-2-phenylhydrazine29 (133). Yield 45%, mp 165–168 °C (petroleum ether). Lit.42 mp 163–165 °C.
General Procedure for the Preparation of Compounds 86–90, 101, and 102. Example: 2-(2-Fluorophenyl)-1H-indole (86)
A solution of bromo-1-(2-fluorophenyl)ethanone (116) (0.9 g, 0.0041) in N,N-dimethylaniline (1.8 mL) was added dropwise to aniline (1.26 g, 1.2 mL, 0.0135 mol) in the same solvent (2.4 mL). The reaction mixture was heated at 170 °C for 15 min. After cooling to 0 °C, the mixture was made acidic with 1 M HCl (pH ≈ 3–4) and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, acetone/n-hexane = 1:3 as eluent) to furnish 89 (0.3 g, 35%), mp 102–104 °C (from toluene). 1H NMR (DMSO-d6): δ 6.92 (s, 1H), 7.03 (t, J = 7.8 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H), 7.34–7.40 (m, 3H), 7.45 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.93 (t, J = 7.6 Hz, 1H), 11.48 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3412 cm−1. Lit.43 mp 97–98.5 °C.
2-(3-Fluorophenyl)-1H-indole (87)
87 was synthesized similarly to 86, starting from 2-bromo-1-(3-fluorophenyl)ethanone (117). Yield 42%, mp 142–144 °C (from toluene). Lit.44 mp 144 °C.
2-(4-Fluorophenyl)-1H-indole (88)
88 was synthesized similarly to 86, starting from 2-bromo-1-(4-fluorophenyl)ethanone (118). Yield 42%, mp 182–187 °C (from toluene). Lit.44 mp 185 °C.
2-(2-Methoxyphenyl)-1H-indole (89)
89 was synthesized similarly to 86, starting from 2-bromo-1-(2-methoxyphenyl)ethanone28 (119). Yield 41%, mp 75–80 °C (from toluene). Lit.45 mp 78.5–81.0 °C.
2-(3-Methoxyphenyl)-1H-indole (90)
90 was synthesized similarly to 86, starting from 2-bromo-1-(3-methoxyphenyl)ethanone28 (120). Yield 40%, mp 138–141 °C (from toluene). Lit.37 mp 139.4–140.7 °C.
2-(3-Isopropoxy-4-methoxyphenyl)-1H-indole (101)
101 was synthesized similarly to 86, starting from 2-bromo-1-(4-fluorophenyl)-ethanone (121). Yield 12%, mp 137–141 °C (from ethanol). 1H NMR (CDCl3): δ 1.42 (s, 3H), 1.43 (s, 3H), 3.90 (s, 3H), 4.64 (m, 1H), 6.72 (s, 1H), 6.95 (d, J = 8.2 Hz, 1H), 7.10–7.27 (m, 5H), 7.39 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 8.3 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3366 cm−1.
2-(Naphthalen-1-yl)-1H-indole (102)
102 was synthesized similarly to 86, starting from 2-bromo-1-(naphthalen-1-yl)ethanone (122). Yield 28%, mp 95–97 °C (from petroleum ether). Lit.46 mp 97–99°C.
General Procedure for the Preparation of Compounds 95–97, 100, and 104. Example: 2-(p-Tolyl)-1H-indole (100)
n-Butyllithium (4.2 mL, 0.0067 mol, 1.6 M in n-hexane) was added dropwise to a –40 °C solution of 4-methyl-N-(o-tolyl)benzamide (123) (0.5 g, 0.0022 mol) in anhydrous THF (18 mL). After being stirred at –40 °C for 1 h, the reaction mixture was warmed at 0 °C for 1 h and then kept at 25 °C for 12 h. The mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:5 as eluent) to furnish 100 (0.2 g, 43%), mp 217–219 °C (from ethanol). Lit.45 mp 218.0–219.9 °C.
2-((1,1′-Biphenyl)-4-yl)-1H-indole (104)
104 was synthesized similarly to 100, starting from N-(o-tolyl)-(1,1′-biphenyl)-4-carboxamide (124). Yield 22%, mp 296–298 °C (from ethanol). 1H NMR (DMSO-d6): δ 6.97–7.04 (m, 2H), 7.10–7.14 (t, J = 8.0 Hz, 1H), 7.37–7.41 (m, 2H), 7.44–7.56 (m, 3H), 7.75–7.80 (m, 4H), 7.97 (d, J = 8.0 Hz, 2H), 11,61 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3681 cm−1. Lit.47 mp 297–298 °C.
2-Cyclobutyl-1H-indole (95)
95 was synthesized similarly to 100, starting from N-(o-tolyl)cyclobutanecarboxamide (125). Yield 40%, mp 73–76 °C (from toluene). 1H NMR (DMSO-d6): δ 1.86–2.03 (m, 2H), 2.19–2.33 (m, 2H), 2.58–2.54 (m, 2H), 3.57–3.66 (m, 1H), 6.16–6.17 (m, 1H), 6.89–6.93 (m, 1H), 6.98–7.01 (m, 1H), 7.26–7.29 (m, 1H), 7.39–7.41 (m, 1H), 10.90 (broad s, disappeared on treatment with D2O, 1H). IR: ν 3334 cm−1.
2-Cyclopentyl-1H-indole (96)
96 was synthesized similarly to 100, starting from N-(o-tolyl)cyclopentanecarboxamide (126). Yield 40%, mp 80–82 °C (from toluene). Lit.48 mp 84–85 °C.
2-Cyclohexyl-1H-indole (97)
97 was synthesized similarly to 100, starting from N-(o-tolyl)cyclohexanecarboxamide (127). Yield 59%, mp 101–103 °C (from toluene). Lit.49 mp 103–105 °C.
2-(1H-Pyrazol-4-yl)-1H-indole (98)
A mixture of 106 (0.1 g, 0.000 41 mol), 1-Boc-pyrazole-4-boronic acid pinacol ester (0.18 g, 0.000 615 mol), and 1 M potassium carbonate (4.1 mL) in DMF (2 mL) was degassed for 30 min. PdCl2(PPh3)2 (0.003 g, 0. 0041 mmol) was added, and the reaction mixture was placed into the microwave cavity (closed vessel mode, Pmax = 250 psi). A starting microwave irradiation of 250 W was used, the temperature being ramped from 25 to 160 °C, while stirring. Once 160 °C was reached, taking about 2 min, the reaction mixture was held at this temperature for 10 min. The reaction mixture was diluted with water, neutralized with 1 M HCl, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, diethyl ether/methanol/n-hexane = 8:1:2 as eluent) to furnish 79 (0.02 g, 26%), mp 220–225 °C (from ethanol). 1H NMR (DMSO-d6): δ 6.55 (s, 1H), 6.97 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 7.1 Hz, 1H), 7.32 (d, J = 7.9 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.90–8.22 (m, 2H), 11.24 (broad s, disappeared on treatment with D2O, 1H), 13.05 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 2851, 2921, 3111, 3400 cm−1.
3-((3,4,5-Trimethoxyphenyl)thio)-1H-indole-2-carboxylic Acid (107)
107 was synthesized similarly to 14, starting from 105. Yield 45%, mp 240–245 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.33 (broad s, disappeared on treatment with D2O, 1H), 3.56 (s, 6H), 3.57 (s, 3H), 6.42 (s, 2H), 7.06–7.10 (m, 1H), 7.26–7.30 (m, 1H), 7.40–7.42 (m, 1H), 7.47–7.49 (m, 1H), 12.18 (broad s, disappeared on treatment with D2O, 1H), 13.10 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1738, 2574, 3329 cm−1.
2-Iodo-3-((3,4,5-trimethoxyphenyl)thio)-1H-indole (108)
108 was synthesized similarly to 14, starting from 106. Yield 93%, mp 135–140 °C (from ethanol). 1H NMR (CDCl3): δ 3.71 (s, 6H), 3.79 (s, 6H), 6.68 (s, 2H), 7.13–7.22 (m, 2H), 7.37–7.39 (m, 1H), 7.61–7.63 (m, 1H), 8.65 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3331 cm−1.
2-(1H-Indol-2-yl)oxazole (114)
114 was synthesized similarly to 31, using 106 and 2-(tributylstannyl)oxazole.50 Yield 26%, mp 33–35°C (from toluene). 1H NMR (CDCl3): δ 7.16–7.20 (m, 2H), 7.27–7.32 (m, 2H), 7.40–7.43 (m, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.77 (s, 1H), 8.65 (broad s, disappeared on treatment with D2O, 1H), 10.05 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3271 cm−1.
General Procedure for the Preparation of Compounds 116–122. Example: 2-Bromo-1-(2-fluorophenyl)ethanone (116)
A solution of bromine (3.47 g, 1.1 mL, 0.0217 mol) in anhydrous dichloromethane (7 mL) was dropped into a solution of 1-(2-fluorophenyl)-ethanone (3.0 g, 2.6 mL, 0.002 17 mol) in the same solvent (14 mL). The reaction mixture was stirred at 25 °C for 2 h, then diluted with a saturated aqueous solution of sodium hydrogen carbonate and extracted with dichloromethane. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, dichloromethane/petroleum ether = 7:3 as eluent) to furnish 116 as a slurry (0.9 g, 20%).51
2-Bromo-1-(3-fluorophenyl)ethanone (117)
117 was synthesized similarly to 116, starting from 1-(3-fluorophenyl)ethanone. Yield 21% as a slurry. 1H NMR (DMSO-d6): δ 4.97 (s, 2H), 7.53–7.65 (m, 2H), 7.79–7.81 (m, 1H), 7.85–7.87 ppm (m, 1H). IR: ν 1686 cm−1.
2-Bromo-1-(4-fluorophenyl)ethanone (118)
118 was synthesized similarly to 116, starting from 1-(4-fluorophenyl)ethanone. Yield 73%, mp 47–49 °C (from n-hexane). 1H NMR (DMSO-d6): δ 4.93 (s, 2H), 7.37–7.49 (m, 2H), 8.03–8.12 ppm (m, 2H). IR: ν 1694 cm−1. Lit.52 mp 48–49 °C.
2-Bromo-1-(3-isopropoxy-4-methoxyphenyl)ethanone (121)
A solution of 1-(3-hydroxy-4-methoxyphenyl)ethanone (0.25 g, 0.0015 mol) in anhydrous DMF (5 mL) was added to a mixture of 2-iodopropane (0.28 g, 0.2 mL, 0.001 65 mol) and potassium carbonate (0.415 g, 0.003 mol) in the same solvent (5 mL). The mixture was heated at 50 °C for 3 h. After cooling, the mixture was neutralized with 1 M HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 3:7 as eluent) to furnish 1-(3-isopropoxy-4-methoxyphenyl)-ethanone (0.27 g, 86%), mp 53–56 °C (from toluene/n-hexane). Lit53 mp 56 °C. As described above for 116, 1-(3-isopropoxy-4-methoxyphenyl)ethanone was converted to 121, yield 65%, mp 75–80°C (from ethanol).
Bromo-1-(naphthalen-1-yl)ethanone (122)
The compound was synthesized similarly to 116, starting from 1-(naphthalen-1-yl)-ethanone. Yield 90%, mp 175–177 °C (petroleum ether). Lit.54 mp 177–179 °C.
General Procedure for the Preparation of Compounds 123–127. Example N-(o-Tolyl)-4-methylbenzamide (123)
To an ice-cooled solution of o-toluidine (5.0 g, 5 mL, 0.0047 mol) and triethyl-amine in anhydrous THF (160 mL) was added dropwise p-tolyl chloride (8.66 g, 7.4 mL, 0.056 mol). The mixture was stirred at 25 °C for 12 h. The mixture was diluted with a saturated solution of sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was triturated with diethyl ether to furnish 123 (8.05 g, 76%), mp 118–120 °C (from ethanol). Lit.55 mp 121 °C.
N-(o-Tolyl)(1,1′-biphenyl)-4-carboxamide (124)
124 was synthesized similarly to 123, starting from (1,1′-biphenyl)-4-carbonyl chloride. Yield 40%, mp 176–178 °C (from ethanol). 1H NMR (CDCl3): δ 2.39 (s, 3H), 7.14–7.18 (m, 1H), 7.27–7.33 (m, 2H), 7.42–7.44 (m, 1H), 7.45–7.54 (m, 2H), 7.65–7.68 (m, 2H), 7.65–7.77 (m, 3H), 7.98–8.02 (m, 3H). IR: ν 1637–3280 cm−1. Lit.56 mp 179.5–180 °C.
N-(o-Tolyl)cyclobutanecarboxamide (125)
125 was synthesized similarly to 123, starting from cyclobutanecarbonyl chloride. Yield 90%, mp 106–110 °C (from toluene). 1H NMR (CDCl3): δ 1.91–2.07 (m, 2H), 2.23 (s, 3H), 2.26–2.44 (m, 4H), 3.16–3.24 (m, 1H), 6.86 (broad s, disappeared on treatment with D2O, 1H), 7.03–7.07 (m, 1H), 7.16–7.22 (m, 2H), 7.86 ppm (d, J = 7.8 Hz, 1H). IR: ν 1646, 3256 cm−1.
N-(o-Tolyl)cyclopentanecarboxamide (126)
126 was synthesized similarly to 123, starting from cyclopentanecarbonyl chloride. Yield 86%, mp 125–129 °C (from toluene). 1H NMR (DMSO-d6): δ 1.52–1.88 (m, 8H), 2.18 (s, 3H), 2.81–2.88 (m, 1H), 7.05–7.08 (m, 1H), 7.13–7.16 (m, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 9.19 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1648, 3268 cm−1.
N-(o-Tolyl)cyclohexanecarboxamide (127)
127 was synthesized similarly to 123, starting from cyclohexanecarbonyl chloride. Yield 85%, mp 150–155 °C (from toluene). 1H NMR (DMSO-d6): δ 1.18–1.47 (m, 5H), 1.64–1.84 (m, 5H), 2.17 (s, 3H), 2.37–2.42 (m, 1H), 7.05–7.08 (m 1H), 7.12–7.16 (m, 1H), 7.19 (d, J = 7.4 Hz, 1H), 7.32 (d, J = 7.1 Hz, 1H), 9.13 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1649, 3284 cm−1.
General Procedure for the Preparation of Compounds 128 and 129. Example: 1-(1-(4-Chlorophenyl)ethylidene)-2-phenyl-hydrazine (128)
A mixture of phenylhydrazine hydrochloride (2.2 g, 0.015 mol), 4′-chloroacetophenone (1.054 g, 1.3 mL, 0.01 mol), and sodium acetate (1.23 g, 0.015 mol) in ethanol (25 mL) was placed into the microwave cavity (open vessel mode). Microwave irradiation of 250 W was used, the temperature being ramped from 25 to 80 °C. Once 80 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 5 min with stirring. The reaction mixture was cooled to 0 °C, filtered, washed with petroleum ether, and dried to give 128 (3.38 g, 92%), mp 114–116 °C (from ethanol). 1H NMR (CDCl3): δ 2.23 (s, 3H), 6.91 (broad s, disappeared on treatment with D2O, 1H), 7.17–7.19 (m, 2H), 7.28–7.37 (m, 5H), 7.28–7.75 ppm (m, 2H). IR: ν 3349 cm−1. Lit.57 mp 112–113 °C.
1-(1-(4-Methoxyphenyl)ethylidene)-2-phenylhydrazine (129)
129 was synthesized similarly to 128, starting from 4′-methoxyacetophenone (166). Yield 95%, mp 102–104 °C (from ethanol). 1H NMR (CDCl3): δ 2.23 (s, 3H), 3.85 (s, 3H), 6.87 (broad s, disappeared on treatment with D2O, 1H), 6.90–6.94 (m, 2H), 7.17–7.19 (m, 2H), 7.28–7.31 (m, 2H), 7.75–7.77 ppm (m, 2H). IR: ν 3336 cm−1. Lit.58 mp 97–98 °C.
Biology. Tubulin Assembly
The reaction mixtures contained 0.8 M monosodium glutamate (pH 6.6 with HCl in a 2 M stock solution), 10 μM tubulin, and varying concentrations of drug. Following a 15 min preincubation at 30 °C, samples were chilled on ice, GTP to 0.4 mM was added, and turbidity development was followed at 350 nm in a temperature controlled recording spectrophotometer for 20 min at 30 °C. Extent of reaction was measured. Full experimental details were previously reported.59
[3H]Colchicine Binding Assay
The reaction mixtures contained 1.0 μM tubulin, 5.0 μM [3H]colchicine, and 5.0 μM inhibitor and were incubated 10 min at 37 °C. Complete details were described previously.60
Cell Cultures and Treatment
Cell lines were obtained from the American Tissue Culture Collection (ATCC), unless specified otherwise. Cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. In all experiments 300 000 cells were plated in 9 cm2 dishes and exposed to test compound dissolved in DMSO (0.1% final concentration) at the indicated concentrations.
HeLa, HT29, and A549 cell lines were grown at 37 °C in DMEM containing 10 mM glucose supplemented with 10% FBS, 100 units/mL each of penicillin and streptomycin, and 2 mM glutamine. At the onset of each experiment, cells were placed in fresh medium and cultured in the presence of test compounds from 0.01 to 25 μM.
231-MDA and A549 cells were cultured in DMEM supplemented with 10% FBS for 24, 48, and 72 h in a 96-well tissue culture plate at 37 °C and 5% CO2 in the absence or presence of different drug concentrations.
HCT 116, HCT 15, Messa, and Messa/Dx5 were seeded into 96-well plates (Corning Inc., Costar) at a density of 2 × 103 cells/well in a volume of 50 μL of the appropriate tissue culture medium. Compounds were added with different concentrations for the indicated incubation time at 37 °C with 5% CO2.
The human glioblastoma multiforme (GBM) cell line U87MG was purchased from the National Institute for Cancer Research (ICLC) of Genoa (Italy). The U87MG cells (1 × 106) were seeded onto T-75 flasks and cultured in RPMI medium supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin, and 1% nonessential amino acids at 37 °C in humidified atmosphere composed of oxygen (95%) and carbon dioxide (5%). For U87MG cell treatments, the cells were seeded at appropriate densities (5000 cells/well in 96-well plates or 50 000 cells/well in 24-well plates) in complete medium. After 12 or 24 h the medium was replaced either with complete culture medium containing DMSO (untreated control cells) or with complete medium supplemented with the compound 18 or 57 dissolved in DMSO (<1% v/v of medium).
Cell Viability Assay
The methodology for the evaluation of the growth of human MCF-7 breast carcinoma, OVCAR-8, and NCI/ADR-RES cells, obtained from the National Cancer Institute drug screening laboratory, was previously described except that cells were grown for 96 h for IC50 determinations.61
Cell viability in Hela, HT-29, and A549 cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. The test is based on the ability of mitochondrial dehydrogenase to convert, in viable cells, the yellow MTT reagent into a soluble blue formazan dye. Cells were seeded into 96-well plates to a density of 7 × 103/100 μL well. After 24 h of growth to allow attachment of cells to the wells, compounds were added at various concentrations (from 0.01 to 25 μM). After 48 h of growth and after removal of the culture medium, 100 μL/well medium containing 1 mg/mL MTT was added. Cell cultures were further incubated at 37 °C for 2 h in the dark. The solution was then gently aspirated from each well, and the formazan crystals within the cells were dissolved with 100 μL of DMSO. Optical densities were read at 550 nm using a Multiskan Spectrum Thermo Electron Corporation reader. Results were expressed as a percentage relative to vehicle-treated control (0.5% DMSO was added to untreated cells), and the IC50 values were determined by linear and polynomial regression. Experiments were performed in triplicate.
231-MDA and A549 cells were plated at different cellular densities in order to test the compounds on logarithmic phase cells. Test compounds were added after cell adhesion. On the day of the assay, 10 μL of MTT (5 mg/mL) was added to each well, and cells were incubated for 2 h (37 °C, 5% CO2). When dark crystals appeared at the well bottom, culture medium was discarded, and ethanol (100 μL) was added to each well to solubilize the crystals, yielding a purple solution. Absorbance was read with an enzyme-linked immunosorbent assay (ELISA) reader at 570 nm.
The antiproliferative effect of the tubulin polymerization inhibitors on cell proliferation was evaluated against HCT 116, HCT 15, Messa, and Messa/Dx5 tumor cell lines using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) according to the manufacturer’s instructions. The cells, in exponential growth, were incubated for 72 h at different concentrations of the inhibitors. Then an equivalent of the CellTiter-Glo reagent was added, the solution was mixed for 2 min in order to induce cell lysis, and the luminescence was recorded after a further 10 min. GI50 values were calculated using the parameters of a nonlinear regression analysis program (GraphPad Prism statistics software).
The effects of the treatments with 18 or 57 on U87MG cell survival/growth were estimated by a colorimetric MTS conversion assay32 and trypan blue exclusion assay.59 The MTS assay was used to determine IC50 values on U87MG cell survival following 24 h of treatment. U87MG cells were seeded in 96-well plates and incubated with compound concentrations ranging from 0.1 nM to 20 μM (DMSO as control). After 24 h, MTS reagent was added, and the absorbance at 590 nm was measured by a microplate reader (Wallac, Victor 2, 1420 multilabel counter, PerkinElmer). The percentage of proliferating cells after compound exposure was calculated with respect to control cells (100%). Sigmoid dose response curves were performed using GraphPad Prism 4 software (GraphPad Software Inc., San Diego, CA).
The trypan blue exclusion assay was used to examine the effects of compound treatments on U87MG cell survival. U87MG cells were seeded in 24-well plates and exposed to 1 nM to 10 μM compound concentrations for 8, 24, and 48 h. After exposure, the number of viable (white) and dead (blue) cells was counted using a microscope, and the percentage of viable or dead cells was calculated with respect to control cells (100%).
Immunofluorescence (IF) and Microscopy
HeLa epithelial cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS at 37 °C with 5% CO2. All compounds were dissolved in DMSO (0.1% final concentration) and used at the indicated concentrations for 24 h.
HeLa cells were seeded at 3 × 105 cells in culture dishes containing sterile coverslips coated with poly-L-lysine. At the end of the treatment, they were directly fixed in methanol for 10 min at –20 °C and processed for IF with either mouse anti-α-tubulin antibody (Sigma clone B5.1.2, 1:2000 dilution) followed by FITC-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch Laboratories, 115-095-068, 1:200) or with mouse anti-cyclin B1 antibody (Santa Cruz, GNS1, 1:50) followed by Texas Red conjugated horse anti-mouse (Vector, TI-2000, 1:800) secondary antibody. Slides were counterstained with 0.05 μg/mL DAPI (Sigma) and mounted in Vectashield (Vector). Cells were analyzed under an epifluorescence Olympus AX70 microscope with a CCD camera (Diagnostic Instrument, Spot RT slider model 2.3.0). Unfixed cell cultures were observed under an inverted NIKON TE 300 microscope with a 10× objective, and images were acquired using the ACT-1 software and a DMX1200 CCD (resolution 1280 × 1024 pixels).
Flow Cytometric Analysis
Cell cycle distribution was analyzed after incubation with PI (Sigma-Aldrich), whereas apoptosis was analyzed with annexin V-FITC (Immunological Sciences, IK-11120) staining alone or with annexin V-FITC in combination with PI. Cell samples were analyzed in a Coulter Epics XL cytofluorometer (Beckman Coulter) equipped with EXPO 32 ADC software. Data from at least 10 000 cells per sample were acquired.
Evaluation of Mitochondrial Potential
JC-1 (Invitrogen, T3168, Carlsbad, CA) is a cationic dye that exhibits potential-dependent accumulation in mitochondria indicated by a fluorescence emission shift from green (~525 nm) to red (~590 nm). Consequently, mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio and can be quantified by using fluorescence microscopy.33b To evaluate the mitochondrial depolarization induced by drug treatment, U87MG cells were seeded in 96-well plates and stained for 10 min in medium containing JC-1 at a final concentration of 50 μg/mL. After removal of JC-1, fresh medium containing the drug at 100 nM (approximately its IC50 value in U87MG cells) or 30 μM CCCP, a standard mitochondrial potential dissipation drug, was added to the cells. Immediately and 24 h after the treatment, pictures were taken under an Axiovert fluorescent microscope (Zeiss) using the filter set 10, 488010-0000 (Zeiss) (excitation 450–490 nm, emission 515–565 nm). Pictures were then split in the RGB channels (red and green) and analyzed by using the program ImageJ.62 For each treatment, the red/green fluorescence ratio obtained at 24 h was normalized with respect to red/green fluorescence ratio obtained at 0 h.
ROS Production
The generation of ROS was assessed by the fluorogenic probe DCFH2-DA (Molecular Probes, Invitrogen) in U87MG cells after compound exposure (DMSO, H2O2 as control standard). DCFH2-DA is a reduced and acetylated form of fluorescein used as an indicator for ROS in cells.63 This nonfluorescent molecule is readily converted to a green-fluorescent form (FDA) when the acetate group is removed by intracellular esterases and an oxidation by ROS occurs within the cell.
U87MG cells were seeded in 96-wells plate, washed in PBS/10 mM glucose, and loaded with 10 μM DCFH2-DA for 30 min in the dark at 37 °C. Afterward, the cells were washed with the same medium and incubated with or without the compound at 100 nM. Fluorescence increase was estimated in a plate reader at 485 nm (excitation) and 520 nm (emission) after 1, 5, 15, and 60 min (Wallac, Victor 2, 1420 multilabel counter, PerkinElmer). The fluorescence values were normalized between samples for cell number content and assessed by a crystal violet cell staining assay. Crystal violet is an intense stain that binds to cell nuclei and gives an A595 reading that is proportional to cell number. After washing with PBS/10 mM glucose, the cells were incubated with crystal violet for 30 min at room temperature. After washing, a solution of 1% SDS was added to each well, and the plates were shaken mechanically for 1 h at room temperature. A595 was then determined on a plate reader. The measured optical density readings indicated the relative number of cells in each well, and this was used to normalize the absorbance obtained with FDA.
Statistical Analyses
For data analysis and graphic presentations, the nonlinear multipurpose curve-fitting program GraphPad Prism (GraphPad) was used. All data are presented as the mean ± SEM. Statistical analysis was performed by one-way analysis of variance (ANOVA) with Bonferroni’s corrected t test for posthoc pairwise comparisons. P < 0.05 was considered statistically significant.
In Vivo Vascular Disrupting Effects
This study was approved by the institutional ethical committee for the use and care of laboratory animals. Six adult WAG/Rij rats (Iffa Credo, Brussels, Belgium) weighing 250 g received intrahepatic implantation of bifocal rhabdomyosarcoma allografts, which mimics hypervascular human liver metastases as previously described.34 Three rats bearing six liver tumors in total were thus prepared for studying 18 and 57 separately. The growth of tumors was monitored with MRI until their diameter reached about 10 mm for therapy. MRI was carried out with a clinical 3.0 T whole-body MR magnet (Trio; Siemens, Erlangen, Germany) with a maximum gradient capability of 45 mT/m by using an eight-channel phased-array wrist coil (In Vivo, Latham, NY). The rat was gas-anesthetized with 2% isoflurane in a mixture of 20% oxygen and 80% room air, through a mask connected by a tube to a Harvard Apparatus system (Holliston, MA) and placed supine in a plastic holder. The penile vein of the rat was cannulated for MRI contrast agent or VDA compound administration. With intravenous bolus of gadoterate meglumine (Dotarem; Guerbet, Brussels, Belgium) at 0.1 mmol/kg, contrast enhanced T1 weighted MRI (CE-T1WI)35 was performed at baseline at 1 and 24 h post-VDA treatment to document intratumoral vascular disrupting event. After baseline CE-T1WI, a VDA solution of 18 or 57 was intravenously injected at a dose of 15 mg/kg for subsequent evaluation of the therapeutic effects.
LC–MS/MS Analytical Method
Samples from the metabolic stability, solubility, and permeability assays were analyzed under the following conditions: UPLC instrument (Waters) interfaced with a Premiere XE triple quadrupole instrument (Waters). Eluents were the following: phase A consisting of 95% H2O, 5% acetonitrile + 0.1% HCOOH; phase B consisting of 5% H2O, 95% acetonitrile + 0.1% HCOOH. Parameters were as follows: flow rate, 0.6 mL/min; column, Acquity BEH C18, 50 mm × 2.1 mm, 1.7 μm at 50 °C; injection volume, 5 μL. LC–MS/MS analyses were carried out using an ESI(+) interface in multiple reaction monitoring mode.
Metabolic Stability
Compounds 18, 20, 55, and 57 were dissolved in DMSO in duplicate at a final concentration of 1 μM and preincubated for 10 min at 37 °C in potassium phosphate buffer, pH 7.4, 3 mM MgCl2, with rat liver microsomes (Xenotech) at a final concentration of 0.5 mg/mL. After the preincubation period, reactions were started by adding the cofactors mixture (NADP, Glc6P, G6P-DH). Samples were taken at times 0 and 30 min and added to acetonitrile to stop the reaction and centrifuged. Supernatants were analyzed and quantified by LC–MS/MS. A control sample without cofactors was always added in order to check the stability of test compounds in the reaction mixtures. 7-Ethoxycoumarin was used as a reference standard. A fixed concentration of verapamil was added in every sample as an internal standard for LC–MS/MS. The percent of test compound remaining after a 30 min incubation period was calculated from the peak area relative to the area of the compound after the 30 min incubation time.
Aqueous Solubility
The solubilities of compounds 18, 20, 55, and 57 were measured using a high throughput screening assay format. Samples prepared at the target concentration of 200 μM were placed in a 96-well filter plate and incubated at room temperature for 90 min. The plate was then filtered, and solutions were analyzed by LC/MS–UV. Final concentrations are evaluated by comparing the area under the curve of the MeOH stock solution with those of the test compound solutions.
Caco-2 Cell Permeability
Caco-2 cells (ECACC) were cultured in DMEM, 10% FBS, 1% NEAA, 10 mM Hepes buffer, 50 U penicillin, and 50 ug/mL streptomycin and split at confluence by trypsinization. For transport studies, 200 000 cells/well were seeded on Millicell 24-well cell culture plates. After 24 h at 37 °C in a humidified, 5% CO2 atmosphere, the medium was exchanged for enterocyte differentiation medium with additives (Becton Dickinson), which allows Caco-2 cells to establish within 3 days a differentiated enterocyte monolayer. TEER, measured with a Millicell-ERS (Millipore, Corp), must be >1000 Ω. Transport across the Caco-2 monolayer was determined by adding a 10 μM solution of compound in DMEM (1% final concentration of DMSO) to the side from which permeability was to be determined. The A→B transport across Caco-2 monolayer cells was determined by adding the compound to the apical side at pH 6.5. After 2 h, the basolateral side solution at pH 7.4 and the apical and starting solutions were analyzed by LC–MS/MS. In the B→A experiment, the compound was added to the basolateral side and collected on the apical side. Monolayer integrity was assessed with a lucifer yellow assay at the end of each experiment. The apparent permeability (Papp) was calculated with the following equation:
where J is the flux (dX/dt per A) and C0 is the donor concentration (μM) at t = 0; dX/dt is the change in mass (X, nmol) per time (t, s), and A is the filter surface area (cm2).
CYP450 Inhibition
Compounds 18, 20, 55, and 57 were evaluated in a 96-well plate containing an appropriate buffer and a NADPH regenerating system. Cytochrome P450 inhibition experiments were carried out according to the manufacturer’s instructions (BD Biosciences, Franklin Lakes, NJ, U.S.). The compounds were dissolved in a 96-well plate at a 10 μM final concentration in potassium phosphate buffer (pH 7.4) containing an NADPH regenerating system. For all enzyme/substrate pairs, the final cofactor concentrations were 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, and 0.4 U/mL glucose 6-phosphate dehydrogenase. The reaction was initiated by the addition of specific isoenzymes (Supersomes, Gentest) and substrates at 37 °C. Furafylline (for CYP1A2, 100 mM), sulfaphenazole (for CYP2C9, 10 mM), tranylcypromine (for CYP2C19, 500 μM), quinidine (for CYP2D6, 0.5 mM), and ketoconazole (for CYP3A4, 1.66 mM) were employed as control inhibitors in one-third serial dilution. Incubations were carried out for 15 min (0.5 pmol of CYP1A2, 5 mM 3-cyano-7-ethoxycoumarin), 30 min (0.5 pmol of CYP2C19, 25 mM 3-cyano-7-ethoxycoumarin, 1 pmol of CYP3A4, 50 mM 7-benzyloxy-4-(trifluoromethyl)coumarin; 1.5 pmol of CYP2D6, 1.5 mM 3-[2-(N,N-diethyl-N-methylamino)-ethyl]-7-methoxy-4-methylcoumarin), or 45 min (1 pmol of CYP2C9, 75 mM 7-methoxy-4-(trifluoromethyl)coumarin). The reaction was then quenched by adding 75 mL of a mixture containing 80% CH3CN and 20% Tris base (0.5 m), and plates were read on a fluorimeter at the appropriate emission/excitation wavelengths. The percentage inhibition was calculated relative to enzyme samples without inhibitors.
Supplementary Material
Acknowledgments
This research was supported by PRIN 2008 (Grant 200879X9N9), Progetti di Ricerca di Università, Sapienza Università di Roma (Grant 2011), and Bando Futuro in Ricerca 2010 (Grant RBFR10ZJQT). The authors also thank FILAS (Finanziaria Laziale di Sviluppo, Grant 2010) for partial support. A.C. thanks Istituto Pasteur—Fondazione Cenci Bolognetti for his Borsa di Studio per Ricerche in Italia. A.R. thanks Bando Futuro in Ricerca 2010 for her Borsa di Studio. We are grateful to Alessio Bolognesi, Maria Giubettini, and Annalisa Verrico for experimental work in cell cycle analysis. This work was supported in part by funds from AIRC (Grant IG 10164) to P.L.
ABBREVIATIONS USED
- MT
microtubule
- ATI
arylthioindole
- CSA4
combretastatin A-4
- VBL
vinblastine
- PTX
paclitaxel
- VRB
vinorelbine
- SAR
structure–activity relationship
- Pmax
maximum pressure
- BOP
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexa-fluorophosphate
- Pd2(dba)3
tris(dibenzylideneacetone)-dipalladium(0)
- P(Cy)
tricyclohexylphosphine
- Pd(dppf)-Cl2·CH2Cl2
1,1′-bis(diphenylphosphino)ferrocene]-dichloropalladium(II), complex with dichloromethane
- PdCl2(PPh3)2
bis(triphenylphosphine)palladium(II) dichloride
- Pd(OAc)2
palladium(II) acetate
- Pd(PPh3)4
tetrakis-(triphenylphosphine)palladium(0)
- SPhos
2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl
- MI
mitotic index
- DAPI
4′,6-diamidino-2-phenylindole
- Pgp
P-glycoprotein
- FBS
fetal bovine serum
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- DCFH2-DA
6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- ROS
reactive oxygen species
- JC-1
5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzimidazoylcarbocyanine iodide
- FDA
green-fluorescent form of 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- VDA
vascular disrupting agent
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Molecular modeling results, binding modes, inhibition data, cell death data, and elemental analysis results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Honore S, Pasquier E, Braguer D. Undestanding microtubule dynamics for improved cancer therapy. Cell Mol Life Sci. 2005;62:3039–3056. doi: 10.1007/s00018-005-5330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh HP, Liou JP, Mahindroo N. Pharmaceutical design of antimitotic agents based on combretastatins. Curr Pharm Des. 2005;11:1655–1677. doi: 10.2174/1381612053764751. [DOI] [PubMed] [Google Scholar]
- 4.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 5.Teicher BA. Newer cytotoxic agents: attacking cancer broadly. Clin Cancer Res. 2008;14:1610–1617. doi: 10.1158/1078-0432.CCR-07-2249. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- 7.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 8.Lin MC, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding to colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 9.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 10.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 11.Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat Chem Biol. 2007;3:117–125. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 12.Beckers T, Mahboobi S. Natural, semisynthetic and synthetic microtubule inhibitors for cancer therapy. Drugs Future. 2003;28:767–785. [Google Scholar]
- 13.Sridhare M, Macapinlac MJ, Goel S, Verdier-Pinard D, Fojo T, Rothenberg M, Colevas D. The clinical development of new mitotic inhibitors that stabilize the microtubule. Anti-Cancer Drugs. 2004;15:553–555. doi: 10.1097/01.cad.0000131681.21637.b2. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updates. 2007;10:162–181. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 15.De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. J Med Chem. 2004;47:6120–6123. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- 16.De Martino G, Edler MC, La Regina G, Coluccia A, Barbera MC, Barrow D, Nicholson RI, Chiosis G, Brancale A, Hamel E, Artico M, Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. 2. Structure activity relationships and molecular modeling studies. J Med Chem. 2006;49:947–954. doi: 10.1021/jm050809s. [DOI] [PubMed] [Google Scholar]
- 17.La Regina G, Edler MC, Brancale A, Kandil S, Coluccia A, Piscitelli F, Hamel E, De Martino G, Matesanz R, Díaz JF, Scovassi AI, Prosperi E, Lavecchia A, Novellino E, Artico M, Silvestri R. New arythioindoles inhibitors of tubulin polymerization. 3. Biological evaluation, SAR and molecular modeling studies. J Med Chem. 2007;50:2865–2874. doi: 10.1021/jm061479u. [DOI] [PubMed] [Google Scholar]
- 18.La Regina G, Sarkar T, Bai R, Edler MC, Saletti R, Coluccia A, Piscitelli F, Minelli L, Gatti V, Mazzoccoli C, Palermo V, Mazzoni C, Falcone C, Scovassi AI, Giansanti V, Campiglia P, Porta A, Maresca B, Hamel E, Brancale A, Novellino E, Silvestri R. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J Med Chem. 2009;52:7512–7527. doi: 10.1021/jm900016t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.La Regina G, Bai R, Rensen W, Coluccia A, Piscitelli F, Gatti V, Bolognesi A, Lavecchia A, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Mariani M, Santoni A, Brancale A, Ferlini C, Dondio G, Varasi M, Mercurio C, Hamel E, Lavia P, Novellino E, Silvestri R. Design and synthesis of 2-heterocyclyl-3-arylthio-1H-indoles as potent tubulin polymerization and cell growth inhibitors with improved metabolic stability. J Med Chem. 2011;54:8394–8406. doi: 10.1021/jm2012886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 21.Poirier M, Goudreau S, Poulin J, Savoie J, Beaulieu PL. Metal-free coupling of azoles with 2- and 3-haloindoles providing access to novel 2- or 3-(azol-1-yl)indole derivatives. Org Lett. 2010;12:2334–2337. doi: 10.1021/ol100685p. [DOI] [PubMed] [Google Scholar]
- 22.Gavina BD, Forte B, Mantegani S, Varasi M, Vianello P. PCT Int. Appl. WO 2005005414 A2. Preparation of Pyrazolyl-indole Derivatives Active As Kinase Inhibitors. 2005 Jan 20;
- 23.Bergman J, Venemalm L. Efficient synthesis of 2-chloro-, 2-bromo-, and 2-iodoindole. J Org Chem. 1992;57:2495–2497. [Google Scholar]
- 24.Hudkins RL, Diebold JL, Marsh FD. Synthesis of 2-aryl- and 2-vinyl-1H-indoles via palladium-catalyzed cross-coupling of aryl and vinyl halides with 1-carboxy-2-(tributylstannyl)indole. J Org Chem. 1995;60:6218–6220. [Google Scholar]
- 25.Hugon B, Pfeiffer B, Renard P, Prudhomme M. Synthesis of isogranulatimide analogues possessing a pyrrole moiety instead of an imidazole heterocycle. Tetrahedron Lett. 2003;44:3927–3930. [Google Scholar]
- 26.Kolhatkar RB, Ghorai SK, George C, Reith MEA, Dutta AK. Interaction of cis-(6-benzhydrylpiperidin-3-yl)benzylamine analogues with monoamine transporters: structure–activity relationship study of structurally constrained 3,6-disubstituted piperidine analogues of (2,2-diphenylethyl)-[1-(4-fluorobenzyl)piperidin-4-ylmethyl]amine. J Med Chem. 2003;46:2205–2215. doi: 10.1021/jm020561w. [DOI] [PubMed] [Google Scholar]
- 27.(a) Hoover DJ, Witter KG. Heteroaromatic Compounds as PDE10a Inhibitors and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Central Nervous System Diseases. 2008004117. PCT Int Appl WO. 2008 Jan 10;:A1.; (b) Koradin C, Dohle W, Rodriguez AL, Schmid B, Knochel P. Synthesis of polyfunctional indoles and related heterocycles mediated by cesium and potassium bases. Tetrahedron. 2003;59:1571–1587. [Google Scholar]
- 28.This compound was also purchased from Sigma-Aldrich.
- 29.La Regina G, Gatti V, Piscitelli F, Silvestri R. Open vessel and cooling while heating microwave-assisted synthesis of pyridinyl N-aryl hydrazones. ACS Comb Sci. 2011;13:2–6. doi: 10.1021/co100015b. [DOI] [PubMed] [Google Scholar]
- 30.(a) Vakifahmetoglu HM, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]; (b) Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]; (c) Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 31.Rieder C, Cole R. Microtubule disassembly delays the G2-M transition in vertebrates. Curr Biol. 2000;10:1067–1070. doi: 10.1016/s0960-9822(00)00678-3. [DOI] [PubMed] [Google Scholar]
- 32.(a) Mantymaa P, Siitonen T, Guttorm T, Saily M, Kinnula V, Savolainen ER, Koistinen P. Induction of mitochondrial manganese superoxide dismutase confers resistance to apoptosis in acute myeloblastic leukaemia cells exposed to etoposide. Br J Haematol. 2000;108:574–581. doi: 10.1046/j.1365-2141.2000.01852.x. [DOI] [PubMed] [Google Scholar]; (b) Chelli B, Lena A, Vanacore R, Da Pozzo E, Costa B, Rossi L, Salvetti A, Scatena F, Ceruti S, Abbracchio MP, Gremigni V, Martini C. Peripheral benzodiazepine receptor ligands: mitochondrial transmembrane potential depolarization and apoptosis induction in rat C6 glioma cells. Biochem Pharmacol. 2004;68:125–134. doi: 10.1016/j.bcp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Nohl H, Gille L, Staniek K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005; 69: 719–723. (b) Szilágyi, G., Simon, L., Koska, P., Telek, G., Nagy, Z. Visualization of mitochondrial membrane potential and reactive oxygen species via double staining. Neurosci Lett. 2006;399:206–209. doi: 10.1016/j.neulet.2006.01.071. [DOI] [PubMed] [Google Scholar]
- 34.Chen F, Sun X, De Keyzer F, Yu J, Peeters R, Coudyzer W, Vandecaveye V, Bosmans H, Van Hecke P, Landuyt W, Hermans R, Marchal G, Ni Y. Liver tumor model with implanted rhabdomyosarcoma in rats: MR imaging, microangiography, and histopathologic analysis. Radiology. 2006;239:554–562. doi: 10.1148/radiol.2392050277. [DOI] [PubMed] [Google Scholar]
- 35.Wang HJ, Miranda Cona M, Chen F, Li JJ, Yu J, Feng YB, Peeters R, De Keyzer F, Marchal G, Ni Y. Comparison between nonspecific and necrosis-avid Gadolinium contrast agents in therapeutic necrosis of rodent tumors at 3.0T. Invest Radiol. 2011;46:531–538. doi: 10.1097/RLI.0b013e31821a2116. [DOI] [PubMed] [Google Scholar]
- 36.Kerns EH, Di L. Drug-like Properties: Concepts, Structure Design and Methods. Academic Press; Burlington, MA: 2008. Plasma Stability Methods; pp. 329–347. [Google Scholar]
- 37.Ackermann L, Barfuesser S, Potukuchi HK. Copper-catalyzed N-arylation/hydroamin(d)ation domino synthesis of indoles and its application to the preparation of a Chek1/KDR kinase inhibitor pharmacophore. Adv Synth Catal. 2009;351:1064–1072. [Google Scholar]
- 38.Bellina F, Cauteruccio S, Rossi R. Palladium- and copper-mediated direct C-2 arylation of azoles—including free (NH)-imidazole, -benzimidazole and -indole—under base-free and ligand-less conditions. Eur J Org Chem. 2006;6:1379–1382. [Google Scholar]
- 39.Lipinska TM. Total synthesis of new indolo[2,3-a]quinolizine alkaloids sempervirine type, potential pharmaceuticals. Tetrahedron. 2006;62:5736–5747. [Google Scholar]
- 40.Sakai H, Tsutsumi K, Morimoto T, Kakiuchi K. One-pot/four-step/palladium-catalyzed synthesis of indole derivatives: the combination of heterogeneous and homogeneous systems. Adv Synth Catal. 2008;350:2498–2502. [Google Scholar]
- 41.Jin Z, Guo SX, Qiu LL, Wu GP, Fang JX. Well-defined NHC-Pd complex-mediated intermolecular direct annulations for synthesis of functionalized indoles (NHC = N-hetero-cyclic carbene) Appl Organomet Chem. 2011;25:502–507. [Google Scholar]
- 42.Nador F, Moglie Y, Vitale C, Yus M, Alonso F, Radivoy G. Reduction of polycyclic aromatic hydrocarbons promoted by cobalt or manganese nanoparticles. Tetrahedron. 2010;66:4318–4325. [Google Scholar]
- 43.Klioze SS, Ehrgott FJ, Jr, Glamkowski EJ. Synthesis of 7-substituted 1H-indolo[3, 2-d][1, 2]benzoxazepines. J Heterocycl Chem. 1984;21:1257–1259. [Google Scholar]
- 44.Shen M, Leslie BE, Driver TG. Dirhodium(II)-catalyzed intramolecular C–H amination of aryl azides. Angew Chem, Int Ed. 2008;47:5056–5059. doi: 10.1002/anie.200800689. [DOI] [PubMed] [Google Scholar]
- 45.So CM, Lau CP, Kwong FY. Easily accessible and highly tunable indolyl phosphine ligands for Suzuki–Miyaura coupling of aryl chlorides. Org Lett. 2007;9:2795–2798. doi: 10.1021/ol070898y. [DOI] [PubMed] [Google Scholar]
- 46.Kraus GA, Guo H. One-pot synthesis of 2-substituted indoles from 2-aminobenzyl phosphonium salts. A formal total synthesis of arcyriacyanin A. Org Lett. 2008;10:3061–3063. doi: 10.1021/ol801034x. [DOI] [PubMed] [Google Scholar]
- 47.Kissman HM, Farnsworth DW, Witkop B. Fischer indole syntheses with polyphosphoric acid. J Am Chem Soc. 1952;74:3948–3949. [Google Scholar]
- 48.De Cointet P, Pigerol C, Broll M, Eymard P, Werbenec JP. Synthesis and pharmacological properties of 3-acetoacetylindoles and their derivatives. Eur J Med Chem. 1976;11:471–479. [Google Scholar]
- 49.Adkins H, Coonradt HL. Selective hydrogenation of derivatives of pyrrole, indole, carbazole and acridine. J Am Chem Soc. 1941;63:1563–1570. [Google Scholar]
- 50.Slusasrchyk WA, Bolton SA, Herpin T, Bisacchi GS, Pi Z, Priestley ES. PCT. Int. Appl. WO 2004000788 A1. Preparation of Oxazolylureidoanilines as Inhibitors of Serine Proteases Such as Factor VIIa. 2003 Dec 31;
- 51.Kumar S, Gawandi VB, Capito N, Phillips RS. Substituent effects on the reaction of β-benzoylalanines with Pseudomonas fluorescens kynureninase. Biochemistry. 2010;49:7913–7919. doi: 10.1021/bi100955b. [DOI] [PubMed] [Google Scholar]
- 52.Lutz RE, Allison RK, Ashburn G, Bailey PS, Clark MT, Codington JF, Deinet AJ, Freek JA, Jordan RH, Leake NH, Martin TA, Nicodemus KC, Rowlett RJ, Jr, Shearer NH, Jr, Smith JD, Wilson JW., III Antimalarials. α-Phenyl-β-dialkylamino alcohols. J Org Chem. 1947;12:617–703. doi: 10.1021/jo01169a001. [DOI] [PubMed] [Google Scholar]
- 53.McCoubrey A, Iyengar NK. α-Methylbenzylamines. II. 3-Cyclohexyloxy-4-hydroxy-α-methylbenzylamine, its desoxy derivative, and related ethers. J Chem Soc. 1951:3430–3433. [Google Scholar]
- 54.Imran M, Dudhe R, Sharma PK, Khan SA. Synthesis and anticonvulsant activity of 3-chloro-4-substituted phenyl-1-[{4-(1-naphthyl)-1,3-thiazol-2-yl}amino]azetidin-2-ones. Int J Chem Sci. 2007;5:189–200. [Google Scholar]
- 55.(a) Grammaticakis P. Remarks about the preparation and ultraviolet absorption of some o-, m-, and p-methylbenzoylarylamines. Compt Rend. 1962;255:1456–1458. [Google Scholar]; (b) Ma Y, Song C, Chai Q, Ma C, Andrus MB. Palladium-imidazolium N-heterocyclic carbene-catalyzed carbonylative amidation with boronic acids, aryl diazonium ions, and ammonia. Synthesis. 2003;18:2886–2889. [Google Scholar]
- 56.Bachmann WE, Barton MX. The relative proportions of stereoisomeric oximes formed in the oximation of unsymmetrical ketones. J Org Chem. 1938;3:300–311. [Google Scholar]
- 57.Crowther AF, Mann FG, Purdie D. Mechanism of indole formation from phenacylarylamines. I. J Chem Soc. 1943:58–68. [Google Scholar]
- 58.Harej M, Dolenc D. Autoxidation of hydrazones. Some new insights. J Org Chem. 2007;72:7214–7221. doi: 10.1021/jo071091m. [DOI] [PubMed] [Google Scholar]
- 59.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 60.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;35:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 61.Ruan S, Okcu MF, Pong RC, Andreeff M, Levin V, Hsieh JT, Zhang W. Attenuation of WAF1/Cip1 expression by an antisense adenovirus expression vector sensitizes glioblastoma cells to apoptosis induced by chemotherapeutic agents 1,3-bis(2-chloroethyl)-1-nitro-sourea and cisplatin. Clin Cancer Res. 1999;5:197–202. [PubMed] [Google Scholar]
- 62.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with image. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 63.Barzegar A, Moosavi-Movahedi AA. Intracellular ROS protection efficiency and free radical-scavenging activity of curcumin. PLoS One. 2011;6:e26012. doi: 10.1371/journal.pone.0026012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.