

Abstract
Obstructed transport of biological molecules can result in improper release of pharmaceuticals or biologics from biomedical devices. Recent studies have shown that nonionic surfactants, such as Pluronic® F68 (F68), positively alter biomaterial properties, such as mesh size and microcapsule diameter. To further understand the effect of F68 (incorporated at concentrations well above the critical micelle concentration (CMC)) in traditional biomaterials, the transport properties of BSA and riboflavin were investigated in F68-alginate composite hydrogels. Results indicate that small molecule transport (represented by riboflavin) was not significantly hindered by F68 in homogeneously crosslinked hydrogels (up to an 11% decrease in loading capacity and 14% increase in effective diffusion coefficient, Deff), while protein transport in homogeneously crosslinked hydrogels (represented by BSA) was significantly affected (up to a 43% decrease in loading capacity and 40% increase in Deff). For inhomogeneously crosslinked hydrogels (CaCl2 or BaCl2 gelation), the Deff increased up to 50% and 83% for small molecule and proteins, respectively. Variation in the alginate gelation method was shown to affect transport through measurable changes in swelling ratio (30% decrease) and observable changes in crosslinking structure as well as up to a 3.6 and 11.8-fold difference in Deff for riboflavin and BSA, respectively. The change in protein transport properties is a product of mesh size restrictions (10–25 nm estimated by mechanical properties) and BSA-F68 interaction (DLS). Taken as a whole, these results show that incorporation of a nonionic surfactant at concentrations above the CMC can affect device functionality by impeding the transport of large biological molecules.
Keywords: Protein delivery, Hydrogels, Nonionic Surfactants, Alginate, Diffusion
1 Introduction
The effect of nonionic surfactants on protein and small molecule transport within hydrogel matrices is not fully understood despite their widespread use in the design and formulation of drug delivery systems, biomaterials, and tissue engineering scaffolds [1–5], and the known interaction of many surfactants with proteins and small molecules in solution [6–9]. Many investigators have explored the use of surfactants to improve material properties such as pore size, water retention, tensile strength, and hydrogel or fiber uniformity, without accounting for the effect of the surfactant on protein secretion or cellular signaling. For example, in electrospun-scaffold generation for tissue engineering, the addition of a surfactant to a polymer solution can reduce the surface tension and increase the conductivity, improving fiber uniformity and reducing bead formation [5]. Pluronic® F127, a nonionic tri-block copolymer, has been shown to bind poly(ε-caprolactone) fibers at cross-points, maintaining fiber diameter and pore area while improving mechanical properties such as shrinkage, ultimate tensile strength, and burst pressure [10]. The formation of sol-gel bioactive glasses or silica-gelatin hybrid scaffolds for bone regeneration uses nonionic or anionic surfactants at low volume percentages to reduce surface tension and stabilize bubble formation during the foaming process prior to gelation [3]. The pore size within the silica-gelatin hybrid scaffold can be tailored by varying the surfactant concentration [3]. Also, addition of surfactants to polymer solutions for encapsulation reduces microcapsule size and affects porosity, which are important parameters influencing delivery of cell secreted proteins and metabolites in therapeutic devices [4, 11, 12].
Choice of surfactant is important for biocompatibility and specific device improvement. Formulations of the nonionic family of thermo-reversible PEO-PPO-PEO (poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide)) surfactants were developed in the 1980’s for the delivery of anticancer agents [13]. PEO-PPO-PEO nonionic surfactants, such as Pluronics® F68, F108, and F127, are commonly employed in drug delivery systems for pharmaceuticals and proteins [2, 9, 14–17], as injectable gels for drug administration [1, 18, 19], as additives for tissue engineering [4, 11, 20], as vehicles for gene therapy [21], and as cellular membrane stabilizing agents [22]. At concentrations below the critical micelle concentration (CMC for F68 = 0.04 mM or 0.0336% w/v), F68 has been shown to reduce the uptake and release of growth factors and signaling molecules [23, 24]. Above the CMC, fundamental studies evaluating the effect of surfactant-mediated transport limitations in hydrogels have not been reported. Above the CMC, micelles form, which can significantly alter material properties and transport in a hydrogel. Studies on the effect of surfactant addition on hydrogel mechanical properties show various results, which are dependent upon the surfactant type, surfactant concentration, operating temperature, and inherent hydrogel matrix properties [20]. Composite solutions of biopolymers and surfactants are of significant importance as mechanical and transport properties can be precisely tuned with appropriate combinations. Effective transport within both tissue engineering constructs and drug delivery vehicles is critical to the success of such devices [12, 25–27] and a thorough understanding of the effect of scaffold additives, such as nonionic surfactants, is necessary for optimal design. In this study, incorporation of Pluronic® F68 (F68) in an alginate hydrogel is used to understand transport of biological molecules within a surfactant-biopolymer composite hydrogel network.
Due to their tunable degradation kinetics and mechanical properties as well as intrinsic biocompatibility, alginate hydrogels are one of the major scaffold platforms used in tissue engineering [28–31] and drug delivery [32, 33]. Alginate is a natural biopolymer composed of randomly distributed units of 1,4-linked β-mannuronic (M) and α-guluronic acid (G) residues. Alginate gelation occurs when divalent cations such as Ca2+ or Ba2+ ionically interact with COO− and −OH groups in G residues from differing chains [34], creating a three-dimensional matrix described by the egg-box model [35, 36]. Two common methods used to crosslink alginate include an inhomogeneous method using CaCl2 or BaCl2 [37] and a homogeneous method utilizing in situ release of calcium ions from pH-sensitive CaEDTA decomposition [38]. The effect of Pluronic® addition on rheological properties has been explored to enhance the understanding of nonionic surfactant addition to alginate matrix properties [20]. Transport in alginate matrices, as well as other hydrogel formulations, is dependent upon the gelation method and crosslinking density, alginate or polymer concentration, and pore size of the resulting matrix [25, 37, 39, 40].
To fully understand the implication of surfactant additives on device performance, transport of proteins and metabolites in the presence of surfactant must be evaluated. Such studies will impact important applications such as the dispersion of perfluorocarbon droplets [11, 41] for enhanced oxygen delivery to encapsulated islet cells for the treatment of type 1 diabetes [12, 42] or to seeded stem cells to enhance differentiation [43], the encapsulation of biologicals for drug delivery [44], and the mechanical performance of surfactant-incorporated biomaterials and tissue engineering scaffolds [1, 3–5]. Recent research in our laboratory has focused on the use of surfactants at concentrations above the CMC in biomaterials [4, 11, 41, 45] and here, we expand this work to investigate the influence of surfactant (i.e., F68) on both the transport of biological molecules and the effect on hydrogel (i.e., alginate) mechanical properties.
2 Materials and Methods
Cell culture grade sodium alginate (lot#1353824, %G ≥ 60, Mv~240 kDa determined from intrinsic viscosity in 0.1 M sodium chloride [40]) was obtained from Sigma Aldrich (St. Louis, MO). Polymer solutions were made with water from a NanoPure Water System (nanopure water) purified to 18 mΩ/cm. Bovine serum albumin (BSA), HEPES buffer, D(+)-glucose, sodium pyruvate, sodium bicarbonate, L-glutamine, riboflavin, lysozyme, and sodium citrate were obtained from Sigma Aldrich. BCA Protein Assay kits were obtained from Pierce (New York, NY). Dulbecco’s Modified Eagle’s Medium (DMEM), Roswell Park Memorial Institute medium (RPMI 1640), and trypsin/EDTA were obtained from Mediatech, Inc. (Manassas, VA). Heat-inactivated fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Atlanta, GA). NIH 3T3 fibroblasts and RIN-m5F rat insulinoma cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). Adult human epidermal keratinocytes (HEKa), EpiLife®, and EpiLife® defined growth supplement (EDGS) were obtained from Invitrogen (Carlsbad, CA). All additional materials were obtained from Thermo Fisher Scientific (Waltham, MA) unless otherwise noted.
2.1 Preparing alginate hydrogels
2.1.1 Alginate solutions
Sodium alginate solutions were prepared using nanopure water at 1% weight by volume (w/v) with the addition of 0.1% glucose and 10 mM HEPES buffer and stirred for 24 hours at room temperature. Pluronic® F68 was added to the alginate solution and stirred for an additional 24 hours at room temperature.
2.1.2 CaEDTA-GDL-alginate cylinders
Homogeneously crosslinked alginate hydrogels were made by an internal gelation technique described elsewhere [38, 46]. Briefly, 50 mM CaEDTA and 50 mM glucono-δ-lactone (GDL) were mixed with alginate solution and aliquoted into 6- or 12-well non-treated tissue culture plates. Following 12 hours of initial crosslinking, hydrogels were soaked in 100 mM calcium chloride (CaCl2) in a humidified chamber at 25°C (≥90% humidity) for an additional 48 hours. For gels containing surfactant, the same concentration of surfactant was added to the 100 mM CaCl2 to prevent surfactant leaching. Final hydrogel dimensions were measured using calipers.
The mass of water in the hydrogels was calculated by dividing the difference in mass of a swollen hydrogel (mg,s) and mass of dry polymer (mp) in the hydrogel by the mass of dry polymer, (Equation 1). The volume change of alginate during the gelation process is reported as percent shrinkage. Equation 2 calculates the change in the amount of polymer solution used to create gels, Vsol, and the relaxed volume of the resultant hydrogel, Vg,r. The relaxed and swollen volumes of a hydrogel were calculated geometrically using calipers.
| (Equation 2) |
2.1.3 CaCl2 and BaCl2 alginate cylinders
To form hydrogel cylinders by passive diffusion crosslinking, alginate solution was poured into 10 mm dialysis tubing with a molecular weight cut-off of 3–5 kDa and submerged in either 1 M CaCl2 or 0.5 M barium chloride (BaCl2) with a specified F68 concentration for 48 hours. The lower concentration of BaCl2 was used because similar mechanical properties were expected using this concentration. The long cylinder was cut into individual gel cylinders with a thickness of 5 mm using a knife. Final hydrogel dimensions were measured using calipers. Samples were lyophilized for 36 hours in a Labconco FreeZone 6 freeze dry system (Kansas City, MO).
2.2 Protein and small molecule transport
BSA was prepared by dissolution in nanopure water at 25 mg/mL. Riboflavin was dissolved in nanopure water at a concentration of 75 µg/mL.
2.2.1 Loading
Protein and small molecules can be loaded into hydrogels using two methods: in situ loading and imbibing. Riboflavin transport was explored using both methods. In situ loading required the addition of solid riboflavin to both the alginate solution and crosslinking bath at a concentration of 75 µg/mL. For addition by imbibition, fully formed hydrogels were soaked in a solution of 75 µg/mL riboflavin for 24–48 hours in 6- or 12-well non-tissue culture plates in a humidified incubator (≥90% humidity) at 25°C. For experiments with BSA, the same procedure was utilized to imbibe the hydrogels, using a 25 mg/mL BSA solution. In situ loading of BSA was not performed due to the electrostatic interaction between alginate and BSA in solution [47].
2.2.2 Release and quantification
Protein release was monitored similarly to traditional studies which examine drug release from hydrogels [39]. Briefly, following a predetermined time course, the hydrogels were moved among wells of a 6- or 12-well plate containing fresh nanopure water to maintain ambient sink conditions. After 120 minutes, the hydrogels were soaked in 5 mL of 100 mM sodium citrate to dissolve the alginate for complete mass balance analysis. Six replicates were run for each condition to allow for statistical validation. BSA concentration was quantified using a BCA Protein Assay kit by following the provided protocol which measures the color change of bicinchoninic acid due to copper reduction at 562 nm on a µQuant spectrophotometer (BioTek Instruments, Inc., Richmond, VA). The riboflavin concentration was quantified by analyzing 200 µL of sample using a µQuant spectrophotometer at 370 nm and comparing to a standard riboflavin curve. All samples were quantified in triplicate.
Effective diffusion coefficients were calculated from release kinetics analyzed with a three-dimensional diffusion model for polymeric tablets developed by Fu and co-workers [48]. Similar studies have effectively applied this model to calculate diffusion coefficients [40, 49]. Hydrogel disks were geometrically measured using calipers and the average radii and thicknesses were utilized in the model [48], as shown in Equation 3.
| (Equation 3) |
In Equation 3, h is the hydrogel half thickness, r is the radius of the hydrogel, Deff is the effective diffusion coefficient, t is time, Mt is the amount of protein released at time t, M∞ is the amount of protein released at infinite time, and j and k are integers. αj is defined as the roots of the zero-order Bessel function, (Equation 4). βk is defined as (Equation 5). The summations in Equation 3 were evaluated for j=1,2,…,15, and k=0,1,…,150. Deff was determined by minimizing the sum of an objective function, (Equation 6), where represents the theoretical release fraction at time t, and the represents the experimental release fraction at the same time t. Figure 1 is a representative experimental release profile fit with the diffusion model in Equation 3 using the calculated effective diffusion coefficient determined from the objective function minimization described above.
Figure 1.

Representative experimental data with model fit. (■) represents experimental data and (-) represents the model fit for release of riboflavin from a 1% alginate hydrogel containing 1% F68 formed by CaEDTA/GDL gelation.
2.3 Solution viscosity
Alginate solution viscosity was measured using a TA Instruments AR-G2 stress-controlled rheometer (New Castle, DE). Samples were injected into a Couette geometry and were maintained at 25 ± 0.1°C by the built-in Peltier control. Dynamic viscosity measurements were performed using steady-state flow with shear rates ranging from 0.1 to 100 s−1.
2.4 Hydrogel mechanical properties
Alginate hydrogel mechanical properties were measured using a TA Instruments AR2000 stress-controlled rheometer (New Castle, DE). Samples were prepared using the CaEDTA/GDL method (section 2.1.2) in 100 mm Petri dishes. Acrylic parallel plates (60 mm) were used and gels were maintained at 25 ± 0.1°C by the Peltier control. Storage (G’) and loss (G”) moduli were measured using oscillation frequency sweeps covering frequencies from 0.1 to 10 Hz while maintaining an oscillation stress of 1 Pa to remain in the linear viscoelastic region of the hydrogel. The linear viscoelastic region was determined using an oscillation stress sweep at 0.1, 1, and 10 Hz. Batches of six gels were made in triplicate, and each gel was run once on the rheometer.
Hydrogel mesh size, ξR, was estimated from rheological measurements. Equation 7 estimates ξR for F68-containing alginate hydrogels based on the measured shear modulus [50].
| (Equation 7) |
The low-frequency shear modulus, G0, was approximated as the complex shear modulus, G*, measured at 0.1 Hz where (Equation 8). Temperature, T, is 298 K for all estimations and the Boltzmann’s constant, k, is 1.38×10−24 J/K.
2.5 Hydrogel swelling
Hydrogel swelling studies were performed as previously reported [51]. Hydrogel volume in the relaxed state, Vg,r, was defined as the volume of an alginate hydrogel immediately after soaking for 24 hours in 100 mM CaCl2 (or 50 mM BaCl2) solution (derived from [51]). Hydrogel volume in the swollen state, Vg,s, was defined as the volume of an alginate hydrogel after soaking for 24 hours in nanopure water (derived from [51]). Corresponding polymer volume ratios can be calculated as (Equation 9) and (Equation 10), respectively, where Vp is the volume of dry polymer [51].
Based on a method described by Peppas and colleagues [51], the mesh size, ξS, of homogeneously crosslinked hydrogels was estimated using data from swelling experiments. ξS was calculated based on the molecular weight between crosslinks, MC, and end-to-end distance of alginate chains in an unperturbed state, (r20)1/2. MC was calculated from Equation 11. The Flory interaction parameter for alginate was taken as χ=0 .738 [52]. V1 is the molar volume of the solvent (water), v̄ is the specific volume of alginate (0.48 mL/g [53]) and Mn is the number average molecular weight of alginate (estimated by the viscosity average molecular weight [53], not shown).
| (Equation 11) |
| (Equation 12) |
| (Equation 13) |
Using Equation 12, is calculated from MC and the characteristic ratio of the alginate chain, Cn, was taken as 24 [40]. The length of a C-C bond (l = 1.54 Å) and the molecular weight of the alginate repeating unit (Mr =198 g/mole) were used to determine in Equation 12. The mesh size, ξS, was calculated using Equation 13.
2.6 Particle size analysis
Dynamic light scattering was performed using a Brookhaven Instrument Corporation ZetaPlus Particle Size Analyzer (Holtsville, NY) to determine the effect increasing F68 concentration has on the effective diameter of aqueous BSA and lysozyme. Initially, F68 was dissolved in 10 mM sodium chloride at a pH 7.4. BSA was added to the F68 solution at a concentration of 10 mg/mL. To investigate the interaction between F68 and alginate in solution, F68 was added to a 0.5% alginate solution in nanopure water. A 90° angle and a wavelength of 660 nm were used. Size distributions were analyzed using the Brookhaven Instrument Corporation Particle Analyzing software. Effective diameter and the corresponding standard deviations of the analyses are reported.
2.7 Cell encapsulation
Analysis of viability within 1% alginate hydrogels containing 1% F68 was performed through encapsulation of two cell types (primary human keratinocytes, HEKa, and rat insulinomas, RIN-m5f) by coaxial air flow following a previously described protocol [11, 41] using sterile alginate, Pronova SLG100 (NovaMatrix, Sandvika, Norway). Briefly, cell pellets were mixed with alginate solution to resuspend cells at 1×105 cells/mL alginate solution. The mixture was pumped using a peristaltic pump (Cole Parmer, Niles, IL) through a syringe needle (23-gauge for HEKa, 30-gauge for RIN-m5f) fitted with 1/8” luer-lock glass tubing inserted into an air chamber with an air inlet on one side to maintain coaxial air flow. Alginate microcapsules were formed at the tip of the needle and were extruded into a non-tissue culture 6-well plate filled with 50 mM BaCl2, where they were allowed to crosslink for 30 minutes. Capsules were maintained in cell culture medium in 6-well plates on a rocker (Stovall Life Sciences, Greensboro, NC) in a humidified incubator at 37°C and 5% CO2. For the HEKa cells, EpiLife® was supplemented with 1.5 g/L sodium bicarbonate, 1X EDGS, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% FBS. For the RIN-m5f cells, RPMI 1640 was supplemented with 292 mg/L L-glutamine, 10 mM HEPES buffer, 3 g/L sodium bicarbonate, U/mL penicillin, 100 µg/mL streptomycin, and 10% FBS.
2.8 Cell viability
2.8.1 Encapsulation
Cell viability within the encapsulation matrix was investigated using a LIVE/DEAD assay. Encapsulated cells were stained for 10 minutes using acridine orange (LIVE stain, 120 µg/mL cell culture media) and propidium iodide (DEAD stain, 30 µg/mL cell culture media). Capsules were rinsed 5 times in 8 mL of cell culture medium for 2 minutes each prior to imaging to minimize background. Fluorescence imaging was performed on an Olympus IX71 Inverted Epi-fluorescence Microscope (Center Valley, PA, USA) equipped with a Ludl Motorized Z-Stage (Hawthorne, NY, USA), a monochromatic Hamamatsu cooled-CCD Digital Camera (Hamamatsu City, Japan), and a CRI MicroColor trichromatic filter (Woburn, MA, USA). Images were analyzed using ImageJ [54] and are presented as an overlay of both LIVE and DEAD images.
2.8.2 Cell culture
The effect of F68 on cell viability was analyzed by the addition of F68 to cells in traditional culture medium. Cell culture growth and viability were monitored over a 9-day period. NIH 3T3 fibroblasts were obtained from ATCC and grown in tissue culture-treated flasks as directed in the specifications for this cell line. Briefly, cells were maintained using DMEM supplemented with 110 mg/L sodium pyruvate, 3.7 g/L sodium bicarbonate, and 584 mg/L L-glutamine, and 10% FBS. Cells were subcultured every 6 days into fresh medium for maintenance. To investigate the effect of F68 on growth and viability, 2% and 5% w/v F68 was added to the cell culture medium. All biological media solutions were filtered under sterile conditions using a 0.22 µm polyethylene terephthalate (PET) filter (Millipore, Billerica, MA) after F68 addition. Cells were passaged into 12-well tissue-culture treated plates at 1×103 cells/cm2. After two days, media was replaced with F68-containing media. Media containing F68 was changed every two days throughout the experiment. To determine growth and viability, three wells of the 12-well plate were trypsinized every two days and were counted on a hemacytometer using Trypan blue exclusion to determine the total number of cells in each well along with percentage viability. Experiments were run in triplicate. Relative growth rate was determined by subtracting the number of live cells on day 0 from the number of total live cells on the sample day and dividing the result by the number of live cells on day 0.
2.9 Statistical analysis
All samples were analyzed in triplicate unless stated otherwise. Rheological properties at each independent variable, sample loading capacity, and sample release were analyzed for significance using a two-sample equal variance Student’s t-test. All reported values are mean ± standard deviation. Diffusion coefficients were analyzed using a two-sample equal variance Student’s t-test comparison of samples one standard deviation above and below the average release rate for each sample type. Differences were considered statistically significant for p ≤ 0.05 for all tests.
3 Results and Discussion
Transport of biological molecules within nonionic surfactant-containing hydrogels was evaluated using a Pluronic® F68 (F68)-alginate system. The addition of F68 above the CMC allowed for incorporation of nonionic surfactant micelles in the hydrogel which were capable of interacting with imbibed proteins and small molecules. The bulk structure of a hydrogel defines its transport characteristics and biofunctionality. Important parameters for characterizing hydrogel matrices include polymer volume fraction in the swollen state (ν2,s), molecular weight of the polymer chain between two neighboring crosslinking junctions (Mc), and mesh size (ξ) [55]. The effects of F68 micelle incorporation and crosslinking method on the transport properties of the alginate hydrogel were analyzed using these parameters along with release kinetics (Mt/M∞) and calculated effective diffusion coefficients (Deff).
Gelation method influences transport of both small and large molecules by altering the internal structure of the hydrogel, mainly through variations in mesh size [56]. Homogeneously crosslinked alginate hydrogels are achieved by utilizing the decomplexation of CaEDTA using GDL to slowly reduce the pH of the solution, thus releasing the calcium ions [38]. Inhomogeneously crosslinked alginate hydrogels are formed by passive diffusion of Ca2+ or Ba2+ by simply exposing alginate solution to CaCl2 or BaCl2 [37]; however, this method does not allow for homogeneous dispersion of cations throughout the matrix [57], creating variations in crosslinking density which can be observed by examining the cross-section of a hydrogel. Inhomogeneously crosslinked hydrogels have a lower protein or small molecule loading capacity compared to homogeneously crosslinked hydrogels, due to decreased swelling ratios and decreased final hydrogel volume (or increased matrix shrinkage upon gelation), which therefore decreases the amount of protein which can be imbibed. Hydrogels created by crosslinking with BaCl2 retain the least amount of water and BaCl2 has been shown to be a more robust crosslinking method compared to CaCl2, yielding stronger and tighter hydrogel structures for lower cation concentrations [37]. Therefore, the effect of gelation parameters, such as cation concentration and cation delivery (passive diffusion or CaEDTA degradation), should be considered when designing alginate hydrogels for tissue engineering applications.
The effect of surfactant and gelation method on release of riboflavin, a small metabolite (hydrodynamic radius of 0.58 nm [58]), and BSA, a large, multi-domain protein (hydrodynamic radius of 4.5 nm [59]) was investigated. Riboflavin was chosen because it is a small hydrophobic molecule, whose transport parameters mimic those of other relevant small metabolites such as glucose, vitamins, or small-molecule therapeutics. BSA was chosen as a model protein containing multiple tertiary domains including charged, hydrophobic, and globular. Because of its tertiary structural characteristics, BSA closely resembles the structure of biological therapeutics, such as human serum albumin (HSA), and the results of these transport studies are therefore applicable to the delivery and release of such proteins [60]. Results below summarize and discuss the influence of F68 on the transport of both riboflavin and BSA in alginate hydrogels.
3.1 Effect of F68 and loading method on small molecule transport
For transport of small molecules such as riboflavin, no statistical difference related to F68 incorporation was observed for Deff for homogeneously crosslinked alginate hydrogels (CaEDTA/GDL method). Conversely, an increase (up to 23% for CaCl2 and 50% for BaCl2) in Deff for riboflavin was observed for inhomogeneously crosslinked hydrogels (CaCl2 and BaCl2), which is likely due to the wide variation in mesh size caused by the inhomogeneities in the hydrogel structure, observed in Figure 2a. Arrows in Figure 2a indicate changes in optical properties across a cross-section of a 1% alginate hydrogel formed with 1 M CaCl2. Figure 2b shows the change in swelling ratio or water uptake properties for 1% alginate hydrogels formed using various formulations and gelation strategies. These data help explain the 12–24% decrease in the riboflavin loading capacity observed across gelation methods. The release percentage and loading capacity of riboflavin were largely unaffected by the addition of 1 or 2% F68 to an alginate hydrogel regardless of the gelation method. Based on these results, we can conclude that small molecule transport within hydrogels is improved slightly by the addition of F68, as evidenced by the increase in Deff for inhomogeneously crosslinked hydrogels. Use of a higher surfactant concentration or larger molecular weight surfactant, such as Pluronic® F108 or F127, is expected to continue to enhance Deff and improve transport characteristics, as long as the material properties of the hydrogel are not strongly affected by the change in formulation.
Figure 2.


Effect of gelation method on crosslinking density and water uptake. A) Arrows depict changes in optical properties in the radial direction of a cross-section of 1% alginate hydrogel which indicate variations in crosslinking density within the hydrogel (1 M CaCl2). B) Water uptake for alginate hydrogels formed using (□) CaEDTA/GDL, (○) 1 M CaCl2, and (Δ) 0.5 M BaCl2. Addition of F68 has less of an effect than the gelation method on water uptake properties for an alginate hydrogel. Error bars represent standard deviation (p≤0.05, n=5).
Riboflavin was loaded into the homogeneously crosslinked hydrogels by two methods: 1) in situ addition by adding solid riboflavin to the alginate solution prior to crosslinking and 2) imbibition of the riboflavin into a fully crosslinked hydrogel. The in situ method allowed for incorporation of significantly more riboflavin per gram of alginate in the hydrogel (up to a 44% increase), as seen in the comparison of the loading capacities for riboflavin in Table 1. Adding the small molecule to the alginate solution prior to gelation increases the loading capacity without altering the release profile or Deff (Table 1). This is particularly relevant for the delivery of therapeutics. For instance, the creation of alginate microcapsules for the steady release of angiogenic factors has been investigated [61]. Our data support the creation of these capsules by first adding a growth factor, such as VEGF, to the alginate solution and then extruding the capsules into a crosslinking solution rather than forming capsules and imbibing the growth factor later in the process. For the same volume of capsules, more growth factor or protein can be loaded and hence delivered.
Table 1.
Transport properties of riboflavin and BSA in alginate-F68 composite hydrogels.
| Loading Capacity g solute/g dry polymer |
Release % Mt/M∞ (2 hrs) |
Deff (×10−10) m2/s |
Obj. Fxn |
|
|---|---|---|---|---|
| Riboflavin | ||||
| CaEDTA/GDL in situ loading | ||||
| 0% F68 | 72.7 ± 1.7 | 97.9 ± 1.1 | 2.2 ± 0.7 | 0.2 |
| 1% F68 | 64.4 ± 2.9* | 97.7 ± 1.6 | 2.1 ± 0.2 | 0.1 |
| 2% F68 | 64.4 ± 7.2 | 97.4 ± 1.3 | 2.5 ± 0.7 | 0.1 |
| CaEDTA/GDL | ||||
| 0% F68 | 50.4 ± 2.5 | 99.2 ± 1.3 | 1.7 ± 0.1 | 0.1 |
| 1% F68 | 44.8 ± 3.6 | 99.4 ± 0.7 | 1.9 ± 0.2 | 0.2 |
| 2% F68 | 51.9 ± 3.3 | 96.7 ± 0.8* | 1.9 ± 0.2 | 0.1 |
| CaCl2 | ||||
| 0% F68 | 38.6 ± 8.1 | 85.9 ± 7.1 | 6.1 ± 0.1 | 0.3 |
| 1% F68 | 28.8 ± 2.5 | 83.8 ± 2.9 | 7.0 ± 0.3* | 0.1 |
| 2% F68 | 39.5 ± 2.1 | 79.9 ± 2.4 | 7.5 ± 0.4* | 0.1 |
| BaCl2 | ||||
| 0% F68 | 44.1 ± 7.4 | 91.0 ± 3.1 | 5.0 ± 0.8 | 0.3 |
| 1% F68 | 37.7 ± 1.5 | 89.2 ± 4.1 | 6.0 ± 0.5 | 0.1 |
| 2% F68 | 27.6 ± 1.9* | 87.6 ± 1.2 | 7.5 ± 0.5* | 0.1 |
|
Bovine Serum Albumin (BSA) | ||||
| CaEDTA/GDL | ||||
| 0% F68 | 50.9 ± 3.9 | 95.3 ± 3.4 | 0.5 ± 0.1 | 0.2 |
| 1% F68 | 35.1 ± 3.7* | 87.7 ± 6.2 | 0.6 ± 0.1 | 0.0 |
| 2% F68 | 28.9 ± 1.2* | 82.2 ± 3.9* | 0.7 ± 0.1 | 0.0 |
| CaCl2 | ||||
| 0% F68 | 39.5 ± 1.9 | 68.4 ± 2.3 | 3.6 ± 0.6 | 0.4 |
| 1% F68 | 22.4 ± 1.7* | 78.7 ± 4.4* | 4.6 ± 0.7 | 0.1 |
| 2% F68 | 18.5 ± 2.0* | 82.7 ± 2.1* | 6.6 ± 0.2* | 0.1 |
| BaCl2 | ||||
| 0% F68 | 32.3 ± 4.0 | 67.3 ± 4.6 | 5.9 ± 0.3 | 0.0 |
| 1% F68 | 19.6 ± 1.6* | 71.9 ± 2.4 | 5.5 ± 0.1 | 0.2 |
| 2% F68 | 19.2 ± 0.4* | 86.8 ± 2.8* | 7.2 ± 0.5* | 0.2 |
denotes statistical difference from the appropriate 0% F68 control sample (p≤0.05, n=8).
Inhomogeneously crosslinked hydrogels demonstrated an approximate increase in Deff of 200% as compared with homogeneously crosslinked hydrogels. The increase in Deff could be due to the variation in mesh size in the inhomogeneously crosslinked material. The model used to analyze the data is commonly used to determine Deff, but it assumes transport within a uniform network [48], and therefore the effect of the non-uniform network within the inhomogeneously crosslinked hydrogels may be exaggerated. The loading capacity of an inhomogeneously crosslinked hydrogel may be affected by the change in swelling ratio (Figure 2b) and the tight outer pore structure, which may limit the ability of small molecules to fully diffuse into or out of the matrix [57].
3.2 Effect of F68 and loading method on protein transport
The loading capacity of BSA within the composite hydrogel decreased with increasing F68 concentration independent of the gelation method as shown in Table 1. Similar results would be expected using other PEO-PPO-PEO synthetic surfactants, with a decrease in protein loading expected for a hydrogel containing micelles of a surfactant with a high ratio of hydrophobic PPO units to lipophilic PEO units, such as Pluronic® F108 or F127 [14]. Observable changes show increased water loss upon gelation with increasing surfactant concentration. One way to evaluate this change in volume over the liquid to solid transition is with a shrinkage ratio. Shrinkage, or water loss upon gelation, increases with increasing F68 concentration as shown in Figure 3. Increased shrinkage results in a decrease in final hydrogel volume (and an increase in final crosslinking density), which leads to a change in matrix properties, such as mechanical strength (G’ discussed below) and swelling ratio (Figure 2b) [37]. A smaller hydrogel volume would lead to less water retention and therefore lower protein loading capacity through the imbibement method. These changes in shrinkage upon gelation contribute to the 24–44% increase in the in situ riboflavin loading capacity. Shrinkage measurements translate to lower drug or biological therapeutic loading per gram of delivery vehicle used.
Figure 3.

Matrix shrinkage upon gelation. Water loss upon gelation (matrix shrinkage) increases with increasing F68 incorporation as measured by the relative change of solid hydrogel volume post-crosslinking to liquid alginate volume prior to gelation as depicted by Equation 2. (*) denotes statistical difference from the 0% F68 control. Error bars represent standard deviation (p≤0.05, n=4).
Hydrogel properties and device dimensions play crucial roles in complete release of proteins from a scaffold [25]. Addition of a nonionic surfactant, such as F68, can both improve and inhibit protein release over a two-hour period based on the mechanical properties [40, 56] of the hydrogel. Table 1 summarizes the effect of F68 incorporation in alginate hydrogels on the transport properties of BSA for two gelation methods (inhomogeneous and homogeneous; described above). For inhomogeneously crosslinked hydrogels, addition of F68 facilitated release of BSA (up to 21%) and enhanced Deff (up to 83%) (Table 1), improving the general BSA release properties. In the context of the release of protein therapeutics or signaling molecules, the use of a nonionic surfactant may decrease the time it takes for release. Conversely, homogeneously crosslinked hydrogels containing F68 showed a modest decrease in release percentage compared to alginate-only controls (up to 14%) with no change in Deff (Table 1). The tight pore size of the outer surface of inhomogeneously crosslinked hydrogels as compared to the homogeneously crosslinked hydrogels may contribute to the 30% decrease in overall release percentage observed (Table 1). These results strongly suggest that choice of gelation method is critical in device design and impacts the influence of surfactant addition on protein release.
Observed changes in BSA transport caused by addition of F68 can be explained through two physical measurements: 1) mesh size estimations of alginate and alginate-F68 gels based on swelling (ξS) and mechanical properties (ξR), and 2) the interaction of F68 with proteins using dynamic light scattering (DLS). Mesh size can be approximated using order of magnitude estimations [50, 51] as described by Equations 7 and 13. Both estimation methods yield an average mesh size of 10–25 nm, regardless of F68 incorporation in alginate. Therefore, particles at or above 10 nm will have transport limitations due to mesh size constraints. DLS analysis of BSA-F68 solution in Figure 4a shows an increase in particle size with increasing F68 concentration, suggesting the possibility of conformational changes to the protein structure [8, 62]. The percentage particle size increase with the addition of 2% and 5% F68 was 27.1% and 71.0%, respectively.
Figure 4.


Effect of F68 on protein diameter measured by DLS. A) Increase in F68 concentration increases BSA effective diameter (by 71% in 5% F68). B) Increase in F68 concentration increases lysozyme effective diameter (by 46% in 5% F68). Error bars represent standard deviation (n=4).
The increase in BSA particle size with added F68 may contribute to the decrease in release percentage in homogeneously crosslinked hydrogels through mesh size limitations. The incomplete release of BSA has implications for the use of surfactant-containing hydrogels intended for delivery of protein pharmaceutics. The addition of 1% F68 to BSA solution caused the BSA particle size to increase to 10.3 ± 0.1 nm as shown by DLS, which is within the estimated mesh size range for the alginate hydrogels investigated. This change in protein diameter with the addition of ≥2% F68 may also account for the decrease in the BSA loading capacity observed (described above). Transport limitations due to mesh size restrictions will increase with increasing particle size and therefore a decrease in BSA loading capacity is expected for increasing concentrations of F68 [63].
The increase in BSA effective particle size with added F68 may also be due to a relaxation of the hydrophobic cores of the protein pockets or direct interaction of the free F68 polymer or micelles with hydrophobic sections of the protein [8, 62]. BSA shares 76% sequence homology with human serum albumin [8], and therefore similar trends for transport and particle size would be expected with F68 addition to this pharmaceutical formulation. Due to the complexity of interaction, research investigating the effect of nonionic surfactants on specific protein conformational changes is necessary to understand the implications of the surfactant-protein interaction on the ability of any particular protein to maintain its functionality [8].
To determine if similar behavior is observed for other proteins and enzymes, DLS was used to measure the hydrodynamic diameter of lysozyme (hydrodynamic radius of 2.05 nm [64]) with increasing F68 concentration. It was found that the effective diameter of lysozyme increases with increasing F68 concentration as shown in Figure 4b. The percentage particle size increase with the addition of 2% and 5% F68 was 15.9% and 45.5%, respectively. The reduced effect of F68 on lysozyme particle size as compared to BSA may be due to the lower percentage of hydrophobic domains in the lysozyme molecule [7], as well as the smaller size (14 kDa vs. 67 kDa). At the maximum surfactant concentration measured, lysozyme-F68 particle size is 6.4 ± 0.1 nm, so transport limitations due to mesh size would not be expected for alginate hydrogels containing <5% F68. Constriction of the hydrogel network due to surfactant addition may impact the efficacy of the biomaterial by reducing both protein and nutrient transport within the matrix. Transport is ultimately dependent upon both the initial size of the protein [12, 20, 29, 65] and interaction with surfactant. Hence, changes in protein size due to surfactant addition should be evaluated for specific proteins of interest.
3.3 Rheological properties
Material strength and integrity is critical for biomaterial design and function [40, 65]. Rheological properties of F68-alginate systems were measured to investigate how incorporation of nonionic surfactants affects mechanical integrity of an alginate hydrogel. Solute diffusion is affected by the size and structure of the polymer used to form the hydrogel [66].
The dynamic viscosity of 1% alginate solution increases with increasing F68 as shown in Figure 5a. Addition of 5% F68 to the alginate solution increased the dynamic viscosity by 59%. We believe this is due to formation of some type of complex between F68 and alginate chains, which would increase the effective volume fraction of the polymer in water, leading to an increase in solution dynamic viscosity [67]. Complexation of F68 with alginate chains can be observed via particle size measurements (Figure 5b), which show a steady increase in the effective size of the alginate-F68 assembly with increasing F68 concentration (rather than two disparate populations of sizes, which would correspond to a solution of uncomplexed alginate chains and F68 micelles). The size of the alginate-F68 assembly with 5% F68 is nearly double that of neat alginate chains. We expect a similar type of viscosity increase in solutions of alginate with other types of nonionic surfactants. Solutions of alginate with the anionic surfactant sodium dodecyl sulfate (SDS) [68] and the cationic surfactant cetyltrimethylammonium bromide (CTAB) [69] also display a viscosity increase as compared to neat alginate, although in both of those cases the solution morphology may be quite different than in the nonionic case due to the charged nature of the surfactant. Increases in solution viscosity are significant when considering using a mixed alginate-surfactant solution in creation of a biomedical device. For example, in using alginate solutions for cell encapsulation in a syringe-type process, a significant increase in viscosity will affect final capsule morphology as well as processing parameters [70].
Figure 5.


Effect of F68 on solution properties of alginate. A) Viscosity of a 1% alginate solution increases with increasing F68 incorporation. Samples are 1% alginate solution containing (□) 0%, (○) 1%, (Δ) 2%, and (◊) 5% F68. 5% F68 incorporation increases solution viscosity 59% (n=4). B) Increase in F68 concentration increases alginate solution particle size (by 110% at 5% F68). Error bars represent standard deviation (n=4).
Determining the influence of additives on native mechanical properties of the hydrogel is important for the assessment of the effects on device functionality. The storage modulus (G’) represents the elastic or solid-like behavior of a material. G’ was monitored to determine if F68 addition alters the mechanical integrity of alginate hydrogels. Figure 6a shows a significant decrease in G’, and therefore mechanical strength, with the incorporation of 5% F68 across a wide range of frequencies. Figure 6b shows data collected at a frequency of 1 Hz, clearly demonstrating significant effects for 5% F68 incorporation (reduction in G’ by 57%). For alginate hydrogels, addition of F68 above 2% may lead to undesirable material properties. When designing tissue engineered scaffolds, such as artificial cartilage or soft tissue, it is beneficial to match the mechanical properties of the native tissue [65]. Decreases in mechanical integrity may increase the likelihood of scaffold fracture due to shear or other forces.
Figure 6.


Effect of F68 on storage modulus (G’) of alginate hydrogels. A) Addition of 1% and 2% F68 does not significantly affect G’; however, addition of 5% F68 decreases G’ by 54–60% over the frequencies presented. Samples are 1% alginate solution containing (□) 0%, (○) 1%, (Δ) 2%, and (◊) 5% F68 (n=5). B) G’ measured at 1 Hz. 5% F68 incorporation reduces G’ by 57% over the control (0% F68 incorporation). Error bars represent standard deviation (p≤0.05, n=5).
3.4 Cell viability
To investigate the biocompatibility of F68 with a variety of cell types, cells were cultured in media containing F68 and encapsulated within F68-alginate microcapsules. NIH 3T3 fibroblasts were grown in DMEM supplemented with either 2% or 5% F68 and cell number and viability were monitored over a 9-day period. Figure 7a shows a 42% decrease in relative growth for NIH 3T3 fibroblasts cultured in media containing 5% F68. Throughout the experiment, ≥90% viability was maintained for all samples (data not shown), suggesting minimal F68 toxicity, which agrees with previous studies using a human liver carcinoma cell line (HepG2) [4, 11] and other studies performed using a human microvascular endothelial cells (HMEC-1), keratinocytes (HEKa), and beta cell insulinomas (RIN-m5f) (data not shown). Cells may be exposed to surfactants used in the creation of novel biomedical scaffolds, such as bioactive glasses [3] or electrospun fibers [5, 10], or through hydrogels used for drug delivery [15].
Figure 7.

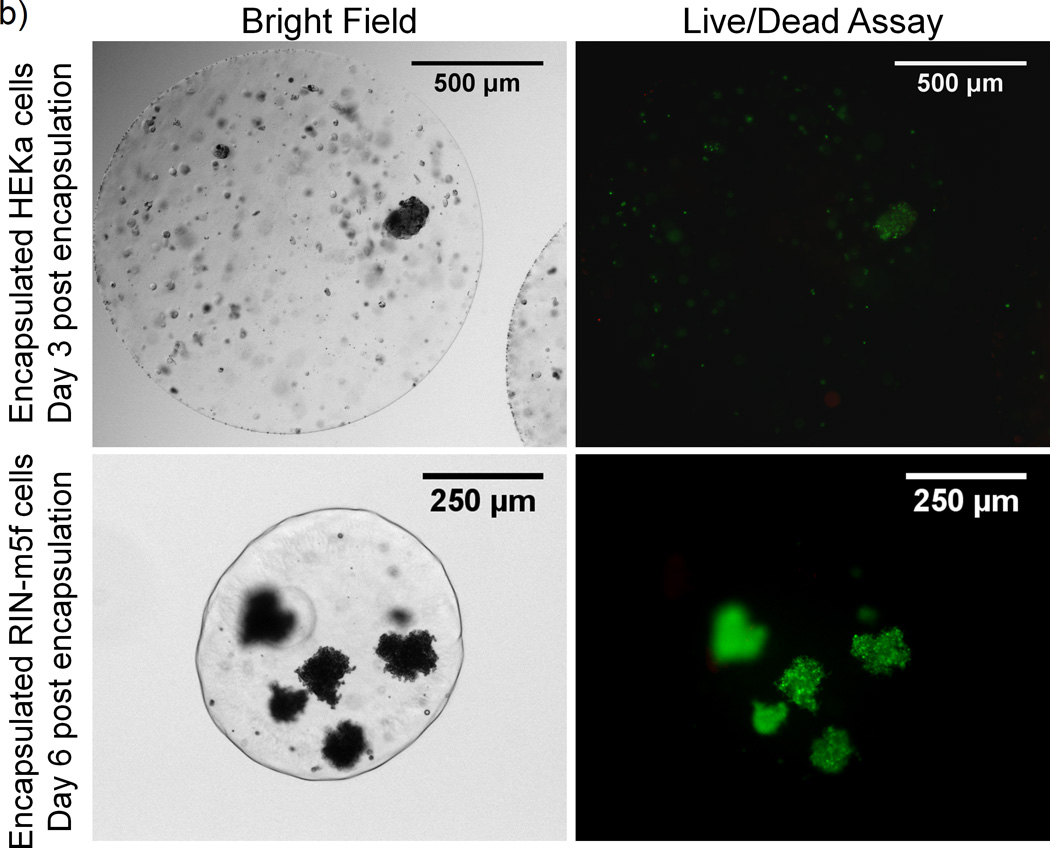
Effect of F68 on cell growth and viability. A) Relative growth of NIH 3T3 fibroblasts cultured in media containing (□) 0%, (○) 2%, or (Δ) 5% F68. After 9 days, 5% F68 incorporation decreases the relative growth rate by 42%. Relative growth is calculated as the relative change in viable cell number on the sample day from day 0. (*) represents time points where 5% F68 incorporation is statistically different from the 0% F68 control and (**) represents time points where both 2% and 5% F68 incorporation are statistically different from the control (p≤0.05, n=9). B) Live/Dead assay of adult human keratinocytes (HEKa) and RIN-m5f beta cell insulinomas (RIN-m5f) encapsulated in 1% alginate with 1% F68. Images show cell viability post-encapsulation by an overlay of the live (green) and dead (red) stains.
To investigate the cytotoxicity of F68 in the gel state on a variety of cell types, HEKa (keratinocytes) and RIN-m5f (β-cell insulinoma) cells were encapsulated in 1% alginate containing 1% F68 and viability was compared to cells encapsulated in 1% alginate without F68. LIVE/DEAD assay results, shown in Figure 7b, support the feasibility of using 1% F68 in alginate biomaterials and concur with previous studies using HepG2s [11] and NIH-3T3s (data not shown). The effect of F68 on the functionality of these cells and other cell types is currently under investigation in our laboratory. Based on cell motility studies with neutrophils [71] and macrophages [72], F68 decreases cell adhesion to the matrix, which may promote cell migration and aggregation within a microcapsule. For applications such as β-cell encapsulation for the treatment of type 1 diabetes [12], aggregation of isolated β-cells in a microcapsule may be encouraged by the use of a biocompatible nonionic surfactant.
4 Conclusions
Nonionic surfactants are often added to traditional hydrogels at high concentrations (i.e., above the CMC) to enable proper dissolution and delivery of relevant biologics. Determining the influence on transport of both proteins and small molecules is critical to device design and function. Here, we investigated the effect of adding the nonionic surfactant Pluronic® F68 (F68) to alginate hydrogels using two typical gelation methods on both small molecule (riboflavin) and protein (BSA) transport. Homogeneous gelation resulted in increased loading capacity and release percentage, with the most significant effects observed for protein transport. F68 addition had minimal effects on riboflavin transport and loading. Conversely, significant effects were observed for BSA where loading capacity was decreased and release was either increased or decreased depending on the gelation method. These effects can be attributed to both changes in material properties (i.e., water retention) and interaction of F68 micelles with BSA in solution. With 5% F68, an increase in dynamic viscosity and decrease in mechanical strength were observed, which has implications for both device processing and function. These results highlight the importance of studying and optimizing transport of biological molecules, particularly proteins, when additives (e.g., nonionic surfactants) are included in hydrogel formulations for cell and drug delivery applications.
Acknowledgements
This work was supported through the National Science Foundation (NSF)-sponsored Center for Hierarchical Manufacturing (CMMI-0531171) and the University of Massachusetts Commercial Ventures and Intellectual Property Office. The authors would also like to acknowledge use of the NSF-funded Materials Research Science and Engineering Center (MRSEC) on Polymers (DMR-0213695) central facilities. W.L.S. acknowledges support from a National Research Service Award T32 GM08515 from the National Institutes of Health (NIH). J.C.W. acknowledges support from the NSF-sponsored Institute for Cellular Engineering IGERT program (DGE-0654128). S.D.H. acknowledges support from the University of Massachusetts Commonwealth College undergraduate research assistantship fellowship.
Contributor Information
Whitney L. Stoppel, Email: wstoppel@ecs.umass.edu.
Joseph C. White, Email: jwhite@ecs.umass.edu.
Sarena D. Horava, Email: shorava@student.umass.edu.
Surita R. Bhatia, Email: sbhatia@ecs.umass.edu.
Susan C. Roberts, Email: sroberts@ecs.umass.edu.
References
- 1.Gutowska A, Jeong B, Jasionowski M. Injectable gels for tissue engineering. The Anatomical Record. 2001;263:342. doi: 10.1002/ar.1115. [DOI] [PubMed] [Google Scholar]
- 2.Kabanov A, Batrakova E, Alakhov V. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. Journal of Controlled Release. 2002;82:189. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 3.Jones JR, Hench LL. Effect of surfactant concentration and composition on the structure and properties of sol-gel-derived bioactive glass foam scaffolds for tissue engineering. Journal of Materials Science. 2003;38:3783. [Google Scholar]
- 4.Khattak SF, Bhatia SR, Roberts SC. Pluronic F127 as a cell encapsulation material: Utilization of membrane-stabilizing agents. Tissue Engineering. 2005;11:974. doi: 10.1089/ten.2005.11.974. [DOI] [PubMed] [Google Scholar]
- 5.Vasita R, Mani G, Agrawal CM, Katti DS. Surface hydrophilization of electrospun PLGA micro-/nano-fibers by blending with Pluronic (R) F-108. Polymer. 2010;51:3706. [Google Scholar]
- 6.Turro NJ, Lei X-G, Ananthapadmanabhan KP, Aronson M. Spectroscopic Probe Analysis of Protein- Surfactant Interactions: The BSA/SDS System. Langmuir. 1995;11:2525. [Google Scholar]
- 7.Valstar A, Brown W, Almgren M. The Lysozyme-Sodium Dodecyl Sulfate System Studied by Dynamic and Static Light Scattering. Langmuir. 1999;15:2366. [Google Scholar]
- 8.Gelamo EL, Silva CHTP, Imasato H, Tabak M. Interaction of bovine (BSA) and human (HSA) serum albumins with ionic surfactants: spectroscopy and modelling. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 2002;1594:84. doi: 10.1016/s0167-4838(01)00287-4. [DOI] [PubMed] [Google Scholar]
- 9.Sharma PK, Reilly MJ, Jones DN, Robinson PM, Bhatia SR. The effect of pharmaceuticals on the nanoscale structure of PEO-PPO-PEO micelles. Colloids and Surfaces B-Biointerfaces. 2008;61:53. doi: 10.1016/j.colsurfb.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Lee SJ, Oh SH, Liu J, Soker S, Atala A, Yoo JJ. The use of thermal treatments to enhance the mechanical properties of electrospun poly([var epsilon]-caprolactone) scaffolds. Biomaterials. 2008;29:1422. doi: 10.1016/j.biomaterials.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 11.Khattak SF, Chin KS, Bhatia SR, Roberts SC. Enhancing oxygen tension and cellular function in alginate cell encapsulation devices through the use of perfluorocarbons. Biotechnology and Bioengineering. 2007;96:156. doi: 10.1002/bit.21151. [DOI] [PubMed] [Google Scholar]
- 12.de Vos P, Bucko M, Gemeiner P, Navratil M, Svitel J, Faas M, Strand BL, Skjak-Braek G, Morch YA, Vikartovska A, Lacik I, Kollarikova G, Orive G, Poncelet D, Pedraz JL, Ansorge-Schumacher MB. Multiscale requirements for bioencapsulation in medicine and biotechnology. Biomaterials. 2009;30:2559. doi: 10.1016/j.biomaterials.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert JC, Hadgraft J, Bye A, Brookes LG. Drug release from Pluronic F-127 gels. International Journal of Pharmaceutics. 1986;32:223. [Google Scholar]
- 14.Fusco S, Borzacchiello A, Netti PA. Perspectives on: PEO-PPO-PEO Triblock Copolymers and their Biomedical Applications. Journal of Bioactive and Compatible Polymers. 2006;21:149. [Google Scholar]
- 15.Moreira T. Influence of oleic acid on the rheology and in vitro release of lumiracoxib from poloxamer gels. Journal of Pharmacy & Pharmaceutical Sciences. 2010;13 doi: 10.18433/j34880. [DOI] [PubMed] [Google Scholar]
- 16.Moghimi SM, Hunter AC. Poloxamers and poloxamines in nanoparticle engineering and experimental medicine. Trends in Biotechnology. 2000;18:412. doi: 10.1016/s0167-7799(00)01485-2. [DOI] [PubMed] [Google Scholar]
- 17.Anderson BC, Pandit NK, Mallapragada SK. Understanding drug release from poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) gels. Journal of Controlled Release. 2001;70:157. doi: 10.1016/s0168-3659(00)00341-2. [DOI] [PubMed] [Google Scholar]
- 18.Guzmán M, García FF, Molpeceres J, Aberturas MR. Polyoxyethylene-polyoxypropylene block copolymer gels as sustained release vehicles for subcutaneous drug administration. International Journal of Pharmaceutics. 1992;80:119. [Google Scholar]
- 19.Lee Y, Chung HJ, Yeo S, Ahn C-H, Lee H, Messersmith PB, Park TG. Thermo-sensitive, injectable, and tissue adhesive sol-gel transition hyaluronic acid/pluronic composite hydrogels prepared from bio-inspired catechol-thiol reaction. Soft Matter. 2010;6:977. [Google Scholar]
- 20.Grassi G, Crevatin A, Farra R, Guarnieri G, Pascotto A, Rehimers B, Lapasin R, Grassi M. Rheological properties of aqueous Pluronic-alginate systems containing liposomes. Journal of Colloid and Interface Science. 2006;301:282. doi: 10.1016/j.jcis.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 21.Kabanov AV, Lemieux P, Vinogradov S, Alakhov V. Pluronic® block copolymers: novel functional molecules for gene therapy. Advanced Drug Delivery Reviews. 2002;54:223. doi: 10.1016/s0169-409x(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 22.Mina EW, Lasagna-Reeves C, Glabe CG, Kayed R. Poloxamer 188 Copolymer Membrane Sealant Rescues Toxicity of Amyloid Oligomers In Vitro. Journal of Molecular Biology. 2009;391:577. doi: 10.1016/j.jmb.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 23.Clarke MSF, Prendergast MA, Terry AV. Plasma membrane ordering agent pluronic F-68 (PF-68) reduces neurotransmitter uptake and release and produces learning and memory deficits in rats. Learning & Memory. 1999;6:634. doi: 10.1101/lm.6.6.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco D, Alonso MJ. Protein encapsulation and release from poly(lactide-co-glycolide) microspheres: effect of the protein and polymer properties and of the co-encapsulation of surfactants. European Journal of Pharmaceutics and Biopharmaceutics. 1998;45:285. doi: 10.1016/s0939-6411(98)00011-3. [DOI] [PubMed] [Google Scholar]
- 25.Peppas N. Controlling protein diffusion in hydrogels. In: Lee VHL, Hashida M, editors. Trends and Future Perspectives in Peptide and Protein Drug Delivery. CRC Press; 1995. p. 23. [Google Scholar]
- 26.Langer RS, Peppas NA. Present and future applications of biomaterials in controlled drug delivery systems. Biomaterials. 1981;2:201. doi: 10.1016/0142-9612(81)90059-4. [DOI] [PubMed] [Google Scholar]
- 27.Batrakova EV, Kabanov AV. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. Journal of Controlled Release. 2008;130:98. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K, Mooney D. Hydrogels for tissue engineering. Chem. Rev. 2001;101:1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 29.Drury J, Mooney D. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24:4337. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 30.Klöck G, Pfeffermann A, Ryser C, Gröhn P, Kuttler B, Hahn H-J, Zimmermann U. Biocompatibility of mannuronic acid-rich alginates. Biomaterials. 1997;18:707. doi: 10.1016/s0142-9612(96)00204-9. [DOI] [PubMed] [Google Scholar]
- 31.Smidsrød O, Skjak-Brk G. Alginate as immobilization matrix for cells. Trends in Biotechnology. 1990;8:71. doi: 10.1016/0167-7799(90)90139-o. [DOI] [PubMed] [Google Scholar]
- 32.Shilpa A, Agrawal SS, Ray AR. Controlled delivery of drugs from alginate matrix. Journal of Macromolecular Science-Polymer Reviews. 2003;C43:187. [Google Scholar]
- 33.Tønnesen HH, Karlsen J. Alginate in Drug Delivery Systems. Drug Development and Industrial Pharmacy. 2002;28:621. doi: 10.1081/ddc-120003853. [DOI] [PubMed] [Google Scholar]
- 34.Smidsrod O, Haug A. Dependence upon the gel-sol state of the ion-exchange properties of alginates. Acta chem. scand. 1972;26 doi: 10.3891/acta.chem.scand.26-2063. [DOI] [PubMed] [Google Scholar]
- 35.Grant GT, Morris ER, Rees DA, Smith PJC, Thom D. Biological Interactions between Polysaccharides and Divalent Cations - Egg-Box Model. Febs Letters. 1973;32:195. [Google Scholar]
- 36.Sikorski P, Mo F, Skjak-Braek G, Stokke BT. Evidence for egg-box-compatible interactions in calciumalginate gels from fiber X-ray diffraction. Biomacromolecules. 2007;8:2098. doi: 10.1021/bm0701503. [DOI] [PubMed] [Google Scholar]
- 37.Bajpai SK, Sharma S. Investigation of swelling/degradation behaviour of alginate beads crosslinked with Ca2+ and Ba2+ ions. Reactive & Functional Polymers. 2004;59:129. [Google Scholar]
- 38.Draget KI, Ostgaard K, Smidsrod O. Homogeneous Alginate Gels - A Technical Approach. Carbohydrate Polymers. 1990;14:159. [Google Scholar]
- 39.Gombotz WR, Wee SF. Protein release from alginate matrices. Advanced Drug Delivery Reviews. 1998;31:267. doi: 10.1016/s0169-409x(97)00124-5. [DOI] [PubMed] [Google Scholar]
- 40.Amsden B, Turner N. Diffusion characteristics of calcium alginate gels. Biotechnology and Bioengineering. 1999;65:605. doi: 10.1002/(sici)1097-0290(19991205)65:5<605::aid-bit14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 41.Chin K, Khattak SF, Bhatia SR, Roberts SC. Hydrogel-perfluorocarbon composite scaffold promotes oxygen transport to immobilized cells. Biotechnology Progress. 2008;24:358. doi: 10.1021/bp070160f. [DOI] [PubMed] [Google Scholar]
- 42.Johnson AS, Fisher RJ, Weir GC, Colton CK. Oxygen consumption and diffusion in assemblages of respiring spheres: Performance enhancement of a bioartificial pancreas. Chemical Engineering Science. 2009;64:4470. [Google Scholar]
- 43.Kimelman-Bleich N, Pelled G, Sheyn D, Kallai I, Zilberman Y, Mizrahi O, Tal Y, Tawackoli W, Gazit Z, Gazit D. The use of a synthetic oxygen carrier-enriched hydrogel to enhance mesenchymal stem cell-based bone formation in vivo. Biomaterials. 2009;30:4639. doi: 10.1016/j.biomaterials.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 44.Chavanpatil MD, Khdair A, Panyam J. Surfactant-polymer nanoparticles: A novel platform for sustained and enhanced cellular delivery of water-soluble molecules. Pharmaceutical Research. 2007;24:803. doi: 10.1007/s11095-006-9203-2. [DOI] [PubMed] [Google Scholar]
- 45.Bhatia SR, Khattak SF, Roberts SC. Polyelectrolytes for cell encapsulation. Current Opinion in Colloid & Interface Science. 2005;10:45. [Google Scholar]
- 46.Skják-Bræk G, Smidsrød O, Larsen B. Tailoring of alginates by enzymatic modification in vitro. International Journal of Biological Macromolecules. 1986;8:330. [Google Scholar]
- 47.Zhao Y, Li F, Carvajal MT, Harris MT. Interactions between bovine serum albumin and alginate: An evaluation of alginate as protein carrier. Journal of Colloid and Interface Science. 2009;332:345. doi: 10.1016/j.jcis.2008.12.048. [DOI] [PubMed] [Google Scholar]
- 48.Fu JC, Hagemeir C, Moyer DL, Ng EW. Unified Mathematical-Model for Diffusion from Drug-Polymer Composite Tablets. Journal of Biomedical Materials Research. 1976;10:743. doi: 10.1002/jbm.820100507. [DOI] [PubMed] [Google Scholar]
- 49.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Advanced Drug Delivery Reviews. 2001;48:139. doi: 10.1016/s0169-409x(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 50.Gennes P-Gd. Scaling concepts in polymer physics. Ithaca, N.Y.: Cornell University Press; 1979. [Google Scholar]
- 51.Peppas NA, Moynihan HJ, Lucht LM. The Structure of Highly Crosslinked poly(2-hydroxyethyl methacrylate) Hydrogels. Journal of Biomedical Materials Research. 1985;19:397. doi: 10.1002/jbm.820190405. [DOI] [PubMed] [Google Scholar]
- 52.Faroongsarng D, Sukonrat P. Thermal behavior of water in the selected starch- and cellulose-based polymeric hydrogels. International Journal of Pharmaceutics. 2008;352:152. doi: 10.1016/j.ijpharm.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 53.Straatmann A, Borchard W. Determination of thermodynamic properties of sodium alginate from bacteria and seaweeds in aqueous solutions. Analytical Ultracentrifugation VI. 2002:64. [Google Scholar]
- 54.Abramoff M, Magelhaes P, Ram S. Image processing with ImageJ. Biophotonics international. 2004;11:36. [Google Scholar]
- 55.Peppas N, Hilt J, Khademhosseini A, Langer R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Advanced Materials. 2006;18:1345. [Google Scholar]
- 56.Liu XD, Yu WY, Zhang Y, Xue WM, Yu WT, Xiong Y, Ma XJ, Chen Y, Yuan Q. Characterization of structure and diffusion behaviour of Ca-alginate beads prepared with external or internal calcium sources. Journal of Microencapsulation. 2002;19:775. doi: 10.1080/0265204021000022743. [DOI] [PubMed] [Google Scholar]
- 57.Strand BL, Morch YA, Espevik T, Skjak-Braek G. Visualization of alginate-poly-L-lysine-alginate microcapsules by confocal laser scanning microscopy. Biotechnology and Bioengineering. 2003;82:386. doi: 10.1002/bit.10577. [DOI] [PubMed] [Google Scholar]
- 58.Shin HS, Kim SY, Lee YM, Lee KH, Kim SJ, Rogers CE. Permeation of solutes through interpenetrating polymer network hydrogels composed of poly(vinyl alcohol) and poly(acrylic acid) Journal of Applied Polymer Science. 1998;69:479. [Google Scholar]
- 59.Böhme U, Scheler U. Effective charge of bovine serum albumin determined by electrophoresis NMR. Chemical Physics Letters. 2007;435:342. [Google Scholar]
- 60.Talmadge JE. The pharmaceutics and delivery of therapeutic polypeptides and proteins. Advanced Drug Delivery Reviews. 1993;10:247. [Google Scholar]
- 61.Cao L, Mooney DJ. Spatiotemporal control over growth factor signaling for therapeutic neovascularization. Advanced Drug Delivery Reviews. 2007;59:1340. doi: 10.1016/j.addr.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Mol Biol. 1998;5:827. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 63.Amsden B. Solute diffusion within hydrogels. Mechanisms and models. Macromolecules. 1998;31:8382. [Google Scholar]
- 64.Wilkins D, Grimshaw S, Receveur V, Dobson C, Jones J, Smith L. Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques†. Biochemistry. 1999;38:16424. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 65.Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zhang J. Physicochemical, foundations and structural design of hydrogels in medicine and biology. Annual Review of Biomedical Engineering. 2000;2:9. doi: 10.1146/annurev.bioeng.2.1.9. [DOI] [PubMed] [Google Scholar]
- 66.Johansson L, Skantze U, Loefroth JE. Diffusion and interaction in gels and solutions. 2. Experimental results on the obstruction effect. Macromolecules. 1991;24:6019. [Google Scholar]
- 67.Macosko CW. Rheology : principles, measurements, and applications. New York, NY: VCH; 1994. [Google Scholar]
- 68.Bu H, Kjøniksen A, Elgsaeter A, Nyström B. Interaction of unmodified and hydrophobically modified alginate with sodium dodecyl sulfate in dilute aqueous solution:: Calorimetric, rheological, and turbidity studies. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2006;278:166. [Google Scholar]
- 69.Yang J, Chen S, Fang Y. Viscosity study of interactions between sodium alginate and CTAB in dilute solutions at different pH values. Carbohydrate Polymers. 2009;75:333. [Google Scholar]
- 70.Pozrikidis C. Modeling and simulation of capsules and biological cells. Boca Raton, Fla. London: CRC; 2003. [Google Scholar]
- 71.Tan J, Saltzman WM. Influence of synthetic polymers on neutrophil migration in three-dimensional collagen gels. Journal of Biomedical Materials Research. 1999;46:465. doi: 10.1002/(sici)1097-4636(19990915)46:4<465::aid-jbm4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 72.Schürch S, Gehr P, Green F, Wallace JA, McIver DJL. Cell--substrate adhesion affects intracellular motility of pulmonary macrophages. Colloids and Surfaces. 1989;42:271. [Google Scholar]