

Abstract
Subventricular zone (SVZ) astrocytes and ependymal cells are both derived from radial glia and may have similar gliotic reactions after stroke. Diminishing SVZ neurogenesis worsens outcomes in mice, yet the effects of stroke on SVZ astrocytes and ependymal cells are poorly understood. We used mouse experimental stroke to determine if SVZ astrocytes and ependymal cells assume similar phenotypes and if stroke impacts their functions. Using lateral ventricular wall whole mount preparations, we show that stroke caused SVZ reactive astrocytosis, disrupting the neuroblast migratory scaffold. Also, SVZ vascular density and neural proliferation increased but apoptosis did not. In contrast to other reports, ependymal denudation and cell division was never observed. Remarkably, however, ependymal cells assumed features of reactive astrocytes post stroke, robustly expressing de novo glial fibrillary acidic protein, enlargening and extending long processes. Unexpectedly, stroke disrupted motile cilia planar cell polarity in ependymal cells. This suggested ciliary function was affected and indeed ventricular surface flow was slower and more turbulent post stroke. Together, these results demonstrate that in response to stroke there is significant SVZ reorganization with implications for both pathophysiology and therapeutic strategies.
Keywords: cerebrospinal fluid, ependymal cells, neurogenesis, planar cell polarity, reactive astrocytes, stroke, subventricular zone
Introduction
Stroke is a major public health challenge, which contributes significantly to the burden of disease in society. No pharmacological intervention has convincingly demonstrated efficacy to improve neurological outcome in stroke patients after brain tissue has infarcted. While conventional therapeutic strategies focus on preventing neuronal damage, stem cell treatment offers a new therapeutic avenue that aims at promoting neuronal repair. Studies demonstrating adult neurogenesis in rodents (Altman and Das 1966; Lois and Alvarez-Buylla 1993) have generated much excitement that endogenous neural stem cells (NSCs) may be utilized for repair (Szele and Chesselet 1996; Eriksson et al. 1998). However, rodent SVZ-mediated repair is minimal, while in nonhuman primates cell divisions in the subventricular zone (SVZ) generate few neurons and may be gliogenic after injury (Lewis 1968; McDermott and Lantos 1990; Kornack and Rakic 2001). NSCs reside in 2 distinct niches in the adult brain, the SVZ lining the lateral ventricles and the hippocampal subgranular zone, where they generate neurons throughout life in a variety of mammals but only neonatally in humans (Reynolds and Weiss 1992; Lois and Alvarez-Buylla 1993; Sanai et al. 2011).
Experimental stroke by middle cerebral artery occlusion (MCAO) in rodents increases SVZ neurogenesis and causes SVZ-derived neuronal precursor cells to migrate toward the ischemic penumbra, where they reduce infarct size and improve neurological function via as yet unknown mechanisms (Zhang et al. 2009; Jin et al. 2010). Some of these neuronal precursor cells can survive, differentiate, and display evidence of synaptic integration (Arvidsson et al. 2002; Parent et al. 2002). Importantly, human stroke also appears to increase SVZ neurogenesis, suggesting that we may be able to augment this endogenous repair mechanism in the clinical setting (Macas et al. 2006). Neuronal precursor recruitment toward ischemic brain tissue has been carefully investigated. For example, several chemokine signaling pathways that regulate neuroblast emigration have been elucidated, reviewed in (Young et al. 2010). Surprisingly, relatively little is known about early changes in the tightly regulated SVZ neurogenic compartment in response to stroke. Therefore, in this study, we looked for cellular and molecular phenotypic changes in 2 related and important cells of the system, astrocytes and ependymal cells.
In the adult SVZ, astrocyte-like NSCs that express glial fibrillary acidic protein (GFAP), generate new neurons (Doetsch et al. 1999; Garcia et al. 2004). However, recent studies suggest that ependymal cells may also contribute to neurogenesis after stroke (Carlen et al. 2009). Ependymal cells form a ciliated epithelial lining on the ventricular surface and have important physiological functions. Ependymal cells can be distinguished from SVZ cells based on their juxtaventricular position and expression of β-catenin in the cell membrane, S100β and vimentin in the cytoplasm, γ-tubulin in the ciliary basal bodies, and acetylated tubulin on motile cilia (Mirzadeh et al. 2008; Mirzadeh, Han, et al. 2010). Ependymal cells regulate the SVZ neurogenic niche (Lim et al. 2000), establish cerebrospinal fluid (CSF) flow over the ventricular surface (Sawamoto et al. 2006), and form a neuroprotective metabolic barrier at the brain CSF interface (Del Bigio 2010). We hypothesized that the drastic change in ependymal cell fate after stroke may be associated with significant cellular and molecular changes, and lead to ependymal dysfunction.
Recent work form the Gotz lab has shown that after brain injury parenchymal astrocytes become reactive and their neurogenic potential increases. Thus, we asked if the increased neurogenic potential of the SVZ after stroke (Arvidsson et al. 2002; Buffo et al. 2008) was associated with reactive astrocytosis. We had evidence that another reactive glial population, microglia, remained remarkably stable in the SVZ in a model of traumatic brain injury even though microglia in the immediately adjacent striatum and corpus callosum became activated (Goings et al. 2006). Using a combination of whole mount preparations of the lateral ventricular wall and coronal brain sections, we provide a detailed examination of SVZ cytoarchitecture and document how some of its cellular and molecular components reorganize in a mouse model of experimental stroke. We report reactive astrocytosis, increased vascularization, and a nonproliferative “reactive” ependymal cell phenotype with altered ciliary function in response to distant ischemic injury.
Materials and Methods
Animals
Male 129sv mice aged 8–12 weeks, weighing 23–28 g were obtained from University of Oxford Biomedical Services Specific Pathogens Free Breeding Unit (Oxford, UK). Animals were maintained in individually ventilated cages on 12-h light/dark cycles, with free access to food and water. Procedures were carried out with University of Oxford Research Ethics Committee approval, in accordance with the Animals (Scientific Procedures) Act of 1986 (UK). All efforts were made to minimize animal suffering and distress.
Experimental Stroke
MCAO was carried out as previously published (Barber et al. 2004). Briefly, under isoflurane anesthesia, a 200-μm silicon-coated nylon suture (Doccol, Redlands, CA) was introduced into the left external carotid artery and advanced up the internal carotid artery to block the origin of the MCA. During surgery, cerebral blood flow (CBF) in the MCA territory was measured by laser Doppler flowmeter (Oxford Optronix, Oxford, UK) and upon MCAO, CBF decreased to less than 30% of baseline values (Supplementary Fig. 1A). After 50 min, the filament was removed and the MCA reperfused. Animals were recovered in heated incubators for 12 h and body temperature kept at 35–36 °C. Fluid balance was maintained by intraperitoneal 5% glucose normal saline injections (0.4 mL daily for 7 days). The 7-day survival group received a single intraperitoneal injection of bromodeoxyuridine (BrdU; 100 mg/kg; Sigma–Aldrich, Gillingham, UK) 2 h before sacrifice. For cumulative labeling, the 14-day group received daily BrdU intraperitoneal injections from Days 4–8. Neurological deficits were assessed 24 h after MCAO in open field using a modified Neurological Deficit Score (Table 1) (Barber et al. 2004).
Table 1.
Modified neurological deficit score
| Score | Neurological deficit |
| 0 | Normal |
| 1 | Consistent forelimb and axial flexion toward the contralateral side when lifted by the tail. |
| 2 | Score 1 plus consistently reduced resistance to lateral push. |
| 3 | Score 2 plus gait toward or weak circling (>50 cm diameter) toward the paretic side. |
| 4 | Score 2 plus small circling (10–50 cm diameter) toward the paretic side. |
| 5 | Score 2 plus tight circling (<10 cm diameter) toward the paretic side. |
Measurement of Stroke Size
Twenty-four hours after MCAO, brain sections at multiple anterior posterior levels (Bregma +1.5 mm, 0 mm, −1.5 mm and −3.0 mm) were incubated in 2% triphenyl tetrazolium (Sigma–Aldrich) for 30 min at 36.5 °C to visualize the infarct area. In a separate group of animals, 7 days after MCAO, fluorescence immunohistochemistry for NeuN and GFAP was carried out on brain sections (Bregma +1.5 mm, 0 mm, −1.5 mm, and −3.0 mm) to similarly assess infarct size. To compensate for tissue swelling, the infarct area was calculated by the indirect method (infarct area = total contralateral hemisphere area − ipsilateral nonstroke area; Lin et al. 1993). The infarct volume was reconstructed according to the Cavalieri principle and expressed as a percentage of the contralateral hemisphere.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling
Sections were rinsed 2 times in Tris buffered saline (TBS), dehydrated and rehydrated in graded isopropanol, rinsed 2 times in TBS, and preincubated with terminal transferase (TdT) buffer (Roche) for 15 min at 37 °C. Positive control sections were treated with 2 mg/mL DNase (Sigma–Aldrich) for 15 min resulting in robust staining. Subsequently, sections were incubated in TdT mixture for 30 min at 37 °C and then rinsed 3 times in TBS.
Whole Mount Preparation and Immunohistochemistry
Following normal saline transcardiac perfusion, the brain was extracted and cut in the midline. The overlying cerebral cortex, medial ventricular wall, and hippocampus were dissected to reveal the lateral ventricular wall (Mirzadeh, Doetsch, et al. 2010). Whole mounts were postfixed in 4% paraformaldehyde with 0.1% Triton-X 100 at 4 °C overnight. Then, they were washed in phosphate buffered saline (PBS) with 0.1% Triton-X 100 (PBS+), blocked in 10% donkey serum in PBS+ at room temperature, and incubated at 4 °C for 48 h in primary antibodies in PBS containing 10% donkey serum and 2% Triton-X 100 to aid antibody penetration. After rinsing in PBS+, whole mounts were incubated for a further 48 h with secondary antibodies, before PBS wash, 4′,6-diamidino-2-phenylindole (Sigma–Aldrich) nuclear counterstain and final phosphate buffer wash. Whole mounts were trimmed to 200–300 μm sections and mounted on Superfrost slides with adhesive spacers and Flurosave mounting medium (Merck, Darmstadt, Germany). Brain fixation and processing for free-floating coronal section immunofluorescence to detect BrdU and other antigens was carried out as described (Kim et al. 2010). Antibodies used are listed in Table 2. Negative controls omitting the primary antibody were routinely performed.
Table 2.
Antibodies used in study
| Antigen | Dilution | Host | Manufacturer, catalog number |
| Acetyl tubulin | 1:1000 | Mouse | Sigma T6793 |
| β-catenin | 1:1000 | Rabbit | Sigma C2206 |
| BrdU | 1:200 | Mouse | DAKO M0744 |
| BrdU | 1:400 | Sheep | Abcam Ab1893 |
| Dcx | 1:100 | Goat | Santa Cruz SC 8066 |
| γ-tubulin | 1:200 | Goat | Santa Cruz SC 7396 |
| GFAP | 1:500 | Mouse | Millipore 557355 |
| GFAP | 1:200 | Rabbit | Sigma G9269 |
| GFAP | 1:400 | Rat mono | Invitrogen 13-0300 |
| Mash1 | 1:100 | Mouse | BD 556604 |
| NeuN | 1:400 | Mouse | Millipore MAB377 |
| PECAM | 1:200 | Rat | BD 557355 |
| PHi3 | 1:400 | Rabbit | Millipore 06-570 |
| S100b | 1:100 | Rabbit | Sigma 2644 |
| Vimentin | 1:200 | Goat | Santa Cruz SC 7558 |
We examined astrocytes, ependymal cells, neuroblasts, and vasculature with immunohistochemistry but no obvious differences were observed between whole mounts of MCAO contralateral hemispheres and sham-operated controls (n = 5, data not shown). Thereafter, contralateral hemispheres (post stroke) were used as controls, unless otherwise stated.
CSF Flow Study
For dynamic CSF flow studies, lateral ventricular walls were dissected and pinned to a coated Petri dish in 36.5 °C Leibovitz's media. Five nanoliters of 2-μm diameter fluorescent microsphere beads (Invitrogen, Paisley, UK) were released over the anterior dorsal ventricular wall with a stereotactic injector (Stoelting, IL). Bead movements were visualized using a Zeiss Stereo Lumar fluorescence microscope (Zeiss AxioCam camera) and imaged over 60 s at 3 frames/s. Three to five rounds of bead release and imaging were performed for each whole mount to obtain optimal flow. A maximum of 30 min elapsed between death and completion of imaging. Negative controls confirmed absent bead flow over the striatum and on ventricular surface following 5-min incubation in 70% ethanol.
Time-lapse images were analyzed using Volocity 4 software (PerkinElmer, Cambridge, UK). For each whole mount, 20 beads (10 beads each per dorsal and ventral streams) were manually tracked by x:y coordinates for at least 10 frames to calculate flow velocity. The paths of flowing beads were plotted on 2D Cartesian plane with Graphis software (Kylebank; www.kylebank.com). We used the Pythagorean equation to determine distance traveled between 2 frames:
d = distance traveled between frame 1 and 2, x2 = x coordinate in frame 2, x1 = x coordinate in frame 1, y2 = y coordinate in frame 2, y1 = y coordinate in frame 1.
Microscopy and Quantification
High-magnification optical sections of whole mounts and coronal sections were obtained with a Zeiss LSM 710 laser scanning confocal microscope using 20× and 40× oil immersion objectives. Whole mounts were examined globally and images were acquired from 4 specific regions of the lateral ventricular wall (Fig. 1B). The adhesion zone (AZ) was not included in the analysis. Experimental groups were coded, and images were acquired and quantified by blinded investigators. Vascular density was measured by counting the number of blood vessel branches in projected 40 × 10 μm confocal z stacks from 4 locations on each whole mount (Fig. 1B). Vessel diameter was measured using Volocity software. To measure average ependymal cell surface area, the area of each 40× confocal field was divided by the number of cells occupying each field. Planar cell polarity (PCP) of each ependymal cell was measured by drawing a vector between the center of the cell and basal body cluster. The vectors were expressed as an angle away from the median value measured and represented using a histogram (Mirzadeh, Han, et al. 2010). Quantification of PHi3+ and BrdU+ cells and colocalization studies were performed in three 40× orthogonal projections of 10 μm confocal z stacks (dorsal, middle, and ventral SVZ) in 5 evenly spaced sections from Bregma +1.5 to −0.5 mm. Terminal Deoxynucleotidyl Transferase dUTP nick End Labeling (TUNEL)+ cells in the entire SVZ were counted under 20× epifluorescence microscope in brain sections at Bregma +1, +0.2, and −0.5 mm (n = 4).
Figure 1.

Reactive SVZ astrocytosis after stroke. (A) Schematic of a mouse brain showing the lateral ventricular wall and SVZ (red). OB olfactory bulb, RMS rostral migratory stream. Adapted from (Doetsch et al. 1997). (B) Dissected whole mount; lateral ventricular wall outlined in red. Orientation as in A. Analyses were performed in 4 boxed areas AD (anterior dorsal), AV (anterior ventral), PD (posterior dorsal), and PV (posterior ventral). AZ is the intraventricular AZ where the striatum and septum are joined and the ventricle is nonpatent. D, dorsal; V, ventral; A, anterior; P, posterior. (C) White boxed area in B: β-catenin immunofluorescence to show the junction between normal ependymal cell layer and AZ devoid of ependymal cells. (D – K) whole mount GFAP immunofluorescence. Niche astrocytes in the SVZ had long processes aligned in the direction of neuroblast chain migration, in the posterior ventral (F) and anterior dorsal (H) SVZ. Seven days post stroke, reactive astrocytosis with hypertrophy and tortuous glial processes appeared across the SVZ (G,J). Some astrocytes in the SVZ were located near blood vessels (I dotted lines) and made contact with them. After stroke, reactive astrocytes near blood vessels (dotted lines) made multiple contacts (arrows K). (L – O) niche astrocytes formed glial tubes which ensheathed neuroblast chains (L,M). Reactive astrocytes had disrupted glial tube structure after stroke (N,O). Scale bar (D,E) 500 μm, (C,F-O) 50 μm.
Statistics
Data are presented as mean ± standard error of the mean. Differences between 2 groups were assessed using Student's t-test and amongst 3 or more groups with analysis of variance (ANOVA) with Bonferroni post hoc test. Groups were considered significantly different when at least a 95% confidence level (P < 0.05) was obtained. Analyses were performed in Microsoft Excel 2007 with XL Toolbox 2.80 (xltoolbox.sourceforge.net) and GraphPad Prism 5.
Results
MCAO Model of Stroke
MCAO caused reproducible cerebral ischemia, infarction, and neurological deficits. Upon successful MCAO, CBF in the left MCA territory decreased by >70% from baseline values, followed by reperfusion after filament removal (Supplementary Fig. 1A). Twenty-four hours following recovery from 50 min of MCAO, neurological assessment showed consistent contralateral motor hemiparesis and asymmetry of movement (Supplementary Fig. 1B). Consistency of the infarct volume produced by 50-min MCAO was assessed and confirmed in triphenyl tetrazolium-stained brain sections, 24 h after MCAO (data not shown). Seven days post-MCAO, loss of NeuN+ cell bodies and initiation of GFAP+ glial scar formation were independently used to assess infarct volume (Supplementary Fig. 1D,E). The infarct volume measured by NeuN (21.1 ± 1.5% of the hemisphere) and GFAP (25.1% ± 2.8%) did not differ significantly from each other (n = 6; Supplementary Fig. 1C – F). The ischemic lesion extended from the medial striatum to the lower cortical layers, consistent with published works on MCAO in the 129sv strain (Endres et al. 1998; Pham et al. 2010). We have shown that direct damage to the SVZ in models of traumatic brain injury dramatically alter the response of SVZ compared with lesions that spare it (Goings et al. 2006). Critically, in our stroke model, the SVZ was not included in the ischemic lesion, and it was separated from the border of the lesion by a rim of healthy appearing NeuN+ cells (Supplementary Fig. 1G). We never observed ischemic lesion in shams or contralateral hemispheres.
Reactive Astrocytosis in the SVZ after Stroke
Whole mounts of the lateral ventricular wall permitted unobstructed “en-face” views of the SVZ and overlying ependyma (Fig. 1A,B). At the ventricular surface, ependymal cells are clearly visualized by the distinct cobblestone cellular arrangement when immunodetected with anti–β-catenin antibodies, which label ependymal plasma membranes (Fig. 1C). The demarcated AZ (Fig. 1B) is a small area of the ventricular wall where the lateral ventricles are nonpatent and devoid of ependymal cells in the whole mount preparation (Figs. 1C and 4M) (Sawamoto et al. 2006).
Figure 4.

Preserved ependymal cells have increased surface area. (A–J) En-face views of whole mount imunofluorescence for ependymal cell membranes (β-catenin [β-cat] red), motile cilia (acetylated tubulin (ace tub), green). Cell nuclei counterstained with DAPI (blue). (A ,B) ependymal cells lining the lateral ventricles form a cobblestone cell layer. (C,E) tufts of motile cilia visualized on ependymal cell surface. (F,G) after stroke ependymal cell loss/denudation is not seen. (H, J) Motile cilia morphology is preserved. (K) Quantification of ependymal cell cross-sectional surface area in high-power confocal images (D, I). *P = 0.01. ANOVA. (L, M) Coronal section: s100β immunofluorescence to show integrity of ependymal cell layer. Scale bar (A–J) 20 μm (L, M) 100 μm.
Although the ischemic lesion and striatal astrocytosis did not reach the SVZ, there was significant reactive astrocytosis in the ventricular wall (n = 8; Fig. 1D–J). Reactive astrocytosis was particularly apparent in the intraventricular AZ because it is devoid of ependyma (arrow, Fig. 1E). GFAP+ astrocytes in the SVZ extend longitudinal processes (Fig. 1F,H) anteroposteriorly to form glial tubes that ensheath neuroblast chains. Seven days post stroke, many astrocytes were hypertrophic and glial processes became tortuous, disrupting the migratory scaffold (Fig. 1G,J). Some also expressed vimentin (Fig. 5P). In controls, protoplasmic astrocytes in the SVZ were frequently located adjacent to blood vessels, although they typically did not wrap processes around multiple vessels (Fig. 1I). Interestingly, numerous large protoplasmic astrocytes in the SVZ extended multiple processes to several blood vessels post stroke (arrows; Fig. 1K). Tangential orientation of the glial processes with respect to neuroblast chains was lost (Fig. 1L–O). However, neuroblasts maintained their overall chain configuration (Fig. 1L,N).
Figure 5.
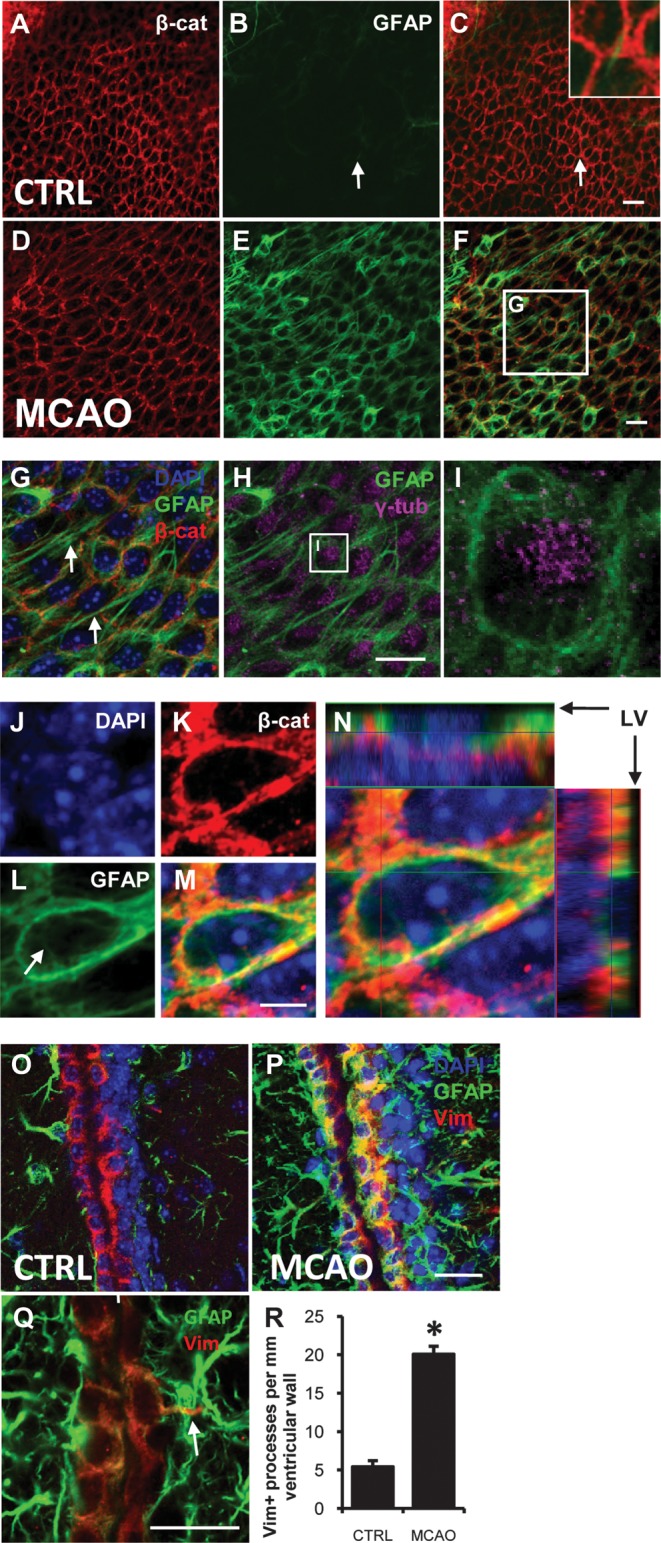
Reactive ependymal cells after stroke. (A–H) single confocal optical sections of whole mounts at the ventricular surface. Immunofluorescence for β-cat (red), GFAP (green), γ-tubulin (γ-tub; magenta) and DAPI counterstain (blue). (A–C) most resting ependymal cells did not express detectable GFAP. In contrast, GFAP + NSCs had apical processes which contact the ventricles (white arrow B ,C; inset). (D –F) after stroke, ependymal cells expressed high levels of GFAP. (G, H) Boxed area in (F). (G) GFP + ependymal cells extended long processes parallel to the ventricular surface (white arrows). (H) Motile cilia basal bodies immunolabeled with γ-tub in GFAP + ependymal cells. (I) Boxed area in (H): cytoplasmic GFAP in close association with ciliary basal bodies. (J–M) High-power single optical sections showing cytoplasmic (arrow L) and cell membrane-associated GFAP + intermediate filaments. (N) Orthogonal projection of confocal z-stack of ependymal cell at ventricular surface. LV lateral ventricle. (O–Q) coronal sections 7 days post stroke. (O, P) immunofluorescence for GFAP (green) and vimentin (Vim; red). (Q) Example of ependymal process extending into SVZ. Scale bars 20 μm except (J–M) is 5 μm. (R) Quantification of Vim + basal processes per mm of SVZ. *P < 0.00001.
Vascular Engorgement and Increased Vascular Density in the Post Stroke SVZ
Since SVZ blood vessels regulate neurogenesis and neuroblast migration (Shen et al. 2004; Tavazoie et al. 2008; Snapyan et al. 2009; Kokovay et al. 2010), we examined the vasculature in the SVZ after stroke. Post stroke vascular engorgement was visible in freshly dissected whole mounts (arrow; Fig. 2D). Whole mounts immunostained for platelet endothelial cell adhesion molecule 1 (PECAM) showed increased vascular density in the SVZ, 7 and 14 days post stroke (Fig. 2B–F,I; n = 4). The number of PECAM+ vessel branches increased at both time points (Fig. 2G; P = 0.03, ANOVA), while the vessel diameter was significantly larger at 7 days (Fig. 2H; P = 0.02, ANOVA).
Figure 2.

Stroke increases vascular density and blood vessel diameter. (A– C) PECAM immunofluorescence of control whole mount SVZ vasculature. (D) After stroke, engorged blood vessels become visible in the ventricular wall (arrow D). (E, F, I) Representative images of increased vascular density 7- and 14-days after stroke. (G) Number of blood vessel branches increased 7- and 14-days after stroke. (H) Increased blood vessel diameter 7-days after stroke. *P < 0.03. Scale bar (A, B, D, E) 500 μm, (C, F, I) 50 μm.
SVZ Proliferation Increased after Stroke but Ependymal Cells Did Not Divide
The number of SVZ cells expressing phosphorylated histone H3 (PHi3), which is specifically phosphorylated during mitosis, increased 7 days post stroke (n = 4; P = 0.02; Fig. 3A–C). To determine which cells increased proliferation, we colabeled for doublecortin (Dcx; neuroblasts) and Mash1 (transit amplifying progenitors; Fig. 3D,E; 499 cells examined). In controls, 44 ± 5.6% of proliferating cells were transit amplifying progenitors (PHi3+/Mash1+); 36 ± 2.0% were neuroblasts (PHi3+/Dcx+); and 8 ± 3.3% colocalized with both Mash1 and Dcx (PHi3+/Mash1+/Dcx+), which were likely young neuroblasts. After stroke, 47 ± 4.3% of proliferating cells were transit-amplifying progenitors; 31 ± 8.8% were neuroblasts; 7 ± 3.7% were young neuroblasts. Thus the percent of proliferating SVZ cell types did not change appreciably after stroke. Also, neural lineage cells (Dcx and/or Mash1-positive cells) constituted the majority of proliferating cells in controls (88 ± 4%) and this did not change significantly after stroke (85 ± 9%). PHi3+/Dcx-/Mash1-cells are probably predominantly microglia (Goings et al. 2006); and they accounted for only 12 ± 4.6% and 15 ± 9.1% of mitotic cells in controls and after stroke, respectively.
Figure 3.

Increased SVZ cell proliferation without ependymal proliferation after stroke. (A, B) Representative PHi3+ cells in the dorsolateral SVZ in control and 7-days after stoke. (C) Number of PHi3+ cells quantified in high-magnification confocal z-stacks was increased after stroke. *P = 0.02. (D) Triple immunofluorescence for PHi3, Dcx, and Mash1 to determine proliferating cell type ratios. (E) Percentage of PHi3+ cells in the SVZ which colocalize with Mash1 and/or Dcx. (F, I) BrdU+ mitotic cells around the lateral ventricle 7 days post stroke (F; single BrdU pulse labeling) and 14-days post-stroke (I; cumulative BrdU labeling). (G, J) Boxed areas in (F) and (I), respectively: BrdU+ cells were closely associated with, but distinct from, s100β+ ependymal cells. DAPI channel left gray for better image contrast. (H, K) Arrowed cell (G) and (J) respectively: Orthogonal projections of high-powered confocal z-stack. BrdU+ cells did not co-localize with the ependymal marker S100β. Scale bars 50 μm.
Ependymal cells have been reported to proliferate after injury (Carlen et al. 2009). However, 7 and 14 days post stroke, we did not observe any proliferating ependymal cells. Seven days after stroke, we gave a pulse injection of BrdU, 2 h later many BrdU+ cells were found in the SVZ (Fig. 3F). Several BrdU+ cells were positioned in close proximity to the ependymal layer (arrow; Fig. 3G). S100β+ ependymal cells and BrdU+ proliferating cells were analyzed in orthogonal projections of confocal z-stacks, and failed to reveal S100β+/BrdU+ double-labeled cells (Fig. 3H; n = 5). If ependymal cells divide after stroke, they may do so only very slowly and therefore, do not label with a single pulse of BrdU. However, 5 days of cumulative BrdU-labeling in the 14-day group also failed to reveal any BrdU+/S100β+ double-labeled ependymal cells (n = 6; Fig. 3I–K). Using different markers for ependymal cells and proliferating cells, we also did not find any PHi-3+/Vim+ double-labeled cells in contact with the ventricle at either time point (data not shown).
Apoptosis can cause reactive astrocytosis, and vice versa, but the number of TUNEL+ cells in the control and 7-days post stroke SVZ remained similar (n = 4; P = 0.3; Supplementary Fig. 2A – C). The absolute numbers of apoptotic cells in the SVZ were very low after stroke (2.5 ± 0.9 cells per section in the SVZ) and similar to the number in controls (Supplementary Fig. 2C). This contrasted with the ischemic striatum where large numbers of TUNEL+ cells were counted (135 ± 24 cells per 20× microscope field; Supplementary Fig. 2C – E).
Ependymal Cells Increased Surface Area but Remained Intact Post Stroke
Ependymal cells can be damaged or die due to neurodegeneration, increased age or stroke (Luo et al. 2008; Carlen et al. 2009). Seven and fourteen days after stroke, the lateral ventricular ependymal cell layer remained intact when visualized en-face on whole mounts with β-catenin immunofluorescence (Fig. 4D,I). Ependymal cells displayed a typical cobblestone appearance with tufts of dense acetylated tubulin + motile cilia (Mirzadeh et al. 2008) at the ventricular surface (Fig. 4A–E). Using a number of different labeling strategies to visualize ependymal cells and their cellular components, we did not observe ependymal denudation or tears at either time point post stroke. The preservation of the ependymal layer was evidenced by the retention of the cobblestone appearance (β-catenin; Figs. 4F,G, 5D–F, and 6B,F–I; n = 20), preservation of motile cilia (acetylated tubulin; Fig. 4H,J; n = 10), and γ-tubulin basal body clusters, which are unique to cells possessing multiple motile cilia (Figs. 5H,I and 6B; n = 10). In coronal sections, s100β-labeling showed a preserved ependymal cell layer around the lateral ventricles (Fig. 4L,M; n = 5). Furthermore, similar to the SVZ, increased apoptosis was not seen in the ependymal layer after stroke (Supplementary Fig. 2).
Figure 6.

Disrupted PCP and CSF flow after stroke. (A, B) whole mount immunofluorescence for β-cat (red) and γ-tub (white) shows ependymal PCP. (A) γ-tub+ cilia basal bodies were systematically localized to one side of the cells (green arrows), downstream of CSF flow direction. (B) After stroke, in the anterior dorsal lateral ventricular wall, ciliary basal bodies were randomly positioned within ependymal cells (yellow arrows). (C) Histogram of PCP orientation vector in controls and 7-days after stroke. P < 0.0001; Two-sample Kolmogorov–Smirnof test. (D, E) Dynamic CSF flow study. Fluorescent beads were deposited on lateral ventricular walls and time-lapse imaged. (D) Last frame of Supplementary Movie 1, section 1 showing CSF flow in control. (E) Last frame of Supplementary Movie 1, section 2 showing CSF flow 7-days post-stroke. Inset. Stroke increased flow turbulence over the anterior dorsal SVZ. Magnified view of the anterior dorsal lateral wall (arrow E, Supplementary Movie 1, section 3). (D′, E′) frames from time-lapse imaging of fluorescent bead movement over the anterior lateral ventricular wall. Please see Supplementary Movie 1. (F –I) Post hoc whole mount immunohistochemistry over area of flow turbulence in E. Some fluorescent beads adhered to ventricular surface (arrow K). Scale bar (B, F, I) 50 μm, (E) 500 μm (J, K) Cartesian plots of bead movement over the anterior ventricular wall. Each trace represents one bead followed over 10 time-lapse frames. (L) Speed of fluorescent bead movement as a read-out for CSF flow and motile ciliary function. *P = 0.02.
In our attempt to examine the ependyma for evidence of post stroke damage, we noticed enlarged en-face ependymal cell surface areas, while individual cilia tufts seemed more dispersed (Fig. 4I,J). Seven days post stroke, ependymal cells ipsilateral to stroke had increased cross-sectional surface area (146 ± 10 μm2) compared with contralateral controls (111 ± 6 μm2; P = 0.02; n = 6; 2630 cells measured; Fig. 4K). The effect persisted and ependymal cells remained enlarged at 14 days post stroke (170 ± 9 μm2; P = 0.01; n = 3; 1149 cells).
Epedymal Cells Expressed Glial Fibrillary Acidic Protein after Stroke
Very few ependymal cells (<5%) contralateral to MCAO and in shams expressed GFAP (Fig. 5A–C; n = 8). Unexpectedly, 7- and 14-days after stroke >85% of ependymal cells expressed high levels of GFAP ipsilateral to the lesion (Fig. 5D–F; n = 8). This de novo GFAP expression was apparent in high-powered single confocal optical sections through the ependymal layer (Fig. 5J–M) and was found in the cytoplasm of ependymal cells (arrow, Fig. 5L). Confocal z stack analysis demonstrated that GFAP filaments were aligned within ependymal cells at the ventricular surface (Fig. 5N) and very few were found between them. We considered the possibility that these apical GFAP+ cells may have been SVZ astrocytes that relocated to the ependymal layer and taken on ependymal morphology after stroke. However, GFAP filaments were found in the cytoplasm of cells with tufts of motile cilia and dense γ-tubulin+ basal bodies consistent with mature ependymal cell phenotype (Fig. 5H,I). GFAP immunohistochemistry was validated using 3 primary anti-GFAP antibodies and 3 fluorophore-conjugated secondary antibodies (Table 2). Negative controls omitting the primary antibody did not fluoresce.
To confirm ependymal GFAP expression, we examined coronal sections and used vimentin as a marker of ependymal cells. Seven days post stroke, vimentin+ ependymal cells expressed GFAP (Fig. 5O,P) and some extended basal processes into the SVZ (Fig. 5Q) (Zhang et al. 2007). The number of vimentin+ ependymal processes, which extend into the SVZ, increased post stroke (20.1 ± 1.8) compared with controls (5.6 ± 1.0; P < 0.0001; Fig. 5R).
Stroke Disrupted Ependymal Planar Cell Polarity and Changed Flow over Ventricular Wall
PCP in ependymal cells is distinguished by the cellular position of motile cilia, their orientations and beat direction, and hence the direction of CSF flow (Mirzadeh, Han, et al. 2010). As expected, γ-tubulin+ ciliary basal bodies were regular and located downstream of CSF flow direction in controls (Fig. 6A) (Mirzadeh, Han, et al. 2010). Seven-days post-stroke, in the anterior dorsal lateral ventricular wall (Fig. 1B, location AD), the polarized cellular position of basal bodies was disrupted; many basal bodies were displaced (yellow arrows, Fig. 6B) and found upstream of CSF flow. Quantification of normal PCP orientation showed a tight distribution around the median angle (Fig. 6C, green bars). In contrast, there was significant increase in PCP dispersion after stroke (Fig. 6C, red bars; n = 3, 302 cells; two-sample Kolmogorov–Smirnov Test, D = 0.32, P < 0.0001). In other regions of the lateral ventricular wall, ependymal PCP was preserved.
We hypothesized that the ependymal changes after stroke may disrupt cilia function and normal CSF flow. To assess CSF flow, we measured fluorescent bead movement over the anterior lateral ventricular wall of freshly dissected whole mounts 7 days post stroke (n = 4). Beads flowed anteriorly in dorsal and ventral streams around the intraventricular AZ (Fig. 6D,E). After stroke, the general direction of CSF flow was unchanged. However, tracking the xy-coordinates of individual bead movement in time-lapse frames (Fig. 6J,K) showed that post stroke, CSF flow rate over the entire ventricular surface decreased by 26% (Fig. 6L; Supplementary Movie 1; n = 4; P = 0.01). Furthermore, in the region of PCP disruption, there were focal pockets wherein fluorescent beads moved in a turbulent whirlpool-like pattern (inset Fig. 6E; Supplementary Movie 1). Post hoc immunostaining and confocal analysis showed the ependymal layer was intact over the turbulent region, thus making it unlikely that the turbulence observed was due to mechanical tissue damage (Fig. 6F–I).
Discussion
This study shows that the MCAO model of stroke alters several cellular components of the SVZ neurogenic niche; astrocytes, blood vessels, and ependymal cells. For the first time, we document post stroke reactive astrocytosis in the SVZ and a disrupted neuroblast migratory scaffold. Blood vessels, which aid neuroblast migration and contribute to the stem cell niche, increased in size and branching complexity after stroke. SVZ neuronal proliferation increased without changes in apoptosis. In contrast to what others have found, we did not detect ependymal cell division after stroke and the ependymal layer remained intact. Unexpectedly though, ependymal cells began to express robust levels of GFAP and also had increased cell surface area. While ependymal motile cilia appeared normal in length and density, they were displaced and PCP was disrupted in the anterior dorsal SVZ. In line with this, functional studies demonstrated more turbulent and decreased CSF flow rates after stroke. Whole mounts provide a unique opportunity to examine the ependymal layer and SVZ, and previous studies have meticulously detailed the lateral ventricular wall in healthy mice (Mirzadeh et al. 2008; Shen et al. 2008; Tavazoie et al. 2008). In the present study, we used whole mounts in histological and functional studies to show multiple dramatic changes in the SVZ neurogenic compartment following stroke.
Stroke Induced SVZ Reactive Astrocytosis Even Though the Lesion Did Include the SVZ
In other studies, we have shown that lesions which directly involve the SVZ dramatically alter its response compared with lesions that spare it (Goings et al. 2006). We therefore used MCAO lesions that encompassed most of the striatum but that did not include the SVZ. Although the SVZ is transiently hypoxic during MCAO (Thored et al. 2007), in our stroke model, SVZ neurogenesis was robust and the small number of apoptotic cells observed in controls increased slightly but not significantly. The relative rates of SVZ proliferation and neurogenesis (tens of thousands of newborn neurons per day) far outweigh the small number of SVZ TUNEL+ cells in controls as well as after stroke, unlikely impacting overall rates of neurogenesis. In contrast, quantification in the striatum after stroke provided a convenient internal positive control that was replete with TUNEL+ dying cells. NeuN+ striatal neurons adjacent to the SVZ appeared plump and healthy, and there were neither reactive astrocytes nor TUNEL+ cells in the medial striatum. Therefore, it was remarkable that SVZ astrocytes assumed a reactive phenotype. This suggested that SVZ astrocytes responded to the distant injury via diffusible, vascular-borne, and/or CSF-borne signals.
SVZ astrocytes normally form glial tubes and provide a physical scaffold for neuroblast migration (Thomas et al. 1996), while their disruption leads to abnormal neurogenesis and migration (Anton et al. 2004; Kaneko et al. 2010). For example, we recently showed that Galectin-3 gene deletion caused a reactive SVZ astrocyte phenotype with thickened GFAP+ processes and concomitant defects in neuroblast migration (Comte et al. 2011). These observations point toward a model whereby multiple mechanisms combine to maintain SVZ glial tube integrity. After stroke, neuroblast migration to the olfactory bulb is decreased (Ohab et al. 2006), and cells tend to move at right angles to their normal rostral direction. We believe that the glial tube dysmorphology we document here may be a contributing factor to these shifts in migration and may increase neuroblast branching and turning (Martinez-Molina et al. 2011) and thereby promote emigration. In addition, reactive astrocytes are deficient in GABA inhibition (Ortinski et al. 2010) and GABA tonically decreases neuroblast migration speed (Bolteus and Bordey 2004) suggesting that after stroke, reactive astrocytes may alter neuroblast migration speed via a GABA-ergic mechanism. In support of this, time-lapse imaging indicates that neuroblasts increase migratory speed as they exit the SVZ and enter the striatum post stroke (Zhang et al. 2009). We showed previously with 2-photon time-lapse microscopy that astrocytes themselves are stationary in the unperturbed SVZ (Nam et al. 2007). However, recent work form the Cunningham laboratory showed SVZ-derived astrocytes migrate into the ischemic striatum (Li et al. 2010), and it is tempting to speculate that this may be related to their reactivity after stroke. Another intriguing question is whether stroke drives SVZ niche astrocytes toward a stem-like phenotype, as has been observed in cortical reactive astrocytes following injury, and whether they have the capacity to function as NSCs in the ischemic striatum (Buffo et al. 2008).
Stroke Increased SVZ Proliferation and Vascular Density
Traumatic brain injury and stroke increases SVZ neural precursor proliferation and newly generated neurons migrate to the ischemic penumbra (Szele and Chesselet 1996; Arvidsson et al. 2002). Similarly, we found in this study that overall numbers of proliferating cells increased, and further showed that the percentage of dividing SVZ neuronal progenitors and neuroblasts did not change. Our studies were not designed to detect quiescent NSCs and we do not know if stem cell self-renewal changed after stroke. Moreover, the high levels of de novo ependymal GFAP expression prevented reliable quantification of ventricle-contacting GFAP+ NSCs, which can be a convenient read-out for changes in the number of stem cells in the system (Doetsch et al. 2002).
The importance of the vascular niche for SVZ neurogenesis and migration is well established at multiple ages (Shen et al. 2008; Snapyan et al. 2009; Nie et al. 2010). Engorged SVZ vasculature and increased angiogenesis were reported following thermocoagulatory cortical lesions (Gotts and Chesselet 2005). Therefore, increased vascular branching and vessel diameter in the post stroke SVZ was not surprising. Since neuroblasts have been reported to follow SVZ blood vessels, it may be that their increased branching also helps promote turning and emigration (Kojima et al. 2010). We also found astrocytes with increased blood vessel contacts post stroke, further raising the possibility they modulate blood flow to the SVZ (Attwell et al. 2010). Since post stroke neurogenesis and angiogenesis are coupled in the ischemic penumbra (Ohab et al. 2006), our data suggest the 2 processes may be coordinated across broad regions of the brain in injury.
Ependymal Cells Acquired Radial Glia and Reactive Astrocyte Features Post Stroke
A subset of GFAP+ SVZ astrocytes are NSCs but whether ependymal cells may be neural precursors remains elusive (Johansson et al. 1999; Gregg and Weiss 2003; Spassky et al. 2005; Gleason et al. 2008). We did not find proliferating ependymal cells 7- and 14-days after stroke, using pulse and long-term BrdU labeling, respectively. This contrasts with post stroke rat ependymal cells that incorporated BrdU and expressed the mitotic radial glia marker phosphorylated vimentin (Zhang et al. 2007). There may be interspecies differences, alternatively ependymal cells may have divided so rarely that even our cumulative labeling was insufficient. Similar cumulative labeling also did not detect BrdU+ ependymal cells after stroke (Carlen et al. 2009). Using a “Foxj1-cre” expressing lentivirus which labels ependymal cells and a subset of SVZ astrocytic stem cells (Jacquet et al. 2009), it was suggested that murine ependymal cells did not divide but delaminated into the SVZ to generate neuroblasts and astrocytes and resulting in ependymal cell loss (Carlen et al. 2009). While we did not fate-map ependymal cells, detailed examination of whole mounts and coronal sections showed the ependymal cell layer was preserved post stroke, without obvious cell loss or denudation.
GFAP is a major structural intermediate filament protein of SVZ astrocytes, reactive astrocytes, and developmental radial glia. SVZ astrocytes and ependymal cells derive from GFAP+ radial glia (Merkle et al. 2004), but postnatal ependymal cells lose GFAP and develop motile cilia (Spassky et al. 2005). Surprisingly, we showed that most ependymal cells expressed high levels of GFAP after stroke. Using whole mounts, GFAP was found at the ventricular surface in ependymal cells identified in single confocal optical sections by ciliary basal bodies and β-catenin expression in the cell membrane. In addition to ependymal cells, β-catenin can be expressed by activated SVZ cells (Cooper and Isacson 2004), reactive astrocytes (White et al. 2009), and SVZ B1 stem cells (Mirzadeh et al. 2008). Thus, it is possible that some of its expression was not ependymal. However, the location of the ependymal cells next to the lateral ventricles makes them easy to distinguish from SVZ cells and this combined with the plasma membrane expression in the typical cobblestone arrangement of ependymal cells suggests β-catenin expression was primarily ependymal. Also, the density of primary cilia is low relative to ependymal cells, and it is unlikely that all of the β-catenin we detect post stroke in the ependymal layer is in them. The GFAP detected in ependymal cells post stroke may have been de novo expression or expression which increased above the threshold of immunohistochemical detectability. Immunoelectron microscopy would provide a useful confirmation of GFAP expression in ependymal cells.
GFAP+ cells have also been reported to appear in the ependyma of aged mice and following neuraminidase denudation (Luo et al. 2008). Under EphB2 regulation, SVZ astrocytes replaced lost ependymal cells and assumed ependymal features, such as adherens junctions, basal bodies, and motile cilia, while low numbers of ependymal cells delaminated into the SVZ and expressed GFAP (Luo et al. 2008; Nomura et al. 2010). After stroke, it is more likely that the GFAP+ cells in the ependymal layer are ependymal cells acquiring features of young or reactive astrocytes. First, the ependymal layer was intact after stroke; even if delamination occurred, astrocytes are unlikely to have replaced >85% of ependymal cells within a week. Second, ependymal GFAP+ cells had cilia and basal bodies typical of mature ependymal cells. It is improbable that astrocytes could form dense basal body clusters and grow motile cilia tufts in only 7 days. Last, ependymal cells have been shown to switch phenotype after neonatal hypoxia and stroke in rats (Ganat et al. 2002).
Vimentin+ ependymal cells that coexpressed GFAP in coronal histological sections provided further proof of our unexpected result. GFAP coexpression with vimentin is required for reactive astrocytosis and scar formation, however, loss of either intermediate filament alone is insufficient to inhibit scar formation (Pekny et al. 1999; Wilhelmsson et al. 2004). It may be that GFAP and vimentin coexpression in ependymal cells post stroke serves a barrier function similar to damage-limiting astrocytic scars (Li et al. 2008). In fact, GFAP and vimentin coexpression increases the physical stiffness of astrocytes (Lu et al. 2011) suggesting it may also alter the mechanical properties of ependymal cells.
Motile Cilia Planar Cell Polarization and CSF Flow Were Disrupted after Stroke
Ependymal cell polarization in the epithelial plane orients ciliary beat directions and hence, CSF flow (Mirzadeh, Han, et al. 2010). We found enlarged GFAP+ ependymal cells in the anterior dorsal ventricular wall that lost their ciliary PCP, after stroke. Whereas ciliary basal bodies were polarized to downstream positions in controls, their planar location shifted dramatically and became randomly distributed post stroke. To our knowledge, this is the first report of acute injury in a nonproliferating tissue disrupting PCP. A possible mechanism for this finding may be that the new GFAP filaments shift basal body locations in ependymal cells. Notably, the kaolin-infusion model of neonatal hydrocephalus also caused ependymal cell stretching and motile cilia displacement (Weller et al. 1978).
The ependymal cell response to stroke was functionally significant; we observed decreased CSF flow speed and increased turbulence 7 days post stroke. Disruption of CSF fluid dynamics may disturb molecular equilibria between CSF and brain parenchyma (Johanson et al. 2011), such as the Slit2 chemorepellant gradient, which mediates SVZ neuroblast migration (Sawamoto et al. 2006). The regulation of other CSF-borne molecules such as insulin-like growth factor, fibroblast growth factor, and sonic hedgehog (Lehtinen and Walsh 2011) are also likely to be affected. These molecules contribute to neurogenic niche maintenance and the SVZ's proximity to the ventricles makes it vulnerable to such imbalances. Tissue swelling regularly occurs after brain trauma, and it may be that increased ependymal cell size was due to this. However, we documented decreases in ipsilateral brain volume after MCAO. This suggests that the specific increase in ependymal cell diameter was not due to tissue swelling. Moreover, post stroke ventricular dilation may account for the increased ependymal cell surface area as the cells are mechanically stretched. This kind of mechanical force may provide a nonmolecular signal, which contributes to the changes observed in the post stroke SVZ.
Conclusions
The roles of SVZ astrocytes, the vasculature and ependyma in the regulation of SVZ neurogenesis are well known, and disruption of their function, such as abnormal glial tube formation and disrupted CSF flow are recognized features which impact neuroblast migration. SVZ-mediated endogenous repair post stroke is likely an intricate and complex process, as highlighted by the multiple cellular changes that we document in this study. Human adult neurogenesis is minimal to absent (Sanai et al. 2011) and our data have uncovered evidence for reactivity in SVZ astrocytes as well as ependymal cells. Rodent neurogenesis is maintained throughout life however and the changes documented here may contribute to mechanisms, which induce neuroblast emigration and the endogenous repair process in this model of stroke. Future work will need to elucidate to what extent these changes serve to facilitate aspects of SVZ-mediated repair.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/
Funding
C.C.Y. was supported by a Rhodes Scholarship. F.G.S. was supported by National Institutes of Health Grant RO1 NS-42253. K.J.B. and A.M.B. were supported by Medical Research Council UK and Foundation Leducq.
Supplementary Material
Acknowledgments
We would like to thank Professor Arturo Alvarez-Buylla, Dr Zaman Mirzadeh, and Dr Hyung-Song Nam for their assistance with the whole mount preparation. We are grateful to Dr Shankar Srinivas and Mr Brad Joyce for their technical expertise with the whole mount time-lapse imaging. We are indebted to members of the Oxford University Biomedical Services for their assistance with the mice. We thank Dr Yongsoo Kim and members of the Szele and Buchan labs for critical discussion of the manuscript. Conflict of Interest: None declared.
References
- Altman J, Das GD. Autoradiographic and histological studies of postnatal neurogenesis. I. A longitudinal investigation of the kinetics, migration and transformation of cells incorporating tritiated thymidine in neonate rats, with special reference to postnatal neurogenesis in some brain regions. J Comp Neurol. 1966;126:337–389. doi: 10.1002/cne.901260302. [DOI] [PubMed] [Google Scholar]
- Anton ES, Ghashghaei HT, Weber JL, McCann C, Fischer TM, Cheung ID, Gassmann M, Messing A, Klein R, Schwab MH, et al. Receptor tyrosine kinase ErbB4 modulates neuroblast migration and placement in the adult forebrain. Nat Neurosci. 2004;7:1319–1328. doi: 10.1038/nn1345. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber PA, Hoyte L, Colbourne F, Buchan AM. Temperature-regulated model of focal ischemia in the mouse: a study with histopathological and behavioral outcomes. Stroke. 2004;35:1720–1725. doi: 10.1161/01.STR.0000129653.22241.d7. [DOI] [PubMed] [Google Scholar]
- Bolteus AJ, Bordey A. GABA release and uptake regulate neuronal precursor migration in the postnatal subventricular zone. J Neurosci. 2004;24:7623–7631. doi: 10.1523/JNEUROSCI.1999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Goritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabe-Heider F, Yeung MS, Naldini L, et al. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009;12:259–267. doi: 10.1038/nn.2268. [DOI] [PubMed] [Google Scholar]
- Comte I, Kim Y, Young CC, van der Harg JM, Hockberger P, Bolam PJ, Poirier F, Szele FG. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J Cell Sci. 2011;124:2438–2447. doi: 10.1242/jcs.079954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Isacson O. Intrastriatal transforming growth factor alpha delivery to a model of Parkinson's disease induces proliferation and migration of endogenous adult neural progenitor cells without differentiation into dopaminergic neurons. J Neurosci. 2004;24:8924–8931. doi: 10.1523/JNEUROSCI.2344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol. 2010;119:55–73. doi: 10.1007/s00401-009-0624-y. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn A-M, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM. Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience. 2002;112:977–991. doi: 10.1016/s0306-4522(02)00060-x. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Gleason D, Fallon JH, Guerra M, Liu JC, Bryant PJ. Ependymal stem cells divide asymmetrically and transfer progeny into the subventricular zone when activated by injury. Neuroscience. 2008;156:81–88. doi: 10.1016/j.neuroscience.2008.06.065. [DOI] [PubMed] [Google Scholar]
- Goings GE, Kozlowski DA, Szele FG. Differential activation of microglia in neurogenic versus non-neurogenic regions of the forebrain. Glia. 2006;54:329–342. doi: 10.1002/glia.20381. [DOI] [PubMed] [Google Scholar]
- Gotts JE, Chesselet MF. Vascular changes in the subventricular zone after distal cortical lesions. Exp Neurol. 2005;194:139–150. doi: 10.1016/j.expneurol.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Gregg C, Weiss S. Generation of functional radial glial cells by embryonic and adult forebrain neural stem cells. J Neurosci. 2003;23:11587–11601. doi: 10.1523/JNEUROSCI.23-37-11587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet BV, Salinas-Mondragon R, Liang H, Therit B, Buie JD, Dykstra M, Campbell K, Ostrowski LE, Brody SL, Ghashghaei HT. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development. 2009;136:4021–4031. doi: 10.1242/dev.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Greenberg DA. Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc Natl Acad Sci U S A. 2010;107:7993–7998. doi: 10.1073/pnas.1000154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson C, Stopa E, McMillan P, Roth D, Funk J, Krinke G. The distributional nexus of choroid plexus to cerebrospinal fluid, ependyma and brain: toxicologic/pathologic phenomena, periventricular destabilization, and lesion spread. Toxicol Pathol. 2011;39:186–212. doi: 10.1177/0192623310394214. [DOI] [PubMed] [Google Scholar]
- Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Marin O, Koike M, Hirota Y, Uchiyama Y, Wu JY, Lu Q, Tessier-Lavigne M, Alvarez-Buylla A, Okano H, et al. New neurons clear the path of astrocytic processes for their rapid migration in the adult brain. Neuron. 2010;67:213–223. doi: 10.1016/j.neuron.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Wang WZ, Comte I, Pastrana E, Tran PB, Brown J, Miller RJ, Doetsch F, Molnar Z, Szele FG. Dopamine stimulation of postnatal murine subventricular zone neurogenesis via the D3 receptor. J Neurochem. 2010;114:750–760. doi: 10.1111/j.1471-4159.2010.06799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T, Hirota Y, Ema M, Takahashi S, Miyoshi I, Okano H, Sawamoto K. Subventricular zone-derived neural progenitor cells migrate along a blood vessel scaffold toward the post-stroke striatum. Stem Cells. 2010;28:545–554. doi: 10.1002/stem.306. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Goderie S, Wang Y, Lotz S, Lin G, Sun Y, Roysam B, Shen Q, Temple S. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell. 2010;7:163–173. doi: 10.1016/j.stem.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc Natl Acad Sci U S A. 2001;98:4752–4757. doi: 10.1073/pnas.081074998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Walsh CA. Neurogenesis at the brain-cerebrospinal fluid interface. Annu Rev Cell Dev Biol. 2011;27:653–679. doi: 10.1146/annurev-cellbio-092910-154026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PD. Mitotic activity in the primate subependymal layer and the genesis of gliomas. Nature. 1968;217:974–975. doi: 10.1038/217974a0. [DOI] [PubMed] [Google Scholar]
- Li L, Harms KM, Ventura PB, Lagace DC, Eisch AJ, Cunningham LA. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia. 2010;58:1610–1619. doi: 10.1002/glia.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, García-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YB, Iandiev I, Hollborn M, Korber N, Ulbricht E, Hirrlinger PG, Pannicke T, Wei EQ, Bringmann A, Wolburg H, et al. Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. Faseb J. 2011;25:624–631. doi: 10.1096/fj.10-163790. [DOI] [PubMed] [Google Scholar]
- Luo J, Shook BA, Daniels SB, Conover JC. Subventricular zone-mediated ependyma repair in the adult mammalian brain. J Neurosci. 2008;28:3804–3813. doi: 10.1523/JNEUROSCI.0224-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas J, Nern C, Plate KH, Momma S. Increased generation of neuronal progenitors after ischemic injury in the aged adult human forebrain. J Neurosci. 2006;26:13114–13119. doi: 10.1523/JNEUROSCI.4667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Molina N, Kim Y, Hockberger P, Szele FG. Rostral migratory stream neuroblasts turn and change directions in stereotypic patterns. Cell Adh Migr. 2011;5:83–95. doi: 10.4161/cam.5.1.13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott KW, Lantos PL. Cell proliferation in the subependymal layer of the postnatal marmoset, Callithrix jacchus. Brain Res Dev Brain Res. 1990;57:269–277. doi: 10.1016/0165-3806(90)90053-2. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci U S A. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Doetsch F, Sawamoto K, Wichterle H, Alvarez-Buylla A. The subventricular zone en-face: wholemount staining and ependymal flow. J Vis Exp. 2010 doi: 10.3791/1938. doi: 10.3791/1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Han YG, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Cilia organize ependymal planar polarity. J Neurosci. 2010;30:2600–2610. doi: 10.1523/JNEUROSCI.3744-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SC, Kim Y, Dryanovski D, Walker A, Goings G, Woolfrey K, Kang SS, Chu C, Chenn A, Erdelyi F, et al. Dynamic features of postnatal subventricular zone cell motility: a two-photon time-lapse study. J Comp Neurol. 2007;505:190–208. doi: 10.1002/cne.21473. [DOI] [PubMed] [Google Scholar]
- Nie K, Molnar Z, Szele FG. Proliferation but not migration is associated with blood vessels during development of the rostral migratory stream. Dev Neurosci. 2010;32:163–172. doi: 10.1159/000301135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Goritz C, Catchpole T, Henkemeyer M, Frisen J. EphB signaling controls lineage plasticity of adult neural stem cell niche cells. Cell Stem Cell. 2010;7:730–743. doi: 10.1016/j.stem.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Valentin VV, Lowenstein DH. Prolonged seizures increase proliferating neuroblasts in the adult rat subventricular zone-olfactory bulb pathway. J Neurosci. 2002;22:3174–3188. doi: 10.1523/JNEUROSCI.22-08-03174.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham M, Helluy X, Braeuninger S, Jakob P, Stoll G, Kleinschnitz C, Bendszus M. Outcome of experimental stroke in C57Bl/6 and Sv/129 mice assessed by multimodal ultra-high field MRI. Exp Transl Stroke Med. 2010;2:6. doi: 10.1186/2040-7378-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JLR, et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell. 2008;3:289–300. doi: 10.1016/j.stem.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapyan M, Lemasson M, Brill MS, Blais M, Massouh M, Ninkovic J, Gravel C, Berthod F, Gotz M, Barker PA, et al. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 2009;29:4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szele FG, Chesselet MF. Cortical lesions induce an increase in cell number and PSA-N-CAM expression in the subventricular zone of adult rats. J Comp Neurol. 1996;368:439–454. doi: 10.1002/(SICI)1096-9861(19960506)368:3<439::AID-CNE9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LB, Gates MA, Steindler DA. Young neurons from the adult subependymal zone proliferate and migrate along an astrocyte, extracellular matrix-rich pathway. Glia. 1996;17:1–14. doi: 10.1002/(SICI)1098-1136(199605)17:1<1::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O. Long-term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke. 2007;38:3032–3039. doi: 10.1161/STROKEAHA.107.488445. [DOI] [PubMed] [Google Scholar]
- Weller RO, Mitchell J, Griffin RL, Gardner MJ. The effects of hydrocephalus upon the developing brain. Histological and quantitative studies of the ependyma and subependyma in hydrocephalic rats. J Neurol Sci. 1978;36:383–402. doi: 10.1016/0022-510x(78)90046-1. [DOI] [PubMed] [Google Scholar]
- White BD, Nathe RJ, Maris DO, Nguyen NK, Goodson JM, Moon RT, Horner PJ. Beta-catenin signaling increases in proliferating NG2+ progenitors and astrocytes during posttraumatic gliogenesis in the adult brain. Stem Cells. 2009;28:297–307. doi: 10.1002/stem.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CC, Brooks KJ, Buchan AM, Szele FG. Cellular and molecular determinants of stroke-induced changes in subventricular zone cell migration. Antioxid Redox Signal. 2010;14:1877–1888. doi: 10.1089/ars.2010.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Gregg SR, Toh Y, Roberts C, Letourneau Y, Buller B, Jia L, Davarani SPN, Zhang ZG. Patterns and dynamics of subventricular zone neuroblast migration in the ischemic striatum of the adult mouse. J Cereb Blood Flow Metab. 2009;29:1240–1250. doi: 10.1038/jcbfm.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Wang Y, LeTourneau Y, Liu XS, Zhang X, Gregg SR, Wang L, Chopp M. Stroke induces ependymal cell transformation into radial glia in the subventricular zone of the adult rodent brain. J Cereb Blood Flow Metab. 2007;27:1201–1212. doi: 10.1038/sj.jcbfm.9600430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.