

Abstract
4-hydroxypanduratin A is a secondary metabolite of Boesenbergia pandurata Schult. (Fingerroot) plant with various pharmacological activities such as neuroprotective, potent antioxidant, antibacterial and antifungal. Flaviviral NS2B/NS3 protease activity is essential for polyprotein processing and viral replication for Japanese Encephalitis Virus (JEV), a major cause of Acute Encephaltis in Asia. Inhibition of formation of this complex by arresting the binding of NS2B with NS3 would reduce the enzyme's activity to meager proportions and hence would prevent further viral proliferation. The automated 3D structure of NS2B protein of the JEV GP78 was predicted based on the sequence-to-structure-to-function paradigm using I-TASSER and the function of NS2B protein was inferred by matching to other known proteins. The stereochemical quality of predicted structure was checked by PROCHECK. The antiviral activity of 4-hydroxypanduratin A against NS2B protein as a potential drug has been elucidated in this paper. Docking simulation analysis showed 4-hydroxypanduratin A as potential inhibitor of NS2B protein/cofactor which is necessary for NS3 protease activity. 220 derivatives of 4-hydroxypanduratin A were virtually screened with rigid criteria of Lipinski's rule of 5 using Autodock4.2. 4-hydroxypanduratin A was found interacting with target hydrophilic domain in NS2B protein by two Hbonds (Gly80 and Asp81) with active residues, several hydrophobic interactions, Log P value of 5.6, inhibition constant (Ki) of 51.07nM and lowest binding energy of -9.95Kcal/Mol. Hence, 4-hydroxypanduratin A targeted to Site 2 will have sufficient profound effect to inhibit protease activity to abrogate viral replication. It could be a promising potential drug candidate for JEV infections using NS2B Site 2 as a Drug target.
Keywords: NS2B/NS3 protease, Japanese Encephalitis Virus, Structure prediction, I-TASSER, Molecular Docking, 4- hydroxypanduratin A
Background
Japanese encephalitis (JE) is one of the major causes of Acute Encephalitis Syndrome in South and East Asian countries. The disease is caused by the Japanese Encephalitis Virus (JEV) which is a plus strand RNA virus belonging to Flaviviridae super family [1]. The Flavivirus genus includes over 70 pathogenic viruses such as Dengue virus (DENV), West Nile virus (WNV), and Yellow fever virus (YFV) etc. JEV strains can be distinguished into 5 distinct genotypes based on analysis of its envelope (E) gene sequences. Of these 5 genotypes Genotype III is most widely distributed in the Indian Subcontinent and among South-East Asian countries [2, 3].
The JE strain selected in this study is the North Indian isolate GP78 which belongs of Genotype III. The ~11kb viral genome of JEV is translated into a single polyprotein which is cleaved into 3 structural and 7 non structural proteins by both host and viral proteases [4–5]. The viral genome is organized into gene sequence as NH2-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4BNS5- COOH. Capsid (C), Membrane (M) and Envelope (E) are structural proteins whereas the non structural proteins are designated as NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5 [6–8]. The NS3 protease in complex with its cofactor NS2B cleaves the polyprotein at intergenic junctions containing dibasic amino acid motifs (Arg-Arg, Lys-Arg or Arg-Lys followed by Ser, Gly, or Ala) [9]. Sequence alignment and similarity studies reveal that the mechanism of polyprotein processing by NS2B/NS3 protease is conserved among Flaviviruses. A 40 residue central hydrophilic domain in Dengue NS2B is essential for the optimum activity of NS2B-NS3 complex. Furthermore, the hydrophobicity profiles of other Flaviviruses including JEV have significant sequence similarity with a central hydrophilic domain surrounded by hydrophobic regions [10].
Natural plant, animal and mineral products with therapeutic properties have been used since time immemorial as drugs for human treatment against various infectious diseases. About 252 drugs prescribed by WHO as basic and essential for humans; 11% are exclusively of plant origin and many synthetic drugs have natural plant precursors [11]. The derivatives of 4- hydroxypanduratin A used are natural plant secondary metabolites of Boesenbergia pandurata (Roxb.) Schltr. (Syn. Kaempferia pandurata Roxb.) (Fingerroot) which is a member of the Zingiberaceae family (ginger). It has been widely used as a medicinal plant, has been reported to possess pharmacological activities such as anti-inflammatory [12], anti-oxidative properties [13], neuroprotective, chemoprotective [14], antioxidant [15] and antimicrobial activity. The 4- hydroxypanduratin A (Figure 1B) has shown promising inhibitory activity against Dengue virus NS2B/NS3 protease [16–17]. Flaviviral NS2B/NS3 protease activity is essential for polyprotein processing and viral replication for Japanese Encephalitis Virus (JEV), a major cause of Acute Encephaltis in Asia. Inhibition of formation of this complex by arresting the binding of NS2B with NS3 would reduce the enzyme's activity to meager proportions and hence would prevent further viral proliferation. In this paper 4-hydroxypanduratin A and its 220 derivatives were docked on the central domain of NS2B consisting of about 40 hydrophilic residues was selected which also forms Site 2 to identify potential lead inhibitor that would prevent the binding of NS2B with NS3. Docking has been used to predict the interactions between ligand and receptor. Since the ligand can bind with the binding site on the receptor molecule in several possible orientations the goal of docking is to screen in favorable interactions against prohibitive ones [18].
Figure 1.
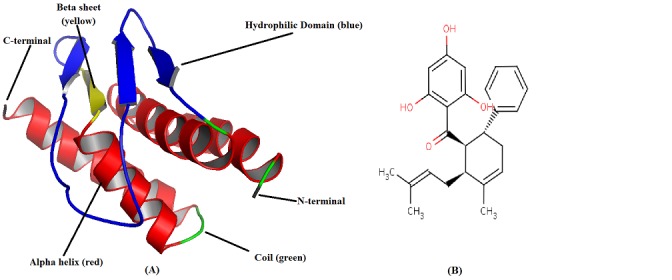
A) Schematic representation of α helices, β sheets and loops in predicted 3D model of NS2B by ab initio, threading and comparative modeling generated by UCSF Chimaera 1.6, B) Chemical structure of 4-hydroxypanduratin A [CID: 636530].
Methodology
Protein selection and structure prediction:
The 3D structure of NS2B protein/cofactor is not reported in the RCSB protein databank. Therefore, the amino acid sequence (128 residues) of NS2B GP78 (AAC27708) was retrieved from GenBank. The sequence was subjected to similarity search using BLASTp against PDB data base but no significant hit with complete query coverage for template to build 3D model of NS2B. However, the best alignment have been identified by using LALIGN in EMBOSS (Figure 2). Homology modeling was used to generate a reliable 3D model of NS2B protein (AAC27708) by MODELLER 9.10 [19]. A good quality model was not obtained even by using multiple templates so the model predicted by MODELLER was poor quality with inappropriate folded conformation. Hence, NS2B structure was predicted using I-TASSER integrated web platform which uses a composite approach for protein modeling combining ab initio, threading and comparative modeling [20]. In the first step, the query sequence was threaded through a non redundant sequence database to identify evolutionary relatives. A profile of homologous sequences was created to predict the secondary structure using PSIPRED [21]. The predicted secondary structure templates were ranked through LOMETS a meta threading server [22]. Templates were judged as per their Z score and top hits were considered for further evaluation.
Figure 2.

Pair-Wise Sequence Alignment using LALIGN in EMBOSS
In the second step, protein structure was built by assembling fragments from different templates while unaligned regions predicted by ab initio modeling [23–25]. To assemble fragments Replica exchange Monte Carlo simulations were performed at different temperatures and low temperature structural trajectories were selected and clustered by SPICKER [26].
In the third step, the 3D model was refined again by performing second round of simulations and the closest PDB structures retrieved by TM-align [27]. The cluster centroids from the first run provided external restraints to remove steric clashes to further refine the model. Finally, the lowest energy structures from each cluster were selected and all atom structural models from Cα traces were built. Accuracy of predicted structure was assessed by C-score [28] and TM-score [29]. NS2B 3D structure predicted by I-TASSER was further verified using PROCHECK.
Ligand selection and preparation:
4-hydroxypanduratin A and its 220 derivatives were retrieved from NCBI PubChem compound database (http://www.ncbi.nlm.nih.gov/pccompound/) and virtually screened on the basis of Lipinski's rule of 5 [30–31]. The ligands were converted into PDB coordinate files using OpenBabel software (www.openbabel.org/). Ligands were prepared by adding hydrogen bonds and neutralization of charged groups. The optimized lignads were subsequently docked against NS2B using Autodock4.2 (http://autodock.scripps.edu/wiki/AutoDock4.2) [32].
Molecular Docking:
Molecular Docking plays a critical role in computational drug design. Docking predicts the preferred orientation of a ligand with the binding site on a receptor. The strength of the interaction between ligand and receptor is measured in terms of experimentally defined inhibition constant Kd. The binding energy of the receptor-ligand interaction can be measured by
Equation 1: ΔGbind = ΔGcomplex- (ΔGligand-ΔGreceptor)
This relationship between ΔG and Kd is shown by Equation 2: ΔGbind = −RT ln Keq = −RT ln Kd
The energy minimized NS2B pdb file was generated by use of Swiss PDB viewer (www.SPDBV.vital-it.ch/). After energy minimization Kollman charges, polar hydrogen atoms and solvation parameters were added to NS2B structure. 3D grid maps for calculating atomic energy potentials for each atom type in the ligand molecule which surround the binding site on the receptor molecule was generated [33–34]. AutoGrid program available with AutoDock 4.2 was used to generate grid maps for the ligands. The grid map was created in such a way that the entire hydrophilic region of NS2B was covered. The box was set to 90Å×78Å×42Å with grid points separated by 0.375Å. Docking was performed in rigid state and Lamarckian genetic algorithm was used to find the most preferred pose where the ligand can bind to the receptor with lowest binding energy. The results of docking studies were visualized using LIGPLOT software (http://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/) and analyzed as per our previous study [35]. A complete drug target identification using molecular modeling and docking studies workflow is followed in this work and given in (Figure 3).
Figure 3.
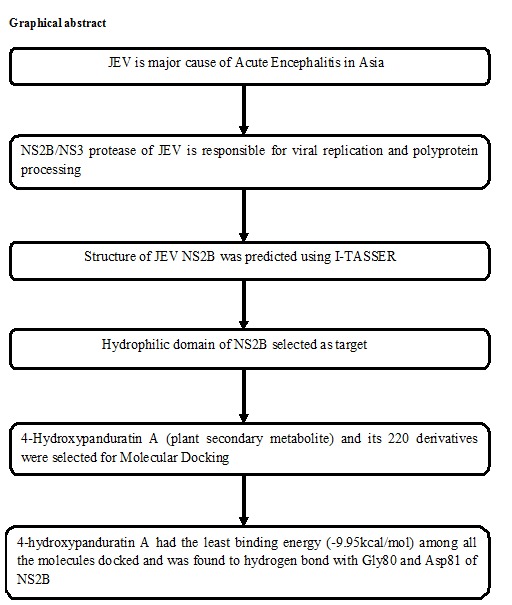
A workflow for complete drug target identification
Analysis and confirmation of Docking Results:
The search for the best ways is to fit ligand (4- hydroxypanduratin A), into NS2B structure, using Autodock4.2 resulted in docking files that contain details including records of docking. The obtained log files were read in ADT (Auto Dock Tool) and Python scripts in MGL tools package were used to analyze the docking results [34]. The similarity of docked structures was measured by computing the RMSD between the coordinates of the atoms and creating clustering of the conformations based on the RMSD values. The lowest binding energy conformations in all clusters were considered as the most favorable docking pose.
Results and Discussion
Structure prediction and validation:
All the information about a protein's biological function cannot be ascertained by mere knowledge of its primary sequence or the secondary structure. It is therefore, essential to know its tertiary structure. Additionally, the 3D structure of NS2B cofactor was not reported in RCSB PDB Data bank. BLASTp similarity search was performed against PDB data base but no significant results with complete query coverage were obtained. Even use of multiple templates could not cover the target protein completely to be modeled. However, the best alignments have been identified by using LALIGN inEMBOSS (Figure 2). 3D model of NS2B protein (AAC27708) was predicted by homology modeling using MODELLER 9.10 [20] with multiple templates. The obtained 3D structure was poor quality with inappropriate folded conformations. Therefore, the automated 3D structure of NS2B cofactor from JEV GP78 was predicted based on the sequence-to-structure-to-function paradigm using I-TASSER (Figure 1A) and the function of NS2B protein was inferred by structurally matching the 3D models with other known proteins [36]. The stereochemical quality of NS2B cofactor structure was checked by PROCHECK [37]. Backbone conformation by evaluation of Psi/Phi angles in Ramachandran plot predicts only two amino acids (Glu24 and Ser68) in disallowed geometry. Ramachandran plot gives 86.5% residues in most favored regions, 9% in additionally allowed regions, 2.7% in generously allowed regions and only 1.8% residues in disallowed regions. Thus, the predicted 3D structure by I-TASSER was of good quality with proper folded conformation.
Docking of 4-hydroxypanduratin A to NS2B:
The active conformation and the molecular alignment of each derivative of 4-hydroxypanduratin A were done using docking program Autodock4.2 into binding pockets of NS2B Site 2 (75- 87). Mutagenesis studies in West Nile Virus NS2B/NS3 protease revealed two regions in NS2B as essential for protease activity. Both of them were found conserved in other Flaviviruses including JEV. Site 1 (59-62) is a 4 residue long region and contain conserved residues Ile60/Val60 and Trp62 while Site 2 (75-87) is about 13 residue long and binds very close to the active site of NS3. This region is believed to be quite flexible and could be targeted by potential inhibitors [38]. The molecular alignment is done according to the electrostatic and structural properties of the active Site 2 of NS2B. The calculated binding energies, based on the docked structures, agree well with the experimentally observed inhibitory activities. Further, the steric, electrostatic and hydrophobic fields were mapped onto the active binding pocket of NS2B to better understand these interactions. Prediction of interactions between small molecules and proteins is a crucial step to decipher many biological processes and plays a critical role in drug discovery. The search algorithm tries to combine the ligand with the rigid protein molecule in several possible orientations. Each such orientation of the ligand within the active site region of the protein molecule is called a pose. The most favorable pose will have the lowest binding energy and therefore maximum stability.
In NS2B protein/cofactor, Site 1 (59–62) contains conserved residues Ile/Val60 and Trp62 which bind to adjacent pockets of NS3. This could be targeted by small aromatic, drug like compounds [38]. Additionally, the displacement of NS2B cofactor from this region is likely to prevent correct folding of the protease and hence lead to inactivation. However, Site 1 region of NS2B cofactor remains tightly associated in both inhibitor-bound and substrate/inhibitor-free crystal structures [39–40]. Therefore, it will be very difficult to develop compounds with high affinity to displace the bound cofactor. Hence, Site 2 in NS2B was targeted to find inhibitory effect of 4- hydroxypanduratin A. In NS2B, Site 2 (75–87) forms a β-loop that binds to a deep pocket in close proximity to active site residues. The Site 2 of NS2B cofactor is more flexible and not associated with the substrate-free protease [39] and hence was found highly accessible. Hence, 4-hydroxypanduratin A and 220 derivatives of 4-hydroxypanduratin A with highest fit value were subsequently analyzed for binding pattern to Site 2 in NS2B using Autodock methods. Among all ligand molecules 4- hydroxypanduratin A was found to be having highest fit value and was subsequently analyzed further. 4-hydroxypanduratin A was found tightly associated with targeted hydrophilic domain (44-84) and interacting with Site 2 in NS2B in the vicinity of Gly80, Ala111, Ile112, Ala105, Asp79, Ala115, Cys101, Tyr119, Leu104, Thr108 and Asp81. Ligplot and PyMol analysis revealed that 4-hydroxypanduratin A formed two hydrogen bonds with Gly80 and Asp81 (Figure 4A & Figure 4B) and 10 hydrophobic contacts with Asp79, Asp81, Cys101, Leu104, Ala105, Thr108, Ala111, Ile112, Ala115 and Tyr119 into NS2B Site 2 (Figure 4A).
Figure 4.
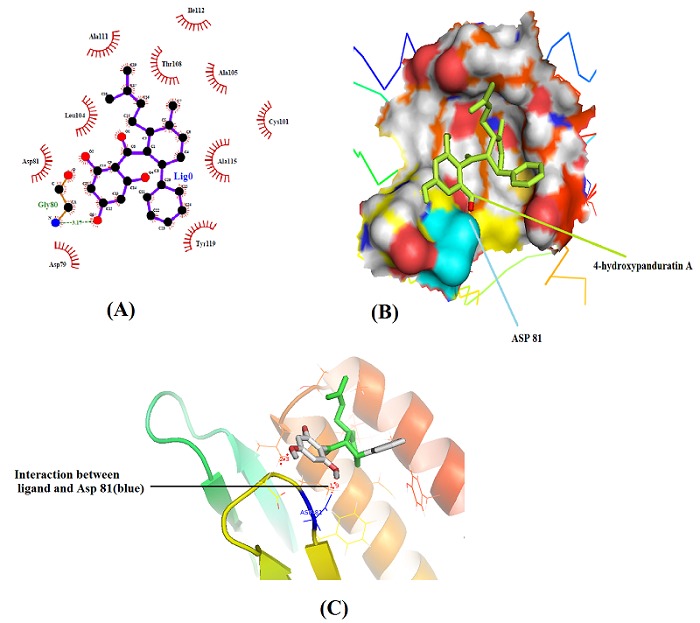
A) A schematic Ligplot of NS2B/4-hydroxypanduratin a complex, B) Binding pocket of NS2B with ligand (light green) bound with Asp81 (cyan) Image generated by Pymol and C) Hydrogen bond between ligand (white) and Aspartate 81 residue (yellow) Image generated by Pymol.
The free binding energy of NS2B/4-hydroxypanduratin A complex was found highest (ΔG = −9.95Kcal/Mol) with LogP value of 5.6 and inhibition constant (Ki) of 51.07nM Table 1 (see supplementary material). Due to the close proximity of Site 2 to the active site, where it forms part of the substrate-binding cleft, it is likely that displacement of Site 2 region will interfere with substrate binding [41]. The inhibitor 4-hydroxypanduratin A bind to NS2B cofactor in Site 2 region with Gly80 and Asp81 and in the vicinity of many hydrophobic contacts (Figure 4B & Figure 4C). Hence, 4-hydroxypanduratin A targeted to Site 2 will have sufficient profound effect to inhibit protease activity to abrogate viral replication. It could be a promising potential drug candidate for JEV infections using NS2B Site 2 as a Drug target.
Conclusion
The present study shows that the molecule 4- hydroxypanduratin A was found to bind with the NS2B cofactor of NS3 with least binding energy among the tested compounds. The free binding energy of NS2B/4- hydroxypanduratin A complex was found highest (ΔG = −9.95Kcal/Mol) with inhibition constant (Ki) of 51.07nM (Table 1). Due to the close proximity of Site 2 to the active site, where it forms part of the substrate binding cleft, it is likely that displacement of Site 2 region will interfere with substrate binding [41]. The inhibitor 4-hydroxypanduratin A bind to NS2B cofactor in Site 2 region with Gly80 and Asp81 and in the vicinity of many hydrophobic contacts (Figure 4A & Figure 4B). Hence, 4-hydroxypanduratin A targeted to Site 2 will have sufficient profound effect to inhibit protease activity to abrogate viral replication. It could be a promising potential drug candidate for JEV infections using NS2B Site 2 as a Drug target.
Supplementary material
Acknowledgments
Authors are grateful to the department of Biotechnology MITS Gwalior for providing computational facility and all the bioinformatics software developers who have developed such wonderful analysis tools.
Footnotes
Citation:Seniya et al, Bioinformation 9(1): 054-060 (2013)
References
- 1.Mancini EJ, et al. Protein Sci. 2007;16:2294. doi: 10.1110/ps.072843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solomon T, et al. J Virol. 2003;77:3091. doi: 10.1128/JVI.77.5.3091-3098.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackenzie JS, et al. Nat Med. 2004;10:S98. doi: 10.1038/nm1144. [DOI] [PubMed] [Google Scholar]
- 4.Vrati S, et al. J Gen Virol. 1999;80:1665. doi: 10.1099/0022-1317-80-7-1665. [DOI] [PubMed] [Google Scholar]
- 5.Chambers TJ, et al. Annu Rev Microbiol. 1990;44:649. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 6.Markoff L. J Virol. 1989;63:3345. doi: 10.1128/jvi.63.8.3345-3352.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowak T, et al. Virology. 1989;169:365. doi: 10.1016/0042-6822(89)90162-1. [DOI] [PubMed] [Google Scholar]
- 8.Rice C, et al. Science. 1985;229:726. doi: 10.1126/science.4023707. [DOI] [PubMed] [Google Scholar]
- 9.Jan LR, et al. J Gen Virol. 1995;76:573. doi: 10.1099/0022-1317-76-3-573. [DOI] [PubMed] [Google Scholar]
- 10.Falgout B, et al. J Virol. 1993;67:2034. doi: 10.1128/jvi.67.4.2034-2042.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rates SM. Toxicon. 2001;39:603. doi: 10.1016/s0041-0101(00)00154-9. [DOI] [PubMed] [Google Scholar]
- 12.Yun JM, et al. Planta Med. 2003;69:1102. doi: 10.1055/s-2003-45190. [DOI] [PubMed] [Google Scholar]
- 13.Tuchinda P, et al. Phytochemistry. 2002;59:169. doi: 10.1016/s0031-9422(01)00451-4. [DOI] [PubMed] [Google Scholar]
- 14.Fahey JW, et al. J Agric Food Chem. 2002;50:7472. doi: 10.1021/jf025692k. [DOI] [PubMed] [Google Scholar]
- 15.Shindo K, et al. Biosci Biotechnol Biochem. 2006;70:2281. doi: 10.1271/bbb.60086. [DOI] [PubMed] [Google Scholar]
- 16.Tan SK, et al. Bioorg Med Chem Lett. 2006;16:3337. doi: 10.1016/j.bmcl.2005.12.075. [DOI] [PubMed] [Google Scholar]
- 17.Frimayanti N, et al. Int J Mol Sci. 2011;12:1089. doi: 10.3390/ijms12021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautam B, et al. Bioinformation. 2012;8:134. doi: 10.6026/97320630008134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sali A, et al. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones DT. J Mol Biol. 1999;292:195. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 22.Wu S, et al. Nucleic Acids Res. 2007;35:3375. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu S, et al. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, et al. Biophys J. 2003;85:1145. doi: 10.1016/S0006-3495(03)74551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu D, et al. Proteins. 2012;80:1715. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. J Comput Chem. 2004;25:865. doi: 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, et al. Nucleic acids research. 2005;33:2302. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, et al. Proc Natl Acad Sci U S A. 2004;101:7594. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. Proteins. 2007;68:1020. [Google Scholar]
- 30.Lipinski CA, et al. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 31.Lipinski CA, et al. Adv Drug Deliv Rev. 2001 [Google Scholar]
- 32.Tambunan USF, S Alamudi. Bioinformation. 2010;5:250. doi: 10.6026/97320630005250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forli S, et al. J Chem Inf Model. 2007;47:1481. doi: 10.1021/ci700036j. [DOI] [PubMed] [Google Scholar]
- 34.Morris GM, et al. J Comput Chem. 2009;16:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seniya C, et al. Bioinformation. 2012;8:678. doi: 10.6026/97320630008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy A, et al. Nat Protoc. 2010;54:725. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laskowski RA, et al. J Biomol NMR. 1996;8:477. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 38.Chappell KJ, et al. J Gen Virol. 2008;89:1010. doi: 10.1099/vir.0.83447-0. [DOI] [PubMed] [Google Scholar]
- 39.Aleshin AE, et al. Protein Sci. 2007;16:795. doi: 10.1110/ps.072753207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erbel P, et al. Nat Struct Mol Biol. 2006;13:372. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- 41.Melino S, et al. FEBS J. 2006;273:3650. doi: 10.1111/j.1742-4658.2006.05369.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.