

Abstract
The initial identification of glucagon as a counter-regulatory hormone to insulin revealed this hormone to be of largely singular physiological and pharmacological purpose. Glucagon agonism, however, has also been shown to exert effects on lipid metabolism, energy balance, body adipose tissue mass and food intake. The ability of glucagon to stimulate energy expenditure, along with its hypolipidemic and satiating effects, in particular, make this hormone an attractive pharmaceutical agent for the treatment of dyslipidemia and obesity. Studies that describe novel preclinical applications of glucagon, alone and in concert with glucagon-like peptide 1 agonism, have revealed potential benefits of glucagon agonism in the treatment of the metabolic syndrome. Collectively, these observations challenge us to thoroughly investigate the physiology and therapeutic potential of insulin's long-known opponent.
Introduction
The isolation of insulin in 1921 by Banting, Best, Collip and Macleod transformed our understanding of the hormonal regulation of glucose metabolism and has provided a life-saving treatment for millions of patients with diabetes mellitus. Of far lesser prominence was the simultaneous description of a pancreatic contaminant observed to elicit a temporary increase in blood glucose levels.1 This contaminant was later studied by Murlin et al.2 and found to oppose insulin in the control of glucose homeostasis, both in healthy animals and in those whose pancreas had been removed. Murlin and colleagues named this new hormone glucagon, as they thought it was a glucose agonist. Later work by Sutherland, Park and Exton described the counter-regulatory actions of glucagon relative to insulin pharmacology. Specifically, the researchers determined that glucagon stimulates hepatic glycogenolysis and gluconeogenesis in hypoglycemic states to restore glucose homeostasis.3,4 The patho-physiological role of hyperglucagonemia and unopposed glucagon action in diabetes mellitus has been emphasized.5–9 By contrast, the possibility that glucagon may also have benefits in the treatment of metabolic disease has received little or no attention. However, new studies that describe novel preclinical applications of glucagon, either alone or in concert with glucagon-like peptide 1 (GLP-1) agonism, have revealed the prospect of harnessing the benefits of glucagon action for the treatment of the metabolic syndrome.10,11
Glucagon
Expression
Glucagon is a hormone produced in the α cells of the pancreatic islets. Encoded by the proglucagon gene, the proglucagon peptide is processed by neuroendocrine convertase-2 (NEC2) to produce the 29-amino acid native glucagon peptide. In addition, the proglucagon gene contains sequences that encode GLP-1, GLP-2, oxyntomodulin and glicentin. These hormones are processed from the proglucagon peptide via NEC1-mediated cleavage in the brain and in L cells of the intestine.12 Studies on the mechanisms of proglucagon transcription have elucidated the minimal promoter region, as well as four enhancer elements.13 Regulation of transcription has been shown to occur through homeodomain transcription factors, influenced by amino acids and cyclic AMP (cAMP) in the pancreas and intestinal cells,14 as well as Wnt signaling in the intestine.15 Insulin is also known to exert inhibitory effects on proglucagon expression in α cells.16,17 Interestingly, this effect is reversed in the intestine, where insulin at pathological concentrations stimulates proglucagon expression, which results in increased GLP-1 production.18
Secretion
Glucagon secretion, similar to that of insulin, is tightly regulated and intimately tied to blood glucose levels (Figure 1). Converse to the inhibition of insulin secretion by hypoglycemia, low levels of blood glucose directly stimulate the pancreatic α cells to secrete glucagon.19 The secretion of glucagon is promoted via the action of voltage-dependent sodium (Na+) and calcium (Ca2+) channels, which maintain action potentials during times of low glucose. Depolarization increases the Ca2+ influx and subsequent glucagon secretion,20 which is supported by the activity of ATP-sensitive potassium (KATP) channels.21 As glucose levels rise, secretion of glucagon is inhibited through the elevation of cytosolic ATP, blockade of KATP channels and termination of the Na+-induced and Ca2+-induced action potentials. This process inhibits Ca2+ influx and ends glucagon secretion. Although the cellular signals regulating glucagon secretion are fairly well-established, the role of glucose, whether directly, or indirectly via β-cell activation, is still a matter of debate. Studies in rats suggest that mediatory, paracrine signaling from the β cell is essential for the inhibition of glucagon secretion by elevated glucose levels.22,23 Investigations in mice and humans, however, suggest that glucose directly inhibits glucagon secretion at concentrations which are too low to stimulate insulin secretion.21 Furthermore, this inhibition has been demonstrated in vitro in both isolated α cells and in intact pancreatic islets.24 However, a study in individuals with type 1 diabetes mellitus suggests that insulin is the primary signal that inhibits glucagon secretion in humans.25 In addition to glucose, several other physiological parameters are known regulators of glucagon secretion, including GLP-1,26 GLP-2,27 fatty acids,28 the autonomic nervous system29 and circulating amino acids.30
Figure 1.
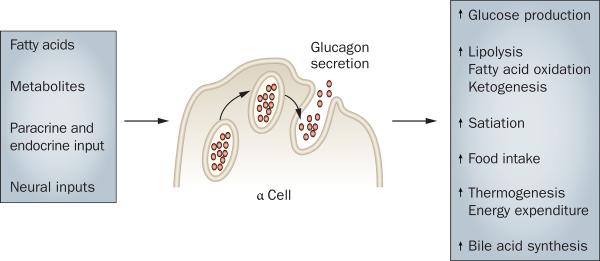
Physiological stimuli and outcomes of glucagon secretion.
Signaling
Glucagon receptor
The effects of glucagon are mediated by the binding of glucagon to its membrane-bound receptor—a seven-transmembrane protein and a member of the class II guanine nucleotide-binding protein (G protein) coupled receptor superfamily.31 Glucagon receptors (encoded by the GCGR gene) are expressed abundantly in the liver and kidney and to a lesser extent in heart, adipocytes, lymphoblasts, spleen, endocrine pancreas, brain, retina, adrenal gland and the gastrointestinal tract.32 Previous studies suggest that glucagon receptors are also expressed in islet α cells.33 In the liver, glucagon receptors are located mainly in hepatocytes, but can also be found on the surface of Kupffer cells.34 Interestingly, glucagon receptors in the pancreas are predominately located on β cells, which, together with the data described above, suggests a bidirectional feedback mechanism.35 Further evidence supporting this hypothesis is the fact that glucagon, at physiological concentrations, was found to stimulate insulin release via these receptors.36
Much knowledge concerning glucagon action has been gained from genetically altered animal models. Glucose homeostasis and pancreatic function has been elaborately studied in mice with a null mutation of the glucagon receptor (Gcgr−/−). As expected, blood glucose levels are markedly lower and glucose tolerance is improved in Gcgr−/− mice compared with wild-type controls.37,38 The improvement in glucose tolerance is not owing to an increase in insulin secretion, but rather to an improvement in insulin sensitivity, as demonstrated by hyperinsulinemic euglycemic clamp studies.39 When fed a high-fat diet (HFD), Gcgr−/− mice do not display diet-induced insulin resistance, which is potentially a result of enhanced insulin sensitivity in these mice. However, after prolonged fasting, Gcgr−/− mice experience severe hypoglycemia,37 which illustrates the essential role of glucagon for the maintenance of blood glucose levels.38Gcgr−/− mice were observed to display an enlargement of the pancreas primarily attributed to α-cell hyperplasia, which indicates glucagon receptor signaling is essential for normal endocrine-cell proliferation.37
Further indication of glucagon's role in endocrine-cell function was demonstrated in transgenic mice that overexpress the glucagon receptor specifically on pancreatic β cells. These mice display enhanced glucagon-stimulated and glucose-stimulated insulin secretion, as well as improved glucose tolerance.40 A reduction in both fasting hyperglycemia and impaired glucose tolerance is observed when these transgenic mice are exposed to a HFD,40 suggesting that enhanced glucagon signaling on β cells improves the function of these cells. The studies that show improvements in glucose homeostasis in mice overexpressing the glucagon receptor on β cells are in contrast to studies in mice treated with streptozotocin, which showed that ablation of glucagon signaling has protective effects. streptozotocin treatment destroys β cells, impairs insulin secretion and induces hyperglycemia in wild-type mice; however, when streptozotocin is administered to Gcgr−/− mice, euglycemia is maintained even after HFD exposure.38 This finding indicates that lack of glucagon receptor signaling results in resistance to streptozotocin-mediated β-cell destruction and hyper glycemia in vivo. The mechanism behind the streptozotocin resistance in Gcgr−/− mice remains unknown; however, given that other reports demonstrate that increased concentration of circulating GLP-1 results in resistance to streptozotocin-induced β-cell destruction, the increased levels of circulating GLP-1 determined in Gcgr−/− mice have been speculated to be an important factor.41,42 These data illustrate that both enhanced, as well as lack of, glucagon receptor signaling have positive effects on glucose homeostasis and pancreatic function and, therefore, further studies are needed to understand the cause of these results.
Signaling pathway and targets
Binding of glucagon to its receptor elicits activation of a heterotrimeric, stimulatory G protein (Gs) in a process dependent on GTP and magnesium.43 In liver cells, the glucagon receptor and Gs proteins have been postulated to be linked in a multimeric configuration, which disengages following activation.44 The activated Gs protein undergoes a conformational change, upon which the GTP-bound subunit Gsα is released from the G-protein complex that comprises two additional subunits, Gsβ and Gsγ. This activation leads to interaction and subsequent stimulation of adenylate cyclase, elevated cAMP levels and enhanced intracellular signaling via Rap guanine nucleotide exchange factor 3 (RAPGEF3; also known as EPAC1), cAMP response element-binding protein (CREB)-regulated transcription coactivator 2 (CRTC2; also known as TORC2) and protein kinase A (PKA). The activation of PKA results in the phosphorylation and nuclear localization of CREB.45,46 Once phosphorylated in the liver, CREB binds to the cAMP response element of target genes, resulting in the recruitment of coactivators, such as hepatic nuclear factor 4α (HNF-4α),47 peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α)48 and the glucocorticoid receptor.47 In addition to this well-described pathway, glucagon has also been implicated in signaling via 5'-AMP-activated protein kinase (AMPK),49 mitogen-activated protein kinase (MAPK) and in a c-Jun N-terminal kinase (JNK)-dependent manner.50
Beyond glucose homeostasis
The role of glucagon in glucose homeostasis has been well-studied and previously reviewed elsewhere.51 However, this pancreatic peptide has additional metabolic effects of notable importance. Glucagon agonism has been shown to regulate lipid metabolism and energy expenditure, as well as reduce histamine-induced cardiac injury during reperfusion.52
Glucagon and lipid metabolism
Plasma lipid homeostasis
Studies in the early 1960s investigated glucagon's action independent of glucose homeostasis. These studies described a lipid-mobilizing effect for glucagon in a range of species.53–57 The regulation of plasma lipids by glucagon was first highlighted in studies which suggested that glucagon acts to decrease plasma cholesterol, total esterified fatty acids and arachidonic acid levels.53,58 As epinephrine, insulin and exogenous glucose all failed to lower levels of plasma cholesterol, the investigators concluded that these effects were not an indirect result of altered carbohydrate metabolism.59 In both canines and humans, the researchers observed a total decrease in levels of plasma cholesterol, as well as plasma total lipid concentrations, within 30 min of intravenous glucagon administration. Intriguingly, this depression was not found for whole-blood total lipid levels, suggesting a repartitioning of lipids from serum to platelet-rich fractions. Incubation of the blood at 37 °C led to partial or full restoration of plasma cholesterol and total lipid levels, further supporting the hypothesis of repartitioning.58 Reports from other groups provided additional support for a role of glucagon in the regulation of plasma lipids.55,56
In the context of triglyceride metabolism, several reports indicated that triglyceride production is decreased in perfused livers treated with glucagon.60–62 Although these studies suggest hepatic lipoprotein metabolism as the site of such regulation, the molecular mechanism by which glucagon achieved this result remained elusive. Building upon these observations, the role of glucagon on lipid suppression was investigated in a rat model of hyperlipidemia.63 This study demonstrated that glucagon sig nificantly decreased levels of serum triglycerides, cholesterol, and VLDL cholesterol in hyperlipidemic rats, as well as in control eulipidemic rats. Furthermore, a decrease in synthesis of hepatic lipoprotein apoproteins was observed,63 complementing earlier studies that described depression of triglyceride synthesis in livers treated with glucagon. A caveat of this study is that it involved supraphysiological concentrations of glucagon administered over a 4-day period. Thus, the relevance of acute signaling by endogenous glucagon remained uncertain. Guettet et al.64–66 continued the investigation of chronic effects of glucagon signaling in the Wistar rat. Their initial studies described a decrease in concentrations of plasma cholesterol, phospholipids and tri glycerides. Interestingly, these reductions were not seen in the liver or in erythrocytes of these rats, which suggested another target tissue as the site of action. Continued investigation of cholesterol turnover revealed increased urinary secretion of cholesterol, as well as elevated transformation to bile acids. Of special importance was the reported observation that the chronic treatment of Wistar rats with twice-daily glucagon (20 μg per day) for 3 weeks reduced blood glucose and insulin concentrations. This report thus indicated that chronic glucagon pharmacology was neither transient nor detrimental to glucose homeostasis.64
Further studies by Guettet et al.65,66 evaluated the effect of chronic glucagon treatment on lipoprotein composition in fed and fasted rats, as well as rats fed a cholesterol-rich diet. Cholesterol, phospholipids and total protein levels were proportionally decreased in chylomicrons, VLDL cholesterol, LDL cholesterol and HDL cholesterol, which suggested a reduction in the number of lipoprotein particles. Triglycerides were reported to be decreased only in the chylomicron and VLDL fractions. Evaluation of the lipoproteins indicated a decline in levels of apolipoprotein E (ApoE) and an increase in ApoB.65 Collectively, these findings suggested glucagon-stimulated targeting of the ApoE-rich lipoproteins to limit the accumulation that typically occurs with high-cholesterol feeding.66 A follow-up study showed that glucagon treatment had no effect on secretion rates of triglyceride-rich particles in either fed or fasted conditions. Conversely, glucagon treatment accelerated the rate of triglyceride removal (fractional catabolic rate constant) from the plasma compartment.67 Taken together, the work of Guettet et al.64–66 revealed that increased glucagon signaling may directly regulate lipid catabolism.
Rudling and Angelin68 extended these findings by the direct study of glucagon's effects on LDL receptors (LDLRs). The investigators found that glucagon administra tion to rats resulted in a dose-dependent increase in binding of LDL cholesterol to the receptor, with no apparent effect on receptor mRNA expression. Concomitant with this increased binding was a decrease in levels of cholesterol, ApoB, and ApoE. Furthermore, these effects were completely antagonized by insulin administration. Rudling and Angelin hypothesized a mechanism such as post-translational receptor modification, whereby glucagon increases LDLR activity without affecting expression levels.68 The findings in rodents were complemented by studies in dairy cattle which showed that subcutaneous injections of glucagon led to variable decreases, some of sizable magnitude, in circulating plasma concentrations of VLDL-triglycerides, HDL1-phospholipids and HDL2-free cholesterol.69 Improved glucose status with decreased nonesterified fatty acids and β-hydoxybutyrate was also reported.70
Genetic manipulation of the glucagon receptor has led to inconclusive results regarding glucagon's role in lipid metabolism. Conarello et al.38 found that Gcgr−/− mice are resistant to HFD-induced liver steatosis; however, Longuet et al.71 showed that Gcgr−/− mice have enhanced susceptibility to hepatic steatosis. Interestingly, these studies used genetically modified null mice generated in an identical manner and of the same background (C57B6). Furthermore, the effect was observed in mice fed a similar diet (45% fat). The length of diet differed between the two studies (8 versus 12 weeks); however, mice fed a HFD for the longer duration did not exhibit steatosis. Taken together, the contribution of glucagon receptor agonism to liver steatosis has yet to be conclusively described.
Longuet et al.71 also demonstrated the importance of glucagon signaling in the adaptive response to fasting. Following exposure to a prolonged fasting period, Gcgr−/− mice showed increased levels of plasma triglycerides and free fatty acids.71 During either glucagon treatment or a prolonged fast, wild-type mice had an increased expression of genes involved in fatty acid oxidation, including Decr2, carnitine O-palmitoyltransferase (Cpt) 1a, Cpt2 and Acadm. These changes in expression were correlated with an increased capacity of the liver to oxidize free fatty acids. By contrast, this increased gene expression was not observed in Gcgr−/− mice, which displayed a reduced oxidation of free fatty acids in both the fed and the fasted state. These data highlight the essential physiological role of glucagon-receptor signaling in the regulation of gene expression during prolonged fasting.
Glucagon-mediated lipolysis and ketogenesis
Glucagon-mediated regulation of lipid metabolism is not limited to plasma triglycerides and cholesterol. Glucagon treatments as low as 10−8 mol/l have been implicated in promoting lipolysis in white adipose tissue.72 This lipolytic effect appears to be independent of sympathetic nervous system innervation,73 as denervation of white adipose tissue does not block glucagon-induced glycerol release, whereas it decreases the release of nonesterified fatty acids. The latter, however, is reported to be the result of re-esterification,73 not a blockade of glucagon's lipolytic effects. Mechanistically, glucagon is known to stimulate the activity of hormone-sensitive lipase (HSL) in adipocytes, resulting in an increase of nonesterified fatty acids in the circulation.74 These fatty acids, usually bound to albumin, are transported to the heart, skeletal muscle, kidney and liver, where they are catabolyzed or, in the case of the liver, alternatively converted to ketone bodies.
Ketone bodies provide up to two-thirds of the energy for the brain during times of glucose deficiency, thus sparing glucose utilization and reducing proteolysis.75 Supporting a role for glucagon in the regulation of keto-genesis, suppression of glucagon secretion via somatostatin prevented the development of ketoacidosis in patients with type 1 diabetes mellitus.76 Pegorier et al.77 elucidated the regulation of fatty acid oxidation and keto-genesis by glucagon in rabbit hepatocytes. The researchers reported increased ketone-body production in the presence of either glucagon or dibutyryl cAMP. Oxidation of exogenous oleate was increased by both glucagon and cAMP, effects which were reduced in the presence of insulin. Furthermore, exposure to glucagon or cAMP completely inhibited lipogenesis and decreased malonyl-CoA concentration and stimulated fatty acid oxidation. These effects were suggested to be driven by a fall in the sensitivity of CPT1 to malonyl-CoA, releasing the inhibition of this rate-limiting enzyme in the transport of fatty acids across the mitochondrial membranes.77,78 The relevance of these studies was confirmed by the observation that glucagon directly controls ketone-body production in primary human hepatocytes.79 Specifically, glucagon increased ketone-body production and fatty acid oxidation, whereas it decreased fatty acid esterification, similar to the effects of cAMP.79
Lipid-induced glucagon resistance
In addition to glucagon's regulation of lipid metabolism, previous studies suggest that lipids may also regulate glucagon signaling. Studies by Charbonneau et al.80 show that rats fed a HFD display hepatic steatosis that is associated with glucagon resistance. Specifically, exercise training in rats was used to increase plasma glucagon levels and improve fatty liver. In trained rats, a negative correlation between liver adiposity and density of the glucagon receptors in the plasma membrane was observed. Additional studies described a reduction in both hepatic glucagon-receptor density and Gsα protein content at the plasma membrane.81,82 The mechanism for this decrease in receptor density was explored in a study which proposed that HFD promoted glucagon receptor proteolysis.81 A reduction in glucagon receptors at the plasma membrane was notable, accompanied by a marked increase in the number of endosomal and lysosomal compartments. These effects were correlated with an increase of protein kinase Cα (PKCα) on the plasma membrane, which is known to phosphorylate G-protein-related kinases. Such action, in turn, is known to inhibit receptor internalization which leads to receptor desensitiza tion.83 The same association seems to be conserved in humans, as glucagon resistance is associated with pathological hyperlipidemia in humans.84
Glucagon and bile acid metabolism
Although the canonical role of bile acids is associated with the absorption of dietary lipids and cholesterol homeostasis, new studies suggest that these acids have additional signaling roles in glucose homeostasis. This emerging function was highlighted in a study by song and Chiang,85 who investigated cholesterol 7-alpha-monooxygenase (CYP7A1) as a possible target of glucagon signaling. Their study demonstrated that glucagon, as well as cAMP, represses CYP7A1 expression in human primary hepatocytes. CYP7A1 is the rate-limiting enzyme in bile acid synthesis, and its expression is tightly regulated as a means to control flux through the pathway.86 Song and Chiang86 further showed that glucagon's regulation of CYP7A1 expression is mediated via PKA signaling to HNF-4α. Given that no direct measure of bile acid synthesis or secretion was available, the direct result on bile acid metabolism remains unknown. The findings of their earlier study,85 however, were complemented by work from Hylemon et al.,87 who showed similar decreases in CYP7A1 expression in response to glucagon or cAMP exposure in rat hepatocytes. These in vitro studies support initial observations that suggest a role for glucagon in the regulation of bile acid metabolism and provide a plausible mechanism for the glucagon-induced decrease in plasma cholesterol levels.
Glucagon and energy expenditure
In addition to regulating glucose and lipid metabolism, glucagon participates in the control of energy expenditure and thermogenesis. Early studies by Davidson et al.88,89 showed that pharmacological infusion of glucagon increased oxygen consumption in rats. This effect was mirrored by a report in human study participants, in whom infusion of a pharmacological dose of glucagon increased resting energy expenditure during acute insulin deficiency produced by the additional infusion of somatostatin.90 Moreover, a physiological dose of glucagon increased energy expenditure in humans during euinsulinemia, and hyperinsulinemia blunted glucagon's thermogenic activity.91 Studies conducted in vitro showed that hyperglucagonemia may increase energy expenditure via stimulation of oxygen consumption and heat production in brown adipose tissue.92,93 Studies in rats confirmed that glucagon administration increased whole-body oxygen consumption, core body temperature, blood flow,94 as well as temperature and mass of brown adipose tissue.95,96 Furthermore, cold exposure was found to increase plasma glucagon levels,97 which implicates glucagon in nonshivering thermogenesis. The stimulation of thermogenic activity in brown adipose tissue by glucagon98 has become even more relevant, as novel findings suggest a renewed importance for brown adipose tissue in human energy metabolism.99
The mechanism by which glucagon-induced thermogenesis is regulated is probably complex and may involve the sympathetic nervous system. Supporting this hypothesis, glucagon-induced increases in oxygen consumption, blood flow and thermogenesis in brown adipose tissue were blocked by the nonselective β adrenergic receptor blocker, propranolol,100,101 whereas denervation of brown adipose tissue blunted the thermogenic action of glucagon.98 Furthermore, although glucagon injection in ducklings was associated with an increase in norepinephrine, blockade of catecholamines via guanethidine sympathectomy (the interruption of the transmission of sympathetic nerve impulses by a chemical agent that blocks the secretion of epinephrine and norepinephrine at postganglionic nerve endings) decreased metabolic rate compared to nonsympathectomized birds treated with glucagon or saline.102 Glucagon might also have a direct effect on thermogenesis, as glucagon incubation of brown adipocytes of rat and mice, but not those of Syrian hamsters, markedly increased oxygen consumption.100 However, the concentration of glucagon required for such in vitro effects has been speculated to be supraphysiological.100
The role of glucagon in mediating cold-induced thermogenic responses has been further questioned by a report that denervated interscapular brown adipose tissue decreased the number of glucagon receptors as compared to the contralateral adipose tissue pad that served as an internal (within animal) control.103 Given that cold exposure is a well-known stimulator of the sympathetic nervous system in brown adipose tissue,104,105 a decrease in glucagon receptor protein or gene expression mitigates the role for glucagon in cold-triggered brown adipose tissue thermogenesis via innervation of the sympathetic nervous system. Taken together, these data suggest a pivotal role for the sympathetic nervous system in glucagon-induced thermogenesis and suggest a thermogenic basis for anti-obesity effects of glucagon.
Glucagon and regulation of food intake
Early studies indicated that glucagon administration diminishes the sense of hunger and decreases food intake in humans106–108 and rats.109 Additional studies in rats confirmed that glucagon specifically decreases meal size owing to increased satiation rather than illness or aversion. Intravenous infusions similarly produced a dose-related decrease in eating.109–113 Evidence for an endogenous role of glucagon in satiation was provided by demonstrations that glucagon concentration increases physiologically during meals, in many situations,114–116 and that antagonism of glucagon by preprandial administration of glucagon antibodies increases the amount of food consumed in one meal (meal size).117,118 Finally, intravenous infusion of a physiological dose of glucagon during meals was shown to reduce meal size in humans.119
A study in rats with both hepatic portal vein and vena cava infusion catheters indicated that both exogenous and endogenous glucagon act in the liver to limit meal size.120 Furthermore, as for several other peripheral signals that control eating, vagal afferents relay the signal to the brain.121 In the case of glucagon, the hepatic branch of the abdominal vagus is critical,122 consistent with the hepatic site of action. Although the inhibitory effect of glucagon on feeding probably arises, at least in part, from a hepatic metabolic action of the hormone, novel studies in sheep suggest that glucagon acts directly in the central nervous system to inhibit food intake.123
Genetically modified animal models also demonstrate that glucagon signaling is involved in regulating food intake and body composition. Gcgr−/− mice display a decreased adipose tissue mass and an increased lean mass despite similar food intake and body weight compared to their wild-type littermates.37 Furthermore, Gcgr−/− mice on a HFD are resistant to diet-induced obesity,38 which can be primarily attributed to a lower food intake compared with controls.
Glucagon-based pharmacological therapy
Glucagon-based drug therapy has largely been restricted to acute emergency use to treat episodes of hypoglycemia in patients with type 1 diabetes mellitus and as an esophageal muscle relaxant to prepare patients for radiological procedures. Nevertheless, glucagon's hypolipidemic, energy expenditure-stimulatory and satiating effects make it an attractive pharmaceutical agent for the treatment of dyslipidemia and obesity. Such chronic uses, however, must be considered carefully in light of glucagon's ability to accelerate the development of glucose intolerance and insulin resistance. Studies in cattle suggest that glucagon may be an effective treatment for fatty liver and dyslipidemia124 without deleterious effects on glucose homestasis, as both single and multiple injections of 5 mg glucagon over 14 days consistently improve the carbohydrate status of dairy cows and decreased concentrations of plasma nonesterified fatty acids.70 Of course, these actions must be verified in nonruminant species.
Glucagon and GLP-1 coagonism
Two prominent studies published in July 2009, have put forth the hypothesis that glucagon agonism may be beneficial in the pharmacological treatment of obesity and, possibly, obesity-associated glucose intolerance.10,11 Interestingly, both studies combined agonists of two proglucagon-derived peptides to elicit these effects in mouse models. Day et al.10 combined the antidiabetic properties of GLP-1 receptor (GLP-1R) agonism with the hypolipidemic properties of glucagon receptor agonism to create a dual agonist, which they conjugated to a polyethylene glycol polymer to prolong pharmacokinetic action. The researchers then compared 1-week administration of this dual agonist with an equimolar amount of a structurally comparable, but selective, GLP-1R agonist in diet-induced obese mice. Both peptides significantly decreased food intake, body weight and adipose tissue mass when compared with saline-injected controls. Significant decreases in blood glucose and insulin concentrations and a profound increase in glucose tolerance were also reported. Intriguingly, the peptide with balanced glucagon and GLP-1 coagonism exhibited significantly greater efficacy, as measured by changes in body weight, adipose tissue mass and glucose homeostasis, compared with the GLP-1R agonist monotherapy.10 Studies conducted over a 1-month period revealed similar effects on body weight, adiposity and blood glucose levels, although cumulative total food intake in this study was not impressively altered. Interestingly, energy expenditure in the 1-month study was significantly increased in the animals treated with the coagonist, with a trend for a decreased respiratory quotient. Lipid metabolism was markedly improved with decreases in levels of total cholesterol, LDL cholesterol, HDL cholesterol and trigylcerides, as well as reversal of liver steatosis and activation of HSL in white adipose tissue.
Similar to the study by Day et al.,10 Pocai et al.11 tested a dual agonist of GLP-1R and the glucagon receptor in diet-induced obese mice. The investigators injected the coagonist daily for 2 weeks and compared these mice with those treated with either a GLP-1R agonist with the same degree of GLP-1R agonism as the coagonist or vehicle. Significant enhancements in measures of body weight and glucose tolerance were reported in mice treated with the coagonist. Results of animals treated only with a GLP-1R agonist were intermediate between those of mice treated with either the coagonist peptide or vehicle, for all measured parameters.11 Also, similar to the study by Day and colleagues,10 Pocai et al.11 showed decreases in liver steatosis, total cholesterol and triglyceride levels in the coagonist-treated animals, with the single-agonist-treated mice again displaying intermediate values in comparison with the other two groups.
Together, these two studies exemplify a novel and potentially important new direction for therapy of obesity and metabolic disease (Figure 2). An advantage of the described approach is the use of single-agent peptides that represent two full hormone-receptor agonists that elicit additive therapeutic effects, while at the same time adverse effects can be minimized. These studies come with the caveat that the glucagon agonism caused by the coagonist peptide far exceeds the actions elicited by endogenous glucagon. Furthermore, these studies did not compare coagonism with glucagon monotherapy; any effect observed is, therefore, a possible, and probable, interaction between the two receptor agonists. Taken together, interpretation of glucagon physiology on the basis of these findings must be tempered. Possibly of greater importance, these studies challenge our deeply rooted understanding of the full physiology of a pancreatic hormone that partners with insulin in the management of glucose and lipid homeostasis and body weight.
Figure 2.

Individual and synergystic effects of glucagon-like peptide 1 and glucagon in obesity and pharmacotherapy.
The fact that excessive glucagon receptor agonism leads to glucose intolerance and insulin dysregulation is well-established. Observations of elevated glucagon concentration in insulin-resistant individuals, as well as animal models of insulin resistance, have supported this view. The studies of Day et al.10 and Pocai et al.11 build upon a foundation of less evident metabolic actions of glucagon reported in this Review. They provide a rationale to reconsider whether glucagon administered at modest concentrations and frequency of exposure constructively contributes to proper glucose and lipid metabolism directly by acting at specific target tissues and indirectly by modulating body weight. The prospect of using glucagon in combination with other agents to address obesity-associated glucose intolerance and insulin resistance, as well as dyslipidemia, is something worthy of scholarly consideration.
Conclusions
The limited data available on the pharmacology of chronically administered glucagon is the result of a focus on acute glucagon action and its disadvantageous diabetogenic properties that render it poorly suited for the treatment of obesity and metabolic diseases. Development of stable, soluble glucagon agonists of varying pharmacokinetics and of glucagon-like co agonists in conjunction with GLP-1, which are suitable for pharmacological study, however, provide an unprecedented opportunity to explore the full physiological character and pharmacological potential of this fascinating hormone. When viewed in combination with historical observations, the first reports resulting from studies of these agonistic peptides suggest that glucagon may potently regulate glucose metabolism and lipid homeostasis, as well as energy balance, body adipose tissue mass and food intake. Clearly, excessive and unopposed glucagon action is catabolic and must be avoided if chronic use is considered. Nevertheless, collectively, novel observations challenge us to thoroughly investigate and broadly take into consideration the physiology and therapeutic potential of this long-known counter-regulatory hormone to insulin.
Key points
-
■
In addition to its well-known effects on glycemia, increased glucagon signaling directly regulates triglyceride, free fatty acid, apolipoprotein and bile acid metabolism
-
■
Glucagon action can be inhibited via receptor desensitization by excess dietary fat intake
-
■
Energy expenditure and thermogenesis are increased by glucagon agonism
-
■
Glucagon administration stimulates satiety and decreases food intake
-
■
Glucagon action, in combination with incretins such as glucagon-like peptide 1, may be a crucial tool in the treatment of the metabolic syndrome
Footnotes
Competing interests
N. Geary declares an association with the following company: Novo Nordisk. R. DiMarchi declares an association with the following company: Marcadia Biotech. See the article online for full details of the relationships. The other authors declare no competing interests.
Author contributions
All authors researched the data for the article, provided a substantial contribution to discussions of the content, contributed equally to writing the article and reviewed and/or edited the manuscript before submission.
References
- 1.Banting FG, Best CH. The internal secretion of the pancreas. J. Lab. Clin. Med. 1922;7:251–266. [PubMed] [Google Scholar]
- 2.Murlin JR, Clough HD, Gibbs CBF, Stokes AM. Aqueous extracts of the pancreas. I. Influence on the carbohydrate metabolism of depancreatized animals. J. Biol. Chem. 1923;56:253–296. [Google Scholar]
- 3.Exton JH, Park CR. The role of cyclic AMP in the control of liver metabolism. Adv. Enzyme Regul. 1968;6:391–407. doi: 10.1016/0065-2571(68)90024-1. [DOI] [PubMed] [Google Scholar]
- 4.Robison GA, Butcher RW, Sutherland EW. Cyclic AMP. Annu. Rev. Biochem. 1968;37:149–174. doi: 10.1146/annurev.bi.37.070168.001053. [DOI] [PubMed] [Google Scholar]
- 5.Gu W, et al. Long-term inhibition of the glucagon receptor with a monoclonal antibody in mice causes sustained improvement in glycemic control, with reversible alpha-cell hyperplasia and hyperglucagonemia. J. Pharmacol. Exp. Ther. 2009;331:871–881. doi: 10.1124/jpet.109.157685. [DOI] [PubMed] [Google Scholar]
- 6.Wang MY, et al. Leptin therapy in insulin-deficient type I diabetes. Proc. Natl Acad. Sci. USA. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown RJ, Sinaii N, Rother KI. Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care. 2008;31:1403–1404. doi: 10.2337/dc08-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr. Rev. 2007;28:253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- 9.Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 10.Day JW, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 11.Pocai A, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 13.Herzig S, Fuzesi L, Knepel W. Heterodimeric Pbx-Prep1 homeodomain protein binding to the glucagon gene restricting transcription in a cell type-dependent manner. J. Biol. Chem. 2000;275:27989–27999. doi: 10.1074/jbc.M003345200. [DOI] [PubMed] [Google Scholar]
- 14.Gevrey JC, et al. Protein hydrolysates stimulate proglucagon gene transcription in intestinal endocrine cells via two elements related to cyclic AMP response element. Diabetologia. 2004;47:926–936. doi: 10.1007/s00125-004-1380-0. [DOI] [PubMed] [Google Scholar]
- 15.Yi F, Brubaker PL, Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. J. Biol. Chem. 2005;280:1457–1464. doi: 10.1074/jbc.M411487200. [DOI] [PubMed] [Google Scholar]
- 16.Philippe J. Insulin regulation of the glucagon gene is mediated by an insulin-responsive DNA element. Proc. Natl Acad. Sci. USA. 1991;88:7224–7227. doi: 10.1073/pnas.88.16.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Artner I, et al. MafB: an activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes. 2006;55:297–304. doi: 10.2337/diabetes.55.02.06.db05-0946. [DOI] [PubMed] [Google Scholar]
- 18.Yi F, et al. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology. 2008;149:2341–2351. doi: 10.1210/en.2007-1142. [DOI] [PubMed] [Google Scholar]
- 19.Quesada I, Todorova MG, Soria B. Different metabolic responses in alpha-, beta-, and delta-cells of the islet of Langerhans monitored by redox confocal microscopy. Biophys. J. 2006;90:2641–2650. doi: 10.1529/biophysj.105.069906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gromada J, et al. Adrenaline stimulates glucagon secretion in pancreatic A-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. J. Gen. Physiol. 1997;110:217–228. doi: 10.1085/jgp.110.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald PE, et al. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol. 2007;5:e143. doi: 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54:1808–1815. doi: 10.2337/diabetes.54.6.1808. [DOI] [PubMed] [Google Scholar]
- 23.Olsen HL, et al. Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells. Endocrinology. 2005;146:4861–4870. doi: 10.1210/en.2005-0800. [DOI] [PubMed] [Google Scholar]
- 24.Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes. 2005;54:1789–1797. doi: 10.2337/diabetes.54.6.1789. [DOI] [PubMed] [Google Scholar]
- 25.Cooperberg BA, Cryer PE. Beta-cell-mediated signaling predominates over direct alpha-cell signaling in the regulation of glucagon secretion in humans. Diabetes Care. 2009;32:2275–2280. doi: 10.2337/dc09-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunning BE, Foley JE, Ahrén B. Alpha cell function in health and disease: influence of glucagon-like peptide-1. Diabetologia. 2005;48:1700–1713. doi: 10.1007/s00125-005-1878-0. [DOI] [PubMed] [Google Scholar]
- 27.Meier JJ, Kjems LL, veldhuis JD, Lefèbvre P, Butler PC. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes. 2006;55:1051–1056. doi: 10.2337/diabetes.55.04.06.db05-1449. [DOI] [PubMed] [Google Scholar]
- 28.Bollheimer LC, et al. Stimulatory short-term effects of free fatty acids on glucagon secretion at low to normal glucose concentrations. Metabolism. 2004;53:1443–1448. doi: 10.1016/j.metabol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Ahrén B. Autonomic regulation of islet hormone secretion—implications for health and disease. Diabetologia. 2000;43:393–410. doi: 10.1007/s001250051322. [DOI] [PubMed] [Google Scholar]
- 30.Dumonteil E, et al. Glucose regulates proinsulin and prosomatostatin but not proglucagon messenger ribonucleic acid levels in rat pancreatic islets. Endocrinology. 2000;141:174–180. doi: 10.1210/endo.141.1.7230. [DOI] [PubMed] [Google Scholar]
- 31.Mayo KE, et al. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 32.Svoboda M, Tastenoy M, vertongen P, Robberecht P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell Endocrinol. 1994;105:131–137. doi: 10.1016/0303-7207(94)90162-7. [DOI] [PubMed] [Google Scholar]
- 33.Kedees MH, Grigoryan M, Guz Y, Teitelman G. Differential expression of glucagon and glucagon-like peptide 1 receptors in mouse pancreatic alpha and beta cells in two models of alpha cell hyperplasia. Mol. Cell Endocrinol. 2009;311:69–76. doi: 10.1016/j.mce.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.watanabe J, Kanai K, Kanamura S. Glucagon receptors in endothelial and Kupffer cells of mouse liver. J. Histochem. Cytochem. 1988;36:1081–1089. doi: 10.1177/36.9.2841370. [DOI] [PubMed] [Google Scholar]
- 35.Kieffer TJ, Heller RS, Unson CG, weir GC, Habener JF. Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology. 1996;137:5119–5125. doi: 10.1210/endo.137.11.8895386. [DOI] [PubMed] [Google Scholar]
- 36.Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia. 2000;43:1012–1019. doi: 10.1007/s001250051484. [DOI] [PubMed] [Google Scholar]
- 37.Gelling R. w., et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl Acad. Sci. USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conarello SL, et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia. 2007;50:142–150. doi: 10.1007/s00125-006-0481-3. [DOI] [PubMed] [Google Scholar]
- 39.Sørensen H, et al. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes. 2006;55:3463–3469. doi: 10.2337/db06-0307. [DOI] [PubMed] [Google Scholar]
- 40.Gelling R. w., et al. Pancreatic beta-cell overexpression of the glucagon receptor gene results in enhanced beta-cell function and mass. Am. J. Physiol. Endocrinol. Metab. 2009;297:E695–E707. doi: 10.1152/ajpendo.00082.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conarello SL, et al. Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc. Natl Acad. Sci. USA. 2003;100:6825–6830. doi: 10.1073/pnas.0631828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pospisilik JA, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes. 2003;52:741–750. doi: 10.2337/diabetes.52.3.741. [DOI] [PubMed] [Google Scholar]
- 43.Rodbell M, Birnbaumer L, Pohl SL, Krans HM. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J. Biol. Chem. 1971;246:1877–1882. [PubMed] [Google Scholar]
- 44.Rodbell M. The complex regulation of receptor- coupled G-proteins. Adv. Enzyme Regul. 1997;37:427–435. doi: 10.1016/s0065-2571(96)00020-9. [DOI] [PubMed] [Google Scholar]
- 45.Jelinek LJ, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259:1614–1616. doi: 10.1126/science.8384375. [DOI] [PubMed] [Google Scholar]
- 46.Koo SH, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 47.Yoon JC, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 48.Herzig S, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 49.Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J. Biol. Chem. 2004;279:54103–54109. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Ishac EJ, Dent P, Kunos G, Gao B. Effects of ethanol on mitogen-activated protein kinase and stress-activated protein kinase cascades in normal and regenerating liver. Biochem. J. 1998;334(Pt 3):669–676. doi: 10.1042/bj3340669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003;284:E671–E678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 52.Rosic M, et al. Glucagon effects on ischemic vasodilatation in the isolated rat heart. J. Biomed. Biotechnol. 2010;2010:231832. doi: 10.1155/2010/231832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caren R, Corbo L. Glucagon and cholesterol metabolism. Metabolism. 1960;9:938–945. [PubMed] [Google Scholar]
- 54.Salter JM, Ezrin C, Laidlaw JC, Gornall AG. Metabolic effects of glucagon in human subjects. Metabolism. 1960;9:753–768. [PubMed] [Google Scholar]
- 55.Paloyan E, Harper PV., Jr. Glucagon as a regulating factor of plasma lipids. Metabolism. 1961;10:315–323. [PubMed] [Google Scholar]
- 56.Amatuzio DS, Grande F, Wada S. Effect of glucagon on the serum lipids in essential hyperlipemia and in hypercholesterolemia. Metabolism. 1962;11:1240–1249. [PubMed] [Google Scholar]
- 57.De Oya M, Prigge WF, Swenson DE, Grande F. Role of glucagon on fatty liver production in birds. Am. J. Physiol. 1971;221:25–30. doi: 10.1152/ajplegacy.1971.221.1.25. [DOI] [PubMed] [Google Scholar]
- 58.Caren R, Corbo L. Transfer of plasma lipid to platelets by action of glucagon. Metabolism. 1970;19:598–607. doi: 10.1016/0026-0495(70)90016-8. [DOI] [PubMed] [Google Scholar]
- 59.Caren R, Corbo L. Glucagon and plasma arachidonic acid. Metabolism. 1965;14:684–692. doi: 10.1016/0026-0495(65)90051-x. [DOI] [PubMed] [Google Scholar]
- 60.De Oya M, Prigge WF, Grande F. Suppression by hepatectomy of glucagon-induced hypertriglyceridemia in geese. Proc. Soc. Exp. Biol. Med. 1971;136:107–110. doi: 10.3181/00379727-136-35205. [DOI] [PubMed] [Google Scholar]
- 61.Heimberg M, Weinstein I, Kohout M. The effects of glucagon, dibutyryl cyclic adenosine 3',5'-monophosphate, and concentration of free fatty acid on hepatic lipid metabolism. J. Biol. Chem. 1969;244:5131–5139. [PubMed] [Google Scholar]
- 62.Penhos JC, Wu CH, Daunas J, Reitman M, Levine R. Effect of glucagon on the metabolism of lipids and on urea formation by the perfused rat liver. Diabetes. 1966;15:740–748. doi: 10.2337/diab.15.10.740. [DOI] [PubMed] [Google Scholar]
- 63.Eaton RP. Hypolipemic action of glucagon in experimental endogenous lipemia in the rat. J. Lipid Res. 1973;14:312–318. [PubMed] [Google Scholar]
- 64.Guettet C, Mathé D, Riottot M, Lutton C. Effects of chronic glucagon administration on cholesterol and bile acid metabolism. Biochim. Biophys. Acta. 1988;963:215–223. doi: 10.1016/0005-2760(88)90283-4. [DOI] [PubMed] [Google Scholar]
- 65.Guettet C, Mathé D, Navarro N, Lecuyer B. Effects of chronic glucagon administration on rat lipoprotein composition. Biochim. Biophys. Acta. 1989;1005:233–238. doi: 10.1016/0005-2760(89)90042-8. [DOI] [PubMed] [Google Scholar]
- 66.Guettet C, et al. Effect of chronic glucagon administration on lipoprotein composition in normally fed, fasted and cholesterol-fed rats. Lipids. 1991;26:451–458. doi: 10.1007/BF02536072. [DOI] [PubMed] [Google Scholar]
- 67.Guettet C, Rostaqui N, Navarro N, Lecuyer B, Mathe D. Effect of chronic glucagon administration on the metabolism of triacylglycerol-rich lipoproteins in rats fed a high sucrose diet. J. Nutr. 1991;121:24–30. doi: 10.1093/jn/121.1.24. [DOI] [PubMed] [Google Scholar]
- 68.Rudling M, Angelin B. Stimulation of rat hepatic low density lipoprotein receptors by glucagon. Evidence of a novel regulatory mechanism in vivo. J. Clin. Invest. 1993;91:2796–2805. doi: 10.1172/JCI116522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bobe G, Ametaj BN, Young J. w., Beitz DC. Effects of exogenous glucagon on lipids in lipoproteins and liver of lactating dairy cows. J. Dairy Sci. 2003;86:2895–2903. doi: 10.3168/jds.S0022-0302(03)73886-7. [DOI] [PubMed] [Google Scholar]
- 70.Bobe G, Sonon RN, Ametaj BN, Young JW, Beitz DC. Metabolic responses of lactating dairy cows to single and multiple subcutaneous injections of glucagon. J. Dairy Sci. 2003;86:2072–2081. doi: 10.3168/jds.S0022-0302(03)73796-5. [DOI] [PubMed] [Google Scholar]
- 71.Longuet C, et al. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008;8:359–371. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richter w. O., Robl H, Schwandt P. Human glucagon and vasoactive intestinal polypeptide (VIP) stimulate free fatty acid release from human adipose tissue in vitro. Peptides. 1989;10:333–335. doi: 10.1016/0196-9781(89)90039-9. [DOI] [PubMed] [Google Scholar]
- 73.Lefebvre P, Luyckx A, Bacq ZM. Effects of denervation on the metabolism and the response to glucagon of white adipose tissue of rats. Horm. Metab. Res. 1973;5:245–250. doi: 10.1055/s-0028-1093959. [DOI] [PubMed] [Google Scholar]
- 74.Perea A, Clemente F, Martinell J, villanueva-Peñacarrillo ML, valverde I. Physiological effect of glucagon in human isolated adipocytes. Horm. Metab. Res. 1995;27:372–375. doi: 10.1055/s-2007-979981. [DOI] [PubMed] [Google Scholar]
- 75.Nair KS, welle SL, Halliday D, Campbell RG. Effect of beta-hydroxybutyrate on whole-body leucine kinetics and fractional mixed skeletal muscle protein synthesis in humans. J. Clin. Invest. 1988;82:198–205. doi: 10.1172/JCI113570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerich JE, et al. Prevention of human diabetic ketoacidosis by somatostatin. Evidence for an essential role of glucagon. N. Engl. J. Med. 1975;292:985–989. doi: 10.1056/NEJM197505082921901. [DOI] [PubMed] [Google Scholar]
- 77.Pegorier JP, et al. Induction of ketogenesis and fatty acid oxidation by glucagon and cyclic AMP in cultured hepatocytes from rabbit fetuses. Evidence for a decreased sensitivity of carnitine palmitoyltransferase I to malonyl-CoA inhibition after glucagon or cyclic AMP treatment. Biochem. J. 1989;264:93–100. doi: 10.1042/bj2640093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Prip-Buus C, Pegorier JP, Duee PH, Kohl C, Girard J. Evidence that the sensitivity of carnitine palmitoyltransferase I to inhibition by malonyl-CoA is an important site of regulation of hepatic fatty acid oxidation in the fetal and newborn rabbit. Perinatal development and effects of pancreatic hormones in cultured rabbit hepatocytes. Biochem. J. 1990;269:409–415. doi: 10.1042/bj2690409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.vons C, et al. Regulation of fatty-acid metabolism by pancreatic hormones in cultured human hepatocytes. Hepatology. 1991;13:1126–1130. [PubMed] [Google Scholar]
- 80.Charbonneau A, Couturier K, Gauthier MS, Lavoie JM. Evidence of hepatic glucagon resistance associated with hepatic steatosis: reversal effect of training. Int. J. Sports Med. 2005;26:432–441. doi: 10.1055/s-2004-821225. [DOI] [PubMed] [Google Scholar]
- 81.Charbonneau A, Unson CG, Lavoie JM. High-fat diet-induced hepatic steatosis reduces glucagon receptor content in rat hepatocytes: potential interaction with acute exercise. J. Physiol. 2007;579(Pt 1):255–267. doi: 10.1113/jphysiol.2006.121954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Charbonneau A, Melancon A, Lavoie C, Lavoie JM. Alterations in hepatic glucagon receptor density and in Gsalpha and Gialpha2 protein content with diet-induced hepatic steatosis: effects of acute exercise. Am. J. Physiol. Endocrinol. Metab. 2005;289:E8–E14. doi: 10.1152/ajpendo.00570.2004. [DOI] [PubMed] [Google Scholar]
- 83.Savage A, Zeng L, Houslay MD. A role for protein kinase C-mediated phosphorylation in eliciting glucagon desensitization in rat hepatocytes. Biochem. J. 1995;307(Pt 1):281–285. doi: 10.1042/bj3070281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eaton RP, Schade DS. Glucagon resistance as a hormonal basis for endogenous hyperlipaemia. Lancet. 1973;1:973–974. doi: 10.1016/s0140-6736(73)91605-x. [DOI] [PubMed] [Google Scholar]
- 85.Song KH, Chiang JY. Glucagon and cAMP inhibit cholesterol 7alpha-hydroxylase (CYP7A1) gene expression in human hepatocytes: discordant regulation of bile acid synthesis and gluconeogenesis. Hepatology. 2006;43:117–125. doi: 10.1002/hep.20919. [DOI] [PubMed] [Google Scholar]
- 86.Chiang JY. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr. Rev. 2002;23:443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 87.Hylemon PB, et al. Hormonal regulation of cholesterol 7 alpha-hydroxylase mRNA levels and transcriptional activity in primary rat hepatocyte cultures. J. Biol. Chem. 1992;267:16866–16871. [PubMed] [Google Scholar]
- 88.Davidson IW, Salter JM, Best CH. The effect of glucagon on the metabolic rate of rats. Am. J. Clin. Nutr. 1960;8:540–546. [Google Scholar]
- 89.Davidson IW, Salter JM, Best CH. Calorigenic action of glucagon. Nature. 1957;180:1124. doi: 10.1038/1801124a0. [DOI] [PubMed] [Google Scholar]
- 90.Nair KS. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J. Clin. Endocrinol. Metab. 1987;64:896–901. doi: 10.1210/jcem-64-5-896. [DOI] [PubMed] [Google Scholar]
- 91.Calles-Escandón J. Insulin dissociates hepatic glucose cycling and glucagon-induced thermogenesis in man. Metabolism. 1994;43:1000–1005. doi: 10.1016/0026-0495(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 92.Joel CD. Stimulation of metabolism of rat brown adipose tissue by addition of lipolytic hormones in vitro. J. Biol. Chem. 1966;241:814–821. [PubMed] [Google Scholar]
- 93.Kuroshima A, Yahata T. Thermogenic responses of brown adipocytes to noradrenaline and glucagon in heat-acclimated and cold-acclimated rats. Jpn. J. Physiol. 1979;29:683–690. doi: 10.2170/jjphysiol.29.683. [DOI] [PubMed] [Google Scholar]
- 94.Yahata T, Habara Y, Kuroshima A. Effects of glucagon and noradrenaline on the blood flow through brown adipose tissue in temperature-acclimated rats. Jpn. J. Physiol. 1983;33:367–376. doi: 10.2170/jjphysiol.33.367. [DOI] [PubMed] [Google Scholar]
- 95.Doi K, Kuroshima A. Modified metabolic responsiveness to glucagon in cold-acclimated and heat-acclimated rats. Life Sci. 1982;30:785–791. doi: 10.1016/0024-3205(82)90614-2. [DOI] [PubMed] [Google Scholar]
- 96.Billington CJ, Briggs JE, Link JG, Levine AS. Glucagon in physiological concentrations stimulates brown fat thermogenesis in vivo. Am. J. Physiol. 1991;261(Pt 2):R501–R507. doi: 10.1152/ajpregu.1991.261.2.R501. [DOI] [PubMed] [Google Scholar]
- 97.Edwards CI, Howland RJ. Adaptive changes in insulin and glucagon secretion during cold acclimation in the rat. Am. J. Physiol. 1986;250(Pt 1):E669–E676. doi: 10.1152/ajpendo.1986.250.6.E669. [DOI] [PubMed] [Google Scholar]
- 98.Billington CJ, Bartness TJ, Briggs J, Levine AS, Morley JE. Glucagon stimulation of brown adipose tissue growth and thermogenesis. Am. J. Physiol. 1987;252(Pt 2):R160–R165. doi: 10.1152/ajpregu.1987.252.1.R160. [DOI] [PubMed] [Google Scholar]
- 99.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dicker A, Zhao J, Cannon B, Nedergaard J. Apparent thermogenic effect of injected glucagon is not due to a direct effect on brown fat cells. Am. J. Physiol. 1998;275(Pt 2):R1674–R1682. doi: 10.1152/ajpregu.1998.275.5.R1674. [DOI] [PubMed] [Google Scholar]
- 101.Heim T, Hull D. The effect of propranalol on the calorigenic response in brown adipose tissue of new-born rabbits to catecholamines, glucagon, corticotrophin and cold exposure. J. Physiol. 1966;187:271–283. doi: 10.1113/jphysiol.1966.sp008088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Filali-Zegzouti Y, et al. Role of catecholamines in glucagon-induced thermogenesis. J. Neural Transm. 2005;112:481–489. doi: 10.1007/s00702-004-0199-7. [DOI] [PubMed] [Google Scholar]
- 103.Morales A, et al. Sympathetic control of glucagon receptor mRNA levels in brown adipose tissue of cold-exposed rats. Mol. Cell Biochem. 2000;208:139–142. doi: 10.1023/a:1007058525309. [DOI] [PubMed] [Google Scholar]
- 104.Brito NA, Brito MN, Bartness TJ. Differential sympathetic drive to adipose tissues after food deprivation, cold exposure or glucoprivation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;294:R1445–R1452. doi: 10.1152/ajpregu.00068.2008. [DOI] [PubMed] [Google Scholar]
- 105.Young JB, Saville E, Rothwell NJ, Stock MJ, Landsberg L. Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. J. Clin. Invest. 1982;69:1061–1071. doi: 10.1172/JCI110541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stunkard AJ, van Itallie TB, Reis BB. The mechanism of satiety: effect of glucagon on gastric hunger contractions in man. Proc. Soc. Exp. Biol. Med. 1955;89:258–261. doi: 10.3181/00379727-89-21776. [DOI] [PubMed] [Google Scholar]
- 107.Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of glucagon on food intake and body weight in man. J. Appl. Physiol. 1957;11:419–421. doi: 10.1152/jappl.1957.11.3.419. [DOI] [PubMed] [Google Scholar]
- 108.Penick SB, Hinkle LE., Jr. Depression of food intake induced in healthy subjects by glucagon. N. Engl. J. Med. 1961;264:893–897. doi: 10.1056/NEJM196105042641801. [DOI] [PubMed] [Google Scholar]
- 109.Martin JR, Novin D. Decreased feeding in rats following hepatic-portal infusion of glucagon. Physiol. Behav. 1977;19:461–466. doi: 10.1016/0031-9384(77)90218-9. [DOI] [PubMed] [Google Scholar]
- 110.Weick BG, Ritter S. Dose-related suppression of feeding by intraportal glucagon infusion in the rat. Am. J. Physiol. 1986;250(Pt 2):R676–R681. doi: 10.1152/ajpregu.1986.250.4.R676. [DOI] [PubMed] [Google Scholar]
- 111.Salter JM. Metabolic effects of glucagon in the Wistar rat. Am. J. Clin. Nutr. 1960;8:535–539. [Google Scholar]
- 112.Holloway SA, Stevenson JA. Effect of glucagon on food intake and weight gain in the young rat. Can. J. Physiol. Pharmacol. 1964;42:867–869. doi: 10.1139/y64-098. [DOI] [PubMed] [Google Scholar]
- 113.Le Sauter J, Geary N. Hepatic portal glucagon infusion decreases spontaneous meal size in rats. Am. J. Physiol. 1991;261(Pt 2):R154–R161. doi: 10.1152/ajpregu.1991.261.1.R154. [DOI] [PubMed] [Google Scholar]
- 114.de Jong A, Strubbe JH, Steffens AB. Hypothalamic influence on insulin and glucagon release in the rat. Am. J. Physiol. 1977;233:E380–E388. doi: 10.1152/ajpendo.1977.233.5.E380. [DOI] [PubMed] [Google Scholar]
- 115.Langhans W, Pantel K, Müller-Schell W, Eggenberger E, Scharrer E. Hepatic handling of pancreatic glucagon and glucose during meals in rats. Am. J. Physiol. 1984;247(Pt 2):R827–R832. doi: 10.1152/ajpregu.1984.247.5.R827. [DOI] [PubMed] [Google Scholar]
- 116.Unger RH, Orci L. Physiology and pathophysiology of glucagon. Physiol. Rev. 1976;56:778–826. doi: 10.1152/physrev.1976.56.4.778. [DOI] [PubMed] [Google Scholar]
- 117.Le Sauter J, Noh U, Geary N. Hepatic portal infusion of glucagon antibodies increases spontaneous meal size in rats. Am. J. Physiol. 1991;261(Pt 2):R162–R165. doi: 10.1152/ajpregu.1991.261.1.R162. [DOI] [PubMed] [Google Scholar]
- 118.Langhans W, Zeiger U, Scharrer E, Geary N. Stimulation of feeding in rats by intraperitoneal injection of antibodies to glucagon. Science. 1982;218:894–896. doi: 10.1126/science.7134979. [DOI] [PubMed] [Google Scholar]
- 119.Geary N, Kissileff HR, Pi-Sunyer FX, Hinton v. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am. J. Physiol. 1992;262(Pt 2):R975–R980. doi: 10.1152/ajpregu.1992.262.6.R975. [DOI] [PubMed] [Google Scholar]
- 120.Geary N, Le Sauter J, Noh U. Glucagon acts in the liver to control spontaneous meal size in rats. Am. J. Physiol. 1993;264(Pt 2):R116–R122. doi: 10.1152/ajpregu.1993.264.1.R116. [DOI] [PubMed] [Google Scholar]
- 121.Martin JR, Novin D, vanderweele DA. Loss of glucagon suppression of feeding after vagotomy in rats. Am. J. Physiol. 1978;234:E314–E318. doi: 10.1152/ajpendo.1978.234.3.E314. [DOI] [PubMed] [Google Scholar]
- 122.Geary N, Smith GP. Selective hepatic vagotomy blocks pancreatic glucagon's satiety effect. Physiol. Behav. 1983;31:391–394. doi: 10.1016/0031-9384(83)90207-x. [DOI] [PubMed] [Google Scholar]
- 123.Kurose Y, et al. Effects of central administration of glucagon on feed intake and endocrine responses in sheep. Anim. Sci. J. 2009;80:686–690. doi: 10.1111/j.1740-0929.2009.00685.x. [DOI] [PubMed] [Google Scholar]
- 124.Bobe G, Ametaj BN, Young JW, Beitz DC. Potential treatment of fatty liver with 14-day subcutaneous injections of glucagon. J. Dairy Sci. 2003;86:3138–3147. doi: 10.3168/jds.S0022-0302(03)73915-0. [DOI] [PubMed] [Google Scholar]