

Abstract
Identification of genes that are coexpressed across various tissues and environmental stresses is biologically interesting, since they may play coordinated role in similar biological processes. Genes with correlated expression patterns can be best identified by using coexpression network analysis of transcriptome data. In the present study, we analyzed the temporal-spatial coordination of gene expression in root, leaf and panicle of rice under drought stress and constructed network using WGCNA and Cytoscape. Total of 2199 differentially expressed genes (DEGs) were identified in at least three or more tissues, wherein 88 genes have coordinated expression profile among all the six tissues under drought stress. These 88 highly coordinated genes were further subjected to module identification in the coexpression network. Based on chief topological properties we identified 18 hub genes such as ABC transporter, ATP-binding protein, dehydrin, protein phosphatase 2C, LTPL153 - Protease inhibitor, phosphatidylethanolaminebinding protein, lactose permease-related, NADP-dependent malic enzyme, etc. Motif enrichment analysis showed the presence of ABRE cis-elements in the promoters of > 62% of the coordinately expressed genes. Our results suggest that drought stress mediated upregulated gene expression was coordinated through an ABA-dependent signaling pathway across tissues, at least for the subset of genes identified in this study, while down regulation appears to be regulated by tissue specific pathways in rice.
Keywords: Coexpression, Drought stress, Hub gene, Rice, Transcriptome, WGCNA
Background
It is well documented that transcriptionally coexpressed genes tend to be functionally related and may interact with each other at physiological or molecular level. Recently, number of comprehensive method have been developed and applied to construct networks from diverse high-throughput data sources such as microarray, next generation sequencing (NGS), chromatin immunoprecipitation (ChIP) and protein-protein interaction assays [1]. These high-throughput techniques have made it possible to analyze thousands of genes in one shot. Availability of these datasets in public domain is useful resource to accelerate incremental hypothesis generation via reanalyzing the data by addressing new questions.
Gene networks are the basis of biological complexity and have become the core area of research in systems biology. These networks are modeled as graph where, node represents the functional unit such as gene, protein, metabolite, etc., and edges are dependencies or interaction between the nodes. In case of the expression data of transcripts, the interaction may be the expression co-relation between the paired genes that is generally measured in terms of Pearson co-relation coefficient (PCC). It has been shown that PCC value with large magnitude are highly coexpressed often a result from direct coregulation [2]. Several user-friendly tools have been developed to build coexpression network based on PCC values. The WGCNA (Weighted gene co-expression network analysis) is one of the tools for coexpression network that supports the assembly of both signed and unsigned network.
In signed network positively and negatively correlated nodes are clustered in separate modules, where as unsigned network finds correlation by their absolute value. Biological networks exhibit scale free topology, where connectivities between one node with other nodes are of major concern [3]. Nodes with high degree of connectivity are called hub nodes, and the edge deletion of a hub gene will have consequences on architecture and biological interpretations of the network [4]. Hence, prioritization of genes by selecting highly connected node as hub node is a facile approach for better understanding and interpretation of the network and overall biological complexity.
The system biology approach has accelerated unraveling the knowledge in the area of plants stress biology [5]. The response of plants to abiotic stresses is a very complex process [6]. Hence, biological data on abiotic stress related genes and QTLs available in public databases of great importance to understand abiotic stresses [7–9]. Plant response to abiotic stresses depends upon the developmental stage at which the plant experiences the stress, the rate of stress development and duration of stress. Some pathways of stress tolerance are conserved across tissues, while others may be tissue specific [10].
In the present study, we systematically reanalyzed the drought stress responsive transcriptome data available for three main tissues including leaf, root, and young panicle in rice [11]. Wang et al. (2011) imposed drought stress by withholding water at three different stages: 4-tiller (tillering) stage, panicle elongation stage, and booting stage. Leaf, root and young panicle (only at booting stage) were sampled at leaf relative water content of about 65-75%, and used for transcriptome analysis. We used the transcriptome data of Wang et al. 2011 for computational and coexpression analysis to: (i) identify differentially expressed genes in coordinated manner across all the tissues, (ii) define module of genes sharing similar expression pattern, eventually defining global biological pathways and (iii) hub gene identification. Our study provides novel insight in to gene coordination under drought stress that was not revealed in previous studies with conventional differential gene expression analysis.
Methodology
Microarray data analysis:
Microarray data for temporal and spatial expression patterns of drought stress response of a drought tolerant indica rice line DK151 (GSE26280) was downloaded from NCBI GEO database (www.ncbi.nlm.nih.gov/geo/). This dataset consists of transcriptome data for three tissues at three different developmental stages of rice viz. root at tillering stage, leaf at tillering stage, root at panicle elongation stage, leaf at panicle elongation, young panicle at booting stage, and leaf at booting stages with three biological replicate (total 36 samples). All CEL files were analyzed by R (version 2.6.1) statistical programming environment, using affy package of BioConductor (http://www.bioconductor.org/) [12]. Normalization was performed by the robust multichip analysis algorithm (RMA), ttest used to calculate the p-value of the expression change of each probe and differentially expressed genes (DEGs) were identified using the limma package [13]. We selected DEGs with p-values <0.05, and fold-change values >2 for up and < 2 for down regulated genes. Probe sets were mapped to the MSU Rice Genome Annotation Project gene set (release 6.1). Hierarchical clustering of significantly coordinated differentially expressed genes was carried out by average linkage and euclidean distance as a measurement of similarity using EPICLUST; a module of Expression Profiler (http://www.bioinf.ebc.ee/EP/EP/EPCLUST/).
Gene Ontology enrichment analysis:
Significantly enriched GO categories for all set of differentially expressed genes was carried out by GOEAST, Gene Ontology enrichment analysis tool (http://omicslab.genetics.ac.cn/GOEAST) by selecting Hypergeometric statistical test and threshold for False Discovery Rate (FDR) adjusted p-value ≤ 0.05 [14].
Gene coexpression network:
Correlation network approach is being increasingly used in bioinformatics applications and successfully applied in various biological contexts [15]. WGCNA is a system biology method to build robust network and module identification of highly correlated genes with module membership measures using the topological overlap measure (TOM) [16]. We used signed version of the scale free topology fitting index and only considered those parameter values that lead to a network satisfying scale-free topology (signed R2>0.8, β = 22) [3]. Expression matrix was created on the basis of correlated expression pattern between the genes, calculated by PCC value. Further, the coexpression network of highly coordinated genes among most of the tissue was visualized and analyzed by Cytoscape (version 2.8.3) [17]. Cytoscape is an open source bioinformatics platform for visualizing molecular interaction networks and biological pathways (http://www.cytoscape.org/).
MCODE clustering:
Clusters of well interconnected genes were identified by Molecular Complex Detection (MCODE) algorithm [18]. The algorithm identified modules with network scoring parameter and the degree cutoff was set to 2. Another important parameter, K-Core value set as default and the depth from the seed was set to 100 that calculate the distance between the seed node and other cluster members.
Identification of over-represented motifs:
Over represented cis-motif in 2 kb upstream sequence from translational initiation codon of coordinated genes that are expressed across tissues was performed using the Osiris programme (http://www.bioinformatics2.wsu.edu/cgibin/Osiris/cgi/home.pl) [19].
Results & Discussion
Signed WGCNA modules in temporal-spatial data set:
Differential expression analysis identified 8244 DEGs (2 fold up or down) in at least one of the tissues under drought condition. Further analysis revealed that 2199 DEGs showing coexpression in ≥ 3 tissue samples under drought stress. We then generated signed coexpression network of these 2199 DEGs (Figure 1a). Though the sample size was small, the network created in this study satisfied the scale free topology [20]. Eleven modules were clustered from the whole network using topological overlap measure (TOM) (Figure 1b). These analyses led to the identification of blue (0.76) and turquoise (0.75) colored modules, with highest significance value. The WGCNA approach was used to create a TOM plot by using gene expression data of the blue and turquoise module (Figure 1c, d).
Figure 1.
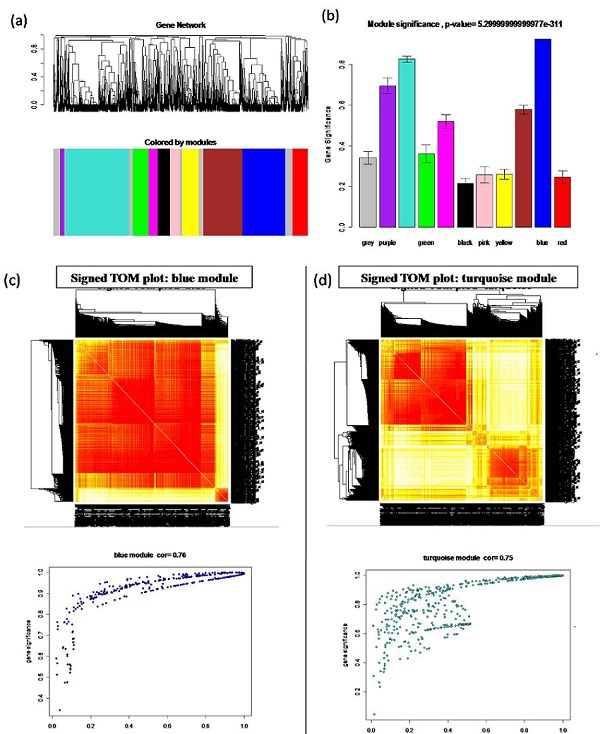
Identification of coexpression network modules using spatial-temporal dataset of rice under drought stress. (a) Hierarchical clustering of the Topological Overlap Measure (TOM) matrix for the expression data. Branches of the hierarchical cluster tree define 11 modules with assigned color. (b) Bar plots showing modules significance. Note that the blue and turquoise color modules are with highest significance value. Grey was reserved to color genes that are not part of any module. (c) Signed TOM plot (top) and the MDS plot (bottom) of blue module with significant correlation value (r = 0.76). (d) Signed TOM plot (top) and the MDS plot (bottom) of turquoise module with significant correlation value (r = 0.75).
Identification of coordinately regulated DEGs across tissues and developmental stages:
Out of 2199 coexpressed DEGs in ≥ 3 samples, 113 probes were identified to have common expression kinetics (highly coordinated) among all tissues across developmental stages under drought stress. Of these 113 probes, two probes did not map on any annotated genes on the rice genome. Rest of the 109 probes mapped to 95 different annotated genes. Interestingly, among these 95 genes, 88 were upregulated (cluster I), and only three genes were repressed under drought stress in all samples (cluster III) (Figure 2); Table 1 (see supplementary material).
Figure 2.

Hierarchical clustering on expression ratios of the differentially expressed genes obtained in three tissues at three different stages used to identify common expression kinetics among differentially expressed genes. Cluster I and III showed constant expression pattern of genes i.e., induced and repressed, respectively. Clusters II grouped two genes with tissue specific opposite regulation.
Cluster I grouped genes for several dehydrins, late embryogenesis abundant proteins (LEAs), protein phosphatase 2Cs and expressed proteins. Cluster III grouped one gene each encoding invertase/pectin methyl esterase inhibitor protein, MYB transcription factor and receptor protein kinase that were significantly repressed in all the tissues under drought stress. Clusters II grouped two genes with tissue specific opposite regulation; LOC_Os03g20680 (LEA) was repressed in leaf at tillering stage, while LOC_Os01g39020 (HSF protein) was repressed in roots at both tillering and panicle elongation stage, where as these genes were induced in other stages. These results suggest that drought stress mediated upregulated gene expression is coordinated through a common signaling pathway across tissues, at least for the subset of genes identified in this study, while downregulation in general appears to be tissue specific in rice. Gene Ontology enrichment analysis of highly coordinated DEGs showed that predominant DEGs were enriched with response to stress (p-value: 0.00535), response to stimulus (p-value: 0.0791), response to water stimulus (p-value: 1.41E-07), and response to abiotic stimulus (p-value: 0.000632) with high significance (Figure 3); Table 1 (see supplementary material).
Figure 3.
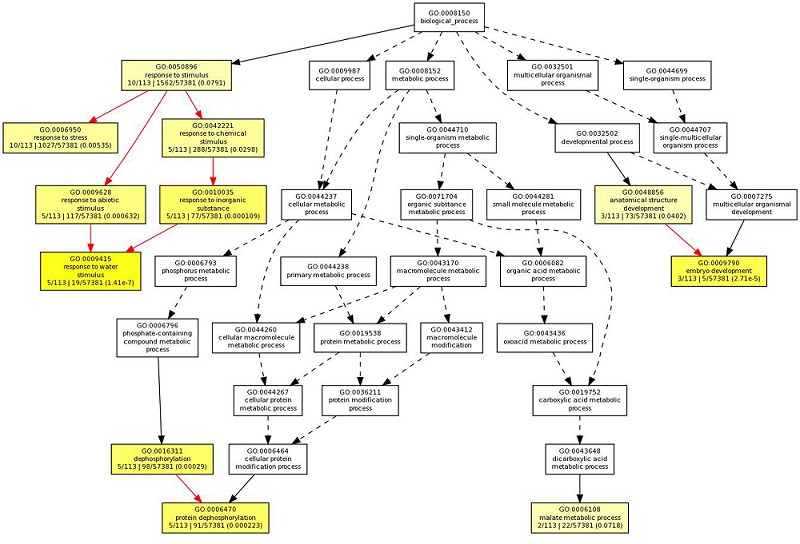
Gene Ontology enrichment analysis of highly coordinated genes in all tissues at each developmental stages showed enrichment of “response to water stimulus” with high significance.
Coexpression network of coordinately upregulated DEGs:
The 107 probes (88 genes and 2 unannotated genes) shown in hierarchical cluster I with coordinated upregulation in all tissues were further subjected for coexpression network construction to identify hub gene (master gene) using Cytoscape (2.8.3). We selected 0.8 as PCC cutoff with which almost all genes were integrated in to the network (Figure 4a). Based on topological parameters like degree of the node, neighborhood connectivity and cluster coefficient value, 18 hub genes were identified (Table 1). Genes with high value of degree (>25), and neighborhood connectivity (>23) are LOC_Os09g39910 (ABC transporter ATP-binding protein), LOC_Os11g26760 (dehydrin), LOC_Os05g47730 (LTPL153 - Protease inhibitor/seed storage/LTP family protein precursor), LOC_Os05g39250 (phosphatidylethanolamine-binding protein), LOC_Os03g07170 (lactose permease), (LOC_Os01g54030) NADP-dependent malic enzyme, Os.55227.1.S1_at (AK107694, unknown protein), etc. were observed as hub genes in network (Figure 4b).
Figure 4.
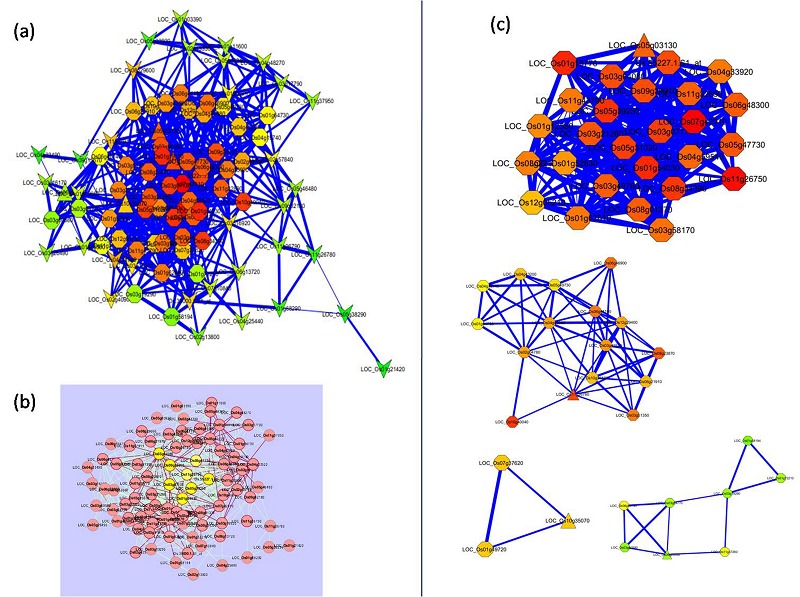
Coexpression network of coordinately upregulated genes across tissues at three developmental stages created by Cytoscape. (a) Seed nodes identified by MCODE in triangle shape, clustered nodes in oval and unclustered nodes are represented in vee shape. Red-yellow-green gradient color represents nodes with high to less neighborhood connectivity. (b) Network representing nodes in yellow color with high degree (>30) as hub genes and their first neighbors highlighted with red colored bordered and edges. (c) Four modules identified by MCODE showed each module with a triangled node (seed node).
MCODE clustering identified four modules which integrated in single turquoise module of WGCNA with high modular membership value, showing the robustness of the network with high significance (Figure 4c). For each of the module one seed gene with high cluster coefficient value showed how well that node was connected with others. Genes lies in largest module with high cluster coefficient values were LOC_Os11g26750 (dehydrin; 0.9), LOC_Os07g42910 (cytochrome C oxidase subunit; 0.7), LOC_Os09g39910 (ABC transporter; 0.65) and LOC_Os01g19770 (mitochondrial import inner membrane translocase subunit Tim17; 0.68) clustered in largest module. Interestingly, gene ontology analysis also showed relatedness of dehydrin gene to the “response to stress” GO term with high significance.
Enrichment of conserved cis-regulatory elements within 2kb upstream sequences from translational start site (ATG) of coordinated genes was investigated. Distribution of TF binding sites on the promoters showed that most predicted TF binding sites on the promoter were between -480 to -1 base from ATG (Supplementary Figure 1). Over-represented motifs analysis revealed that the promoter of more than 55 genes has ABA Responsive (ABRE) cis-elements Table 2 (see supplementary material). Among the five PP2Cs identified in this study, four PP2Cs belongs to ABA responsive Clade A group. These results suggest that ABA dependent pathway may regulate the coordinated upregulation of the genes across tissues and developmental stages under drought in rice.
Conclusion
Correlation analysis facilitates network based gene screening method for identification of candidate genes or targets under drought stress [21]. We analyzed temporal-spatial gene coexpression network of drought stress response in rice and identified 88 coordinated genes. The commonly upregulated genes consisted of several dehydrins, LEA proteins and heat shock proteins, suggesting that protection of cellular machinery is a common theme across tissues and development under drought stress. The network was constructed by using WGCNA tool and further analyzed by Cytoscape. The signed WGCNA network appears to be robust as they retain biologically relevant hub genes and their connections. We showed that genes with high module membership value and neighborhood connectivity (high degree) were valuable for candidate gene identification related to drought response in rice. Hierarchical cluster analysis of differentially expressed genes clearly showed the coordinated expression of genes under drought stress in each tissue and developmental stage. We incorporated gene ontology information and highlighted several stress regulated genes and their neighborhood as candidate drought responsive genes for further biological study that would not have been identified using a standard differential expression analysis. Predominance of ABRE cis-elements in the promoters of coordinately expressed genes suggested that ABA dependent pathway may regulate these genes in response to drought stress across tissues and developmental stages.
Supplementary material
Acknowledgments
We thank Indian Council of Agricultural Research (ICAR) for supporting this work through the National Initiative on Climate Resilient Agriculture (NICRA) project.
Footnotes
Citation:Smita et al, Bioinformation 9(2): 072-078 (2013)
References
- 1.Segal E, et al. Nat Genet. 2003;34:166. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 2.Usadel B, et al. Plant Cell Environ. 2009;32:1633. doi: 10.1111/j.1365-3040.2009.02040.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhang B, S Horvath. Stat Appl Genet Mol Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 4.Horvath S, J Horvath. PLoS Comput Biol. 2008;4:1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cramer GR, et al. BMC Plant Biol. 2011;11:163. doi: 10.1186/1471-2229-11-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atkinson NJ, PE Urwin. J Exp Bot. 2012;63:3523. doi: 10.1093/jxb/ers100. [DOI] [PubMed] [Google Scholar]
- 7.Balaji S, et al. Briefings in bioinformatics. 2007;8:318. [Google Scholar]
- 8.Sundar AS, et al. Bioinformation. 2008;2:431. doi: 10.6026/97320630002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smita S, et al. Database (Oxford) 2011 doi:10.1093/database/BAR037. [Google Scholar]
- 10.Chinnusamy V, et al. J Integr Plant Biol. 2008;50:1187. doi: 10.1111/j.1744-7909.2008.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, et al. BMC Genomics. 2011;12:149. doi: 10.1186/1471-2164-12-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Wong WH. Proc Natl Acad Sci U S A. 2001;98:31. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diboun I, et al. BMC Genomics. 2006;7:252. doi: 10.1186/1471-2164-7-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Q, Wang XJ. Nucleic Acids Res. 2008;36:W358–363. doi: 10.1093/nar/gkn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadarmideen HN, et al. Bioinformation. 2012;8:855. doi: 10.6026/97320630008855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yip AM, S Horvath. BMC Bioinformatics. 2007;8:22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cline MS, et al. Nat Protoc. 2007;2:2366. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bader G, C Hogue. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris RT, et al. Bioinformatics. 2008;24:2915. doi: 10.1093/bioinformatics/btn537. [DOI] [PubMed] [Google Scholar]
- 20.Albert R, AL Barabasi. Phys Rev Lett. 2000;85:5234. doi: 10.1103/PhysRevLett.85.5234. [DOI] [PubMed] [Google Scholar]
- 21.Lenka SK, et al. Plant Biotechnol J. 2011;9:315. doi: 10.1111/j.1467-7652.2010.00560.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.