

Abstract
Thalassospiramides A and B are immunosuppressant cyclic lipopeptides first reported from the marine α-proteobacterium Thalassospira sp. CNJ-328. We describe here the discovery and characterization of an extended family of 14 new analogues from four Tistrella and Thalassospira isolates. These potent calpain 1 protease inhibitors belong to six structure classes in which the length and composition of the acylpeptide side chain varies extensively. Genomic sequence analysis of the thalassospiramide-producing microbes revealed related, genus-specific biosynthetic loci encoding hybrid nonribosomal peptide synthetase/polyketide synthases consistent with thalassospiramide assembly. The bioinformatics analysis of the gene clusters suggests that structural diversity, which ranges from the 803.4 Da thalassospiramide C to the 1291.7 Da thalassospiramide F, results from a complex sequence of reactions involving amino acid substrate channeling and enzymatic multi-module skipping and iteration. Preliminary biochemical analysis of the N-terminal NRPS module from the Thalassospira TtcA megasynthase supports a biosynthetic model in which in cis amino acid activation competes with in trans activation to increase the range of amino acid substrates incorporated at the N-terminus.
Introduction
The multienzymatic thiotemplate mechanism of polyketide and nonribosomal peptide biosynthesis is a common strategy for the construction of complex natural products in microbes.1,2 These biosynthetic assembly lines typically operate in a linear fashion whereby enzymatic substrates are sequentially incorporated into the growing product chain while it remains enzyme bound via thioester linkages throughout the entire synthetic process.3,4 In many cases, such as for the prototypical polyketide synthase (PKS) 6-deoxyerythronolide B synthase5 and the nonribosomal peptide synthetase (NRPS) surfactin synthetase,6 product assembly displays a strict correlation between the enzymatic domain sequence of the megasynthase and the position of malonate/amino acid building blocks in the polyketide/peptide product. This co-linear arrangement has facilitated the engineered biosynthesis of designer metabolites7,8 and the bioinformatics-guided prediction and discovery of new chemical entities from genome-sequenced organisms.9–12 However, the co-linearity rule is not strictly enforced in nature.13,14 Many polyketides, nonribosomal peptides, and hybrids thereof are products of pathways in which enzymatic functions are skipped, repeated, or complimented altogether.15–20 Herein we report an extreme departure from sequential assembly line biosynthesis, associated with the production of the thalassospiramide family of cyclic lipopeptides, which combines many non-canonical strategies for product diversity.
Thalassospiramides A (1) and B (2) were first described by Fenical and co-workers as immunosuppressant agents co-produced by the marine α-proteobacterium Thalassospira sp. CNJ-328.21 These cyclic lipopeptides differ in structure at the N-terminus in which the proteinogenic serine residue in 1 is replaced with the nonstandard phenylalanine-based statine residue 4-amino-3-hydroxy-5-phenylpentanoic acid (Ahppa). We report the characterization of an extended family of 14 new thalasssospiramide molecules from the original producer Thalassospira sp. CNJ-328 and three other marine α-proteobacteria Thalassospira sp. TrichSKD10, Tistrella mobilis KA081020-065 and Tistrella bauzanensis TIO7329. We further describe the genetic and biochemical basis for their biosynthetic diversity through the sequence analysis of their biosynthesis genes and the biochemical characterization of the thalassospiramide synthetase.
Results and Discussion
Isolation and characterization of thalassospiramide lipopeptides from four marine bacteria
We recently reported that the marine α-proteobacteria Tistrella mobilis KA081020-065 and Tistrella bauzanensis TIO7329 from the Red Sea and the Pacific Ocean, respectively, produce the anticancer agent didemnin B.22 Further chemical analysis of these microbes revealed that they produced a second group of unrelated lipopeptides. We first isolated and characterized thalassospiramide A (1) from both strains and noted its several-fold increased production upon iron supplementation of the growth media. We, however, did not detect thalassospiramide B (2), which was previously reported along with 1 as a product of the marine α-proteobacterium Thalassospira sp. CNJ-328.21 Instead, we isolated eight new thalassospiramide analogues (3–10). Inspection of the original producer Thalassospira sp. CNJ-328 and the related strain Thalassospira sp. TrichSKD10, on the other hand, revealed their ability to synthesize not only 1 and 2, but all eight of the new Tistrella thalassospiramides as well as six additional derivatives (11–16) unique to the Thalassospira isolates.
We divided the variants into two structural classes, thalassospiramide A-like and thalassospiramide B-like, distinguished by the identity of the N-terminal amino acid residue. Thalassospiramide A-like molecules (3–11) contain a standard amino acid residue at position one, whereas the thalassospiramide B-like molecules (12–16) have a statine-like amino acid at this position (Figure 1). We further grouped the 16 thalassospiramides into six subclasses based on distinguishing structural features: the A and B groups have a variety of amino acids in the N-terminal residue (shown in blue in Figure 1), the C and D groups are truncated (absence of green structure unit), and the E and F groups are elongated (repetition of the green structure unit). Structure diversity is also evident in the 3Z-decenyl lipid residue that is saturated in several variants (4, 5, 8).
Figure 1.

Structures of A) thalassospiramide A-like and B) thalassospiramide B-like natural products (blue portions indicate N-terminal amino acids used to distinguish the two groups, green portions highlight a repetitive amino acid motif, red portions indicate a common valine residue and black portions are conserved throughout the family). Producing organism(s) shown in brackets include Tistrella mobilis (TM), Tistrella bauzanensis (TB), Thalassospira sp. TrichSKD10 (TT), and Thalassospira sp. CNJ-328 (TC). Several compounds (4,5,8) have a saturated fatty acid at the N-terminus (sat. FA).
We extensively characterized at least one member of each subclass (A–F), including the previously reported 1 and 2, by NMR, MS, and Marfey analyses (see Supporting Information). The stereochemistry of the proteinogenic amino acid residues was assigned using the advanced Marfey method23–25 in which the lipopeptides were first hydrogenated, hydrolyzed, and then reacted with either L- or D-1-fluoro-2,4-dinitrophenyl-5-leucine amide (FDLA, advanced Marfey’s reagent). Analysis by LC-MS revealed that all α-amino acids were in the L configuration and that the hydrogenated 4-amino-5-hydroxypenta-2-enoic acid (Ahpea) was assigned as R. These assignments are all in agreement with the results previously reported for 1 and 2. The stereochemistry shown for the statine units 4-amino-3,5-dihydroxypentanoic acid (Adpa) and 4-amino-3-hydroxy-5-phenylpentanoic acid (Ahppa) is based upon prior assignments for A (1) and B (2) by Fenical and co-workers.21
Two of these compounds, thalassospiramides B1 (12) and D1 (15), were described previously in the patent literature from the α-proteobacterium Oceanospirillum sp. SANK 70992 and were shown to have nanomolar inhibitory action against the cysteine protease calpain 1.26 We thus explored the activity of a selection of our thalassospiramide biosynthetic library against calpain 1 using a fluorescence-based assay (Table 1).27 All compounds tested showed nM inhibitory activity. The truncated thalassospiramide C (7), however, was the most potent at nearly an order of magnitude (3.4±1.2 nM) more active than the others.
Table 1.
Inhibitory activity of selected thalassospiramides against human calpain 1 protease*
| Compound | IC50 (nM) |
|---|---|
| Thalassospiramide A (1) | 56.5 ± 2.4 |
| Thalassospiramide A1 (3) | 42.3 ± 42.3 |
| Thalassospiramide B (2) | 29.0 ± 2.0 |
| Thalassospiramide C (7) | 3.4 ± 1.2 |
| Thalassospiramide D1 (15) | 34.7 ± 2.5 |
| Thalassospiramide E1 (10) | 20.9 ± 1.6 |
| Positive control- Z-Leu-Leu-Tyr-fluoromethylketone | 16.2 ± 2.2 |
Activity of 12 and 15 against human calpain protease 1 was previously reported, with IC50 values of 80 and 39 nM, respectively.26
Identification and analysis of the non-canonical thalassospiramide biosynthetic gene clusters
To gain an understanding of the molecular basis for thalassospiramide chemical diversity, we characterized the associated biosynthetic gene clusters in all four strains. We had previously reported the complete genome sequence of T. mobilis KA081020-065 in the course of our work on didemnin B biosynthesis.22 We compared the T. mobilis sequence with the publically available draft genome of Thalassospira sp. TrichSKD10 (available from https://moore.jcvi.org/moore/) and identified a common gene cluster encoding a hybrid nonribosomal peptide synthetase/polyketide synthase (NRPS/PKS). These were located on plasmid 1 (ORFs 1 and 603) of T. mobilis and contig 7 (nt 128,264–153,704) of Thalassospira sp. TrichSKD10. We also sequenced the draft genomes of T. bauzanensis TIO7329 and Thalassospira sp. CNJ-328 and identified related genus-specific thalassospiramide biosynthetic gene clusters (Figure 2). While the two thalassospiramide biosynthesis loci ttm and ttb from Tistrella share identical gene architecture, they differ in subtle yet distinct ways from the syntenic thalassospiramide biosynthesis loci ttc and ttt from Thalassospira (Figure 2). These key genetic differences correlate to the contrasts in thalassospiramide chemistry observed in each genus (Figure 1). Significantly, the Thalassospira thalassospiramide synthetase contains an additional PKS module (module 1b) that presumably allows for the assembly of the unusual N-terminal statine amino acid residue characteristic of the thalassospiramide B-like molecules particular to Thalassospira. A second striking difference between the two genetic systems is the absence of the vital catalytic adenylation (A) domains that are required for amino acid activation and loading in Tistrella modules 1 and 5. Protein alignment of the sequence bridging the condensation (C) and thiolation (T) domains in the five Tistrella NRPS modules and comparison with the crystal structure of the surfactin NRPS termination module28 revealed that the N-terminal core A domain (~ 400 amino acid residues) is missing for modules 1 and 5, whereas the small C-terminal sub-domain (~ 100 amino acids) persists (see Figures S18 and S19). This loss of two critical enzymatic domains poses an interesting challenge regarding catalysis at these positions and suggests that in trans substrate activation must occur.
Figure 2.
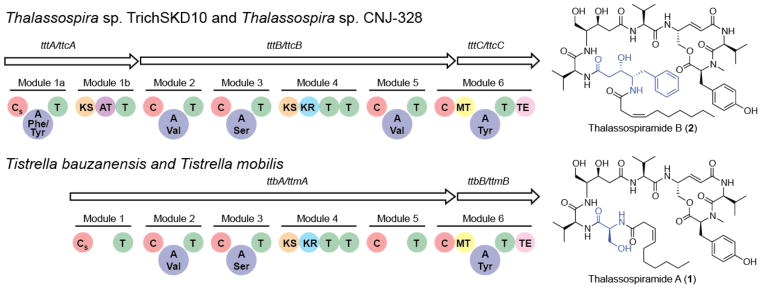
Homologous thalassospiramide gene clusters from Tistrella and Thalassospira. The T. mobilis KA081020-065 and T. bauzanensis TIO7329 thalassospiramide clusters ttm and ttb, respectively, encode the biosynthesis of thalassospiramide A-like molecules only, whereas the Thalassospira sp. CNJ-328 and Thalassospira sp. TrichSKD10 clusters ttc and ttt, respectively, encode the synthesis for both thalassospiramide A and B-like molecules. Domain abbreviations: Starter condensation (CS), condensation (C), adenylation (A), thiolation (T), ketosynthase (KS), acyltransferase (AT) ketoreductase (KR), methyltransferase (MT), thioesterase (TE).
Similarly, the PKS module 4 in both systems is also lacking its substrate activating acyltransferase (AT) domain, while the Thalassospira module 1b PKS harbors an AT domain. We previously observed that the didemnin PKS in T. mobilis also lacks cis AT domains22 and thus suggest that the fatty acid AT FabD may undertake this role in trans for the thalassospiramide and didemnin pathways. Alternatively, the Thalassospira system may uniquely employ the in trans application of the module 1b AT to complement module 4. Bioinformatics analysis of the ketosynthase (KS) domains in module 4 identifies them as trans KS domains and therefore likely capable of interaction with a trans AT.17
Despite these differences, the Thalassospira and Tistrella thalassospiramide genotypes are clearly related and share many common biosynthetic features. For example, all clusters contain a primary condensation domain (CS) consistent with the N-acylation of the N-terminal amino acid of lipopeptides such as in the co-produced didemnin X and Y from T. mobilis.22 Similarly, all clusters feature a separately encoded terminal NRPS module responsible for incorporation of the C-terminal N-methyl tyrosine residue and macrocyclization to yield the 12-membered ring common to all thalassospiramide derivatives.
One of the most salient features of the thalassospiramide system is the apparent mismatch of biosynthetic modules necessary to accommodate all observed molecules. The thalassospiramide megasynthetase possesses six modules in Tistrella and seven modules in Thalassospira, which is consistent with the linear biosynthesis of the thalassospiramide C and D groups, respectively. The other thalassospiramide groups require a previously unprecedented partially iterative usage of the synthetase involving modules 2–4. A further architectural anomaly that could support this hypothesis is the presence of two tandem thiolation domains at the end of module 4 (Figure 2), which may facilitate the shuttling of the peptide biosynthetic intermediate back to module 2 for further cycles of synthesis. Thus in the case of thalassospiramides belonging to the A and B groups, we propose a single repeat of modules 2–4, while the biosynthesis of thalassospiramides E1 and F must undergo yet another repeat. To the best of our knowledge, this type of multi-modular iteration has not been observed before in NRPS biochemistry. Previously reported examples of iteration include single module iteration such as for didemnins X and Y22 and whole peptide oligomerization such as for gramicidin S16 and enterobactin.29,30 We propose that in thalassospiramide biosynthesis the growing peptide chain is actually translocated backward within the cluster from module 4 back to module 2 for a second (or third) round of synthesis.
Biochemical evaluation of the TtcA CAT tridomain
One of the main differences between the biosynthetic clusters for the two genera is the presence of the unique PKS module 1b within the Thalassospira cluster. This discrepancy is ascribed to the synthesis of the C2 portion of the statine-like amino acids at the N-terminus of thalassospiramides in the B-like series. These organisms, however, are still capable of making thalassospiramide Alike molecules, where in fact they dominate the thalassospiramide mixture. To investigate whether module 1a is responsible for incorporating the first amino acid residue observed in any or all of the isolated molecules, we probed the amino acid selectivity of its A-domain. In silico predictions involving antiSMASH31 and NRPSpredictor232 were unreliable and gave different results.
Based on the chemical diversity of amino acid analogues seen for residue 1 (Ser, Val, Phe, Tyr), a promiscuous enzyme specific for four substrates of various polarity seemed unreasonable. Thus we biochemically explored the amino acid selectivity of the module 1a CAT-tridomain of TtcA by the phosphopantetheine ejection assay (Figure 3).33,34 We cloned and overexpressed the TtcA(CAT) as an N-terminal octahistidine tagged fusion protein in Escherichia coli BL21(DE3). The soluble apoprotein was purified by Ni-affinity chromatography and converted to the functional 124 kDa holo enzyme with the phosphopantetheinyl transferase enzyme Sfp. The active enzyme was then incubated with all proteinogenic amino acids in the presence of ATP, then digested with trypsin, and fractionated by HPLC. The fragment containing the active site serine residue posttranslationally modified with CoA was first identified with bodipy (fluorescently) labeled coenzyme A.35 Candidate fractions were then analyzed by FT-ICR MS, and fragmentation of candidate ions allowed identification of phosphopantetheinated peptides (Figure 3). MS3 fragmentation of these ions yielded fragments consisting of phosphopantetheine and the covalently attached amino acid substrate. We found that the TtcA A-domain activated L-phenylalanine (as in thalassospiramides B (2) and A5 (11)) and L-tyrosine (as in thalassospiramides A1 (3) and B1 (12)), but was unable to activate L-serine (as in thalassospiramide A (1) or B2 (13)), L-valine (as in thalassospiramide A4 (6)) or any other amino acids. This result confirms that the TtcA A-domain has narrow amino acid specificity, limited to specific aromatic side-chains.
Figure 3.

Determination of the amino acid selectivity of TtcA(CAT) via the phosphopantetheine ejection assay. Shown are the results for the holo enzyme without addition of amino acids and after treatment of the enzyme with L-phenylalanine or L-tyrosine. All other proteinogenic amino acids, including serine and valine, were not loaded as substrates (see Figures S1–S2).
Upon a second inspection, an intriguing parallel can be drawn between the two thalassospiramide synthetase systems. As with the Tistrella module 1 that lacks a cognate A-domain and thus must be complemented in trans, the Thalassospira module 1a must also interact with a trans A-domain to deliver serine and valine residues that are not activated by the cis Phe/Tyr-specific A-domain (Figure 4). While a trans acting A-domain encoded outside the gene cluster, which competes in the case of the Thalassospira or supplements in the case of the Tistrella thalassospiramide synthetase, is mechanistically plausible, a compelling biosynthetic model may involve internal supplementation from the other cis A-domains in the thalassospiramide megasynthetase.
Figure 4.

Representation of the proposed thalassospiramide biosynthetic pathways in Thalassospira and Tistrella. The pale grey ribbons represent the linear sequence of the gene products, black lines depict the modules located within the same protein, black arrows show the forward/backward movement of the growing peptide chain along the enzyme assembly line and grey arrows show the intermodular movement of amino acid substrates from supplementing A-domains to T-domains.
All observed thalassospiramide analogues at position 1 carry amino acid residues that are also located elsewhere in the molecule, where specific A-domains likely function dually to select and activate a specific amino acid residue as a substrate for its cognate T-domain (cis acting) as well as for the module 1/1a/5 T-domains (trans acting). This approach is reminiscent of the biosynthesis of yersiniabactin, where a single cysteine A-domain loads three different T-domains.36–38 In the case of the thalassospiramides, multiple A-domains would need to be involved. For instance, we propose that the valine-specific module 2 A-domain in the Tistrella thalassospiramide synthetase services T-domains in modules 1, 2, and 5 to produce thalassospiramide A4 (6). While an aminoacyltransferase could be involved in shuttling the amino acids from one module to another as seen in syringomycin biosynthesis,20 it would require a highly promiscuous shuttle or a series of different transferases to account for all of the analogues observed. In Figure 4, we display a proposed 2-dimensional representation of our biosynthetic proposal encompassing in trans amino acid supplementation, module skipping and multi-module iteration to allow for the observed structural diversity represented in the natural thalassospiramide chemical library.
Conclusion
This study illuminates the molecular basis governing the non-canonical biosynthesis of the thalassospiramide family of cyclic lipopeptide calpain I protease inhibitors. While some of these atypical NRPS biochemical features of intra-synthetase trans-A domain activation, module skipping, and multi-module iteration have been previously observed in other biosynthetic systems, the thalassospiramides represent an unprecedented amalgamation of these rare biosynthetic mechanisms. Efforts are ongoing to probe further this intriguing biosynthetic pathway and to learn what makes this multitasking synthetase so peculiar.
Experimental Section
Fermentation, Isolation and Characterization of Thalassospiramides
To isolate thalassospiramides, T. mobilis and T. bauzanensis were grown in GYP media [glucose (10 g/L), yeast extract (4 g/L), peptone (2 g/L) and FeCl3 (0.5 mM at pH 7.4) at 28 °C for 4 days. For each species, 20 x 1 L cultures in 2.8 L Fernbach flasks were grown. T. sp. TrichSKD10 and T. sp. CNJ-328 were grown in a seawater based GYP media at 28 °C for 2 days. For each species, 40 x 1 L cultures were grown for isolation purposes. The same extraction and crude purification was done for all strains. Briefly, an equal volume of EtOAc was added to the bacterial cultures and mixed. The aqueous layer was discarded and the EtOAc was removed under reduced pressure. The crude extract was then subjected to flash C-18 column chromatography, eluting with 30%, 70%, and 100% aqueous MeOH to yield three fractions. The fractions containing thalassospiramides were further purified using reversed phase HPLC (Phenomenex Luna 5μm C18(2), 250 × 100 mm, 100 Å, 3.0 mL/min) with the following conditions. The 70% MeOH fraction from T. mobilis was separated with an isocratic method of 42% CH3CN in water to yield 6 (3.0 mg) and 9 (3.0 mg) at 25.5–27 min and 19.5–21.0 min, respectively. From the same extract 10 (1.0 mg) was obtained using an isocratic method of 38% CH3CN in water and eluted at 74.0–84.0 min. The 70% MeOH fraction from T. bauzanensis was separated with an isocratic method of 51% CH3CN in water to yield 1 (6.0 mg), 3 (3.0 mg), and 7 (3.0 mg) at 10.0–12.0 min, 14.5–16.0 min, and 26.7–28.2 min, respectively. The 100% MeOH fraction from Thalassospira sp. TrichSKD10 was separated with an isocratic method of 50% CH3CN in water to yield 16 (1.2 mg), 2 (2.0 mg), 11 (3.0 mg), and 14 (1.5 mg) at 23.8–25.2 min, 29.2–32.0 min, 36.5–39.5 min, and 46.5–51.5 min, respectively.
To determine the distribution of compounds produced by each strain, a 50 mL culture of each organism was extracted after 3 days using EtOAc (250 mL). Following concentration in vacuo these samples were analyzed and compared by LCMS (1.7μm C18 column, 0.25ml/min, gradient from 5% to 95% CH3CN with 0.1% TFA, 25min).
11D NMR and 2D NMR spectra were obtained with either a 500 MHz or a 700 MHz Varian Inova 500 MHz spectrometer. All samples were dissolved in pyridine except thalassospiramides A4 and C, which were dissolved in DMSO–d6. ESI-TOF-HRMS analysis was conducted with a Bruker micrOTOF instrument (Bruker Daltonics GmbH, Bremen, German). MSn analysis was carried out on a LTQ Velos dual-pressure ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Determination of the Absolute Amino Acid Configurations
To enhance the stability of the thalassospiramides in acid hydrolysis, the Ahpea unit of each compound was converted into 4-amino-5-hydroxy-pentanoic acid (Ahpa). Thalassospiramides A, A1, A4, A5, B, C, D, E, and F (0.5 mg each) were dissolved in MeOH (2 mL), and Pd/C was added to a final concentration of 10%. After stirring for 10 h under an atmosphere of H2, the catalyst was removed by filtration and the resultant product was examined by LC-MS. The hydrogenated product was then hydrolyzed with HCl (6 M, 0.5 mL) in a sealed glass tube at 110 °C for 24 h. The hydrolysate was evaporated to dryness to remove any residual acid, then dissolved in NaHCO3 (1 M, 100 μL), and split into two parts. L-FDLA (3 mg/mL, 50 μL) was added to one part, whereas D-FDLA (3 mg/mL, 50 μL) was added to the other. The two mixtures were incubated at 80 °C for 5 min and were then quenched by addition of HCl (2 M, 50 μL). Finally 50% aqueous CH3CN (200 μL) was added to dissolve the reaction products. The derivative amino acids were analyzed by LC-MS (1.7μm C18 column, 0.25 ml/min, gradient from 5% to 95% CH3CN with 0.1% TFA, 25 min).
Calpain Activity Assay
Calpain 1 (Active Human Calpain 1, BioVision) activity was measured according to the protocol for Calpain Activity Assay Kit (BioVision). Z-Leu-Leu-Tyr-fluoromethylketone (Z-LLY-FMK, BioVision), a known calpain inhibitor, was used as a positive control. Thalassospiramides A, A1, C, D1, E1, and Z-LLY-FMK were dissolved in DMSO to give 1 mM stock solutions, and then serially diluted with MeOH or acetone, depending on solubility. The highest concentration tested was 0.1 mM. First, extraction buffer (85 μL) was added into each assay well of a 96-well plate, followed by addition of calpain 1 (0.1 U). Then, a test compound (1 μL) with desired concentration was added into each well. Reaction buffer (10 μL) and calpain substrate (5 μL) were added into each well and the plate was incubated at 37 °C for 1 h in the dark. After incubation, the samples were recorded by a fluorometer equipped with a 400 nm excitation filter and 505 nm emission filter. A blank assay was run without calpain or a test compound. A negative control was run without a test compound. There were three replicates for each tested concentration. The experiment was repeated for three times.
Gene Cluster Sequencing and Analysis
Sequence data was obtained for both Tistrella bauzanensis and Thalassospira sp. CNJ-328. Sequencing for Tistrella bauzanensis was performed on the Illumina Hiseq2000. A total of 17 million pairs of Illumina reads were obtained from a 170bp and a 300bp paired-end library. Genome assembly was performed using the program Velvet 1.0.15 with the following custom parameters: hash-length = 55 and coverage cut-off = 30. Ion Torrent technology was utilized for the Thalassospira sp. CNJ-328 draft genome. Following sequencing with the Ion PGM sequencer, the contigs were assembled to the putative cluster from Thalassospira sp. TrichSKD10 using the software Geneious (version 5.1.7 created by Biomatters). Sequencing of PCR products was used to close gaps in the resultant sequence for the gene cluster from Thalassospira sp. CNJ-328. The sequence of the cluster in Tistrella mobilis was revised by sequencing PCR products.
The sequences of the thalassospiramide gene clusters can be accessed from the Genbank database with the following accession numbers: T. bauzanensis (KC181865), Thalassospira sp. CNJ-328 (KC181865), and plasmid 1 (cluster consists of ORF 1 and ORF 603) of T. mobilis (CP003237). Draft genome of Thalassospira sp. Trich SKD10 (cluster located at nt 128,264–153,704 on contig 7) can be obtained from the JCVI, Gordon and Betty Moore Foundation Marine Microbial Genome Sequencing Project (https://moore.jcvi.org/moore/).
In silico analysis of the gene clusters by protein-protein BLAST and Pfam analysis was performed.39 This enabled assignment of putative roles for the proteins within the clusters. Prediction of adenylation domain specificity was done using the online software antiSMASH and NRPSpredictor2.31,32 Analysis of condensation and ketosynthase domain functions was performed using the online software NaPDoS.40
Production of TtcA(CAT) by Heterologous Expression
The portion of the gene cluster coding for TtcA(CAT) was amplified by PCR using 5′-dGACCCATGGCTTGCAGAAGCGCACAAACAC-3′ as the forward primer (NcoI site underlined and the translation start site is highlighted in bold) and 5′-dGACAAGCTTTCAGCGTTGGGTCTTTACCGGTT-3′ as the reverse primer (HindIII site underlined and the stop codon is highlighted in bold) from Thalassospira sp. CNJ-328 genomic DNA. The 3.3 kb PCR product was gel purified, digested with NcoI and HindIII followed by a second gel purification, and then ligated (T4 DNA ligase) with NcoI/HindIII digested pHIS841 to generate the expression vector. The construct was transformed into E. coli TOP10 cells and ttcA(CAT) insert checked by sequencing. The construct was then further transformed into E. coli BL21(DE3) for expression. Transformed E. coli was grown in LB with kanamycin (50 μg/mL) at 28 °C until A600nm = 0.4–0.8. Culture was allowed to cool to 18 °C for 30 min and then induced with isopropyl-1-thio- β-D-galactopyranoside (0.5 M, 100 μL). The culture was grown for an additional 20 h and then the cells were pelleted by centrifugation. The cell pellet was re-suspended in phosphate lysis buffer (50 mM sodium phosphate buffer pH 8, 300 mM NaCl, 10 mM imidazole) and incubated at 0 °C for 30 min in the presence of lysozyme (1 mg/mL). The mixture was then sonicated and incubated for a further 15 min at 0 °C with DNAse 1 (1 U/μL). The cellular debris was removed via centrifugation and the resultant supernatant was incubated with Ni-NTA resin (0.5 mL of a 50% slurry in lysis buffer) for 1 h at 0 °C. The mixture was loaded onto a column and the resin was washed with the same phosphate buffer containing increasing concentrations of imidazole (20 mM, 60 mM). The desired proteins were eluted with phosphate buffer containing high concentration of imidazole (250 mM imidazole). The elution fraction was dialyzed with Tris HCl buffer (50 mM at pH 7.5 containing 10% (v/v) glycerol) and was then concentrated to 2.5 mg/mL. Protein samples were stored at −20 °C until use.
Synthesis of Bodipy-CoA (adapted from La Clair et al.)35
DMSO (600 μL) was added to 10 mM Tris HCl buffer pH 7 (3.8 mL), followed by coenzyme A (10 mM, 58 μL). The mixture was allowed to cool at 0 °C in the dark and then bodipy (25 μg/μL in DMSO, 20 μL) was added. The reaction was allowed to stir at 0 °C for 30 min and then after warming to room temperature, mercaptoethanol (12 μL) was added. Reaction mixture was washed with EtOAc (4 × 5 mL) and then the resultant aqueous solution was stored at −80 °C.
In vitro Formation of Aminoacylated TtcA(CAT), Digestion and HPLC Purification
To generate holo-TtcA, 50 mM Tris HCl buffer pH 7.5 with 10% (v/v) glycerol (2.5 μL), MgCl2 (100 mM, 5 μL), tris(2-carboxyethyl)phosphine (2 mM, 1.5 μL), coenzyme A (10 mM, 3.13 μL) and Sfp (170 μM, 1.3 μL) were added to the apo form of TtcA(CAT) (20 μM, 50 μL) and the mixture then incubated at 25 °C for 1 h. To test the amino acid loading capability of the holo enzyme, ATP (100 mM, 3.5 μL), an amino acid (50 mM, 1.4 μL), and 50 mM Tris HCl buffer pH 7.5 with 10% (v/v) glycerol (1.6 μL) were added and the mixture incubated for 30 min at 25 °C. After adjusting the reaction mixture to pH 8 with 1M Tris HCl it was incubated at 25 °C with trypsin (1 μg, Sigma Aldrich proteomics grade) for 5 min. Formic acid (3.5 μL) was then added, and the sample was directly analyzed by HPLC. Using a Higgins Analytical PROTO 300 5 μm C4 300 Å column, the samples were purified using the following 66 min method with 0.1% TFA in both the water and CH3CN solutions: 10% CH3CN from 0–10 min, ramp to 30% by 15 min, ramp to 70% by 55 min, ramp to 90% by 60 min, hold at 90% until 60.1 min, ramp down to 5% by 60.2 min, hold at 5% until 62.6 min, ramp to 95% by 63 min, hold at 95% until 65 min and ramp to 5% by 66 min. 1 min fractions were collected and immediately frozen at −80 °C and then lyophilized. Candidate fractions for analysis were collected based upon retention time of bodipy-CoA holo TtcA(CAT) fragments (HPLC monitored by UV/Vis spectroscopy at 220, 254 and 504nm). The assay was completed with a mixture of all 20 amino acids or with tyrosine/phenylalanine as substrates for the loading step.
Mass Spectrometric Analysis of Protein Samples
Lyophilized HPLC fractions were dissolved in ESI solution (30 μL, containing 50% LC-MS grade MeOH, 49% LC-MS grade water, and 1% formic acid), then gently vortexed for 30 s, and centrifuged for 2 min at 14000 rpm. Analysis was performed using a hybrid 6.4T LTQ-FT (Thermo Electron, North America) mass spectrometer. The instrument first tuned to m/z 816.3 using bovine cytochrome c (Sigma-Aldrich). Samples were introduced by electrospray ionization (1.4–1.5 kV, 0.4 psi) using a Biversa Nanomate 100 (Advion Biosciences) nanospray device in positive ion mode. Detection using the low resolution LTQ was performed using 1000 ms maximum ion accumulation time. An 8000 ms maximum ion accumulation time and a resolution of 100000 were used for FT-ICR-MS detection. MS2 and MS3 analysis for PPant ejection and PPant fragmentation were performed as described previously.42
Active Site Mapping and Substrate Identification of TtcA(CAT)
Candidate active site peaks were identified based on bodipy-retention time and PPant ejection.34 Peak lists were generated from RAW-files with XTRACT software (Thermo Finnigan, Bremen, Germany). The generated peak list was imported into the freeware PAWS to identify the active sites. A match to the active site of the T domain was confirmed by PPant ejection, i.e. MS2 fragmentation of the corresponding active site peak, and PPant fragmentation, i.e. MS3 fragmentation of the ejected PPant ion.43 Loaded substrates on the T domain active site were identified by mass shift of the active site fragment (MS1), the PPant ion (MS2) and the PPant fragments (MS3).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge W. Fenical from UCSD for Thalassospira sp. CNJ-328 and E. Mann from the Skidaway Institute of Oceanography for Thalassospira sp. TrichSKD10. Tistrella bauzanensis TIO7329 was generously provided from the Third Institute of Oceanography, China PRC. J. P. Noel at the Salk Institute for Biological Sciences kindly provided access to the Ion Torrent sequencing instrument. Financial support was provided by the China Ocean Mineral Resources Research and Development Association (DY125-15-T-02), the King Abdullah University of Science and Technology (SA-C0040/UK-C0016 to P.Y.Q.), and the NIH (GM97509 to B.S.M. and P.C.D.).
Footnotes
The authors declare no competing financial interest.
Supporting Information. NMR spectra and characterization tables, annotated MS2 spectra, high resolution MS spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Condurso HL, Bruner SD. Nat Prod Rep. 2012;29:1099. doi: 10.1039/c2np20023f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koglin A, Walsh CT. Nat Prod Rep. 2009;26:987. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hur GH, Vickery CR, Burkart MD. Nat Prod Rep. 2012;29:1074. doi: 10.1039/c2np20025b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pieper R, Luo G, Cane DE, Khosla C. Nature. 1995;378:263. doi: 10.1038/378263a0. [DOI] [PubMed] [Google Scholar]
- 6.Kraas FI, Helmetag V, Wittmann M, Strieker M, Marahiel MA. Chem Biol. 2010;17:872. doi: 10.1016/j.chembiol.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen KT, Ritz D, Gu JQ, Alexander D, Chu M, Miao V, Brian P, Baltz RH. Proc Natl Acad Sci U S A. 2006;103:17462. doi: 10.1073/pnas.0608589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao CM, Katz L, Khosla C. Science. 1994;265:509. doi: 10.1126/science.8036492. [DOI] [PubMed] [Google Scholar]
- 9.Lautru S, Deeth RJ, Bailey LM, Challis GL. Nat Chem Biol. 2005;1:265. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 10.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc Natl Acad Sci U S A. 2007;104:10376. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergmann S, Schümann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. Nat Chem Biol. 2007;3:213. doi: 10.1038/nchembio869. [DOI] [PubMed] [Google Scholar]
- 12.Kersten RD, Yang YL, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. Nat Chem Biol. 2011;7:794. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenzel SC, Müller R. Curr Opin Chem Biol. 2005;9:447. doi: 10.1016/j.cbpa.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Lane AL, Moore BS. Nat Prod Rep. 2011;28:411. doi: 10.1039/c0np90032j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen B, Du L, Sanchez C, Edwards DJ, Chen M, Murrell JM. J Nat Prod. 2002;65:422. doi: 10.1021/np010550q. [DOI] [PubMed] [Google Scholar]
- 16.Hoyer KM, Mahlert C, Marahiel MA. Chem Biol. 2007;14:13. doi: 10.1016/j.chembiol.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piel J. Nat Prod Rep. 2010;27:996. doi: 10.1039/b816430b. [DOI] [PubMed] [Google Scholar]
- 18.Galm U, Wendt-Pienkowski E, Wang L, Huang SX, Unsin C, Tao M, Coughlin JM, Shen B. J Nat Prod. 2011;74:526. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 20.Singh GM, Vaillancourt FH, Yin J, Walsh CT. Chem Biol. 2007;14:31. doi: 10.1016/j.chembiol.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Oh DC, Strangman WK, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2007;9:1525. doi: 10.1021/ol070294u. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Kersten RD, Nam SJ, Lu L, Al-Suwailem AM, Zheng H, Fenical W, Dorrestein PC, Moore BS, Qian PY. J Am Chem Soc. 2012;134:8625. doi: 10.1021/ja301735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marfey P. Carlsberg Res Commun. 1984;49:591. [Google Scholar]
- 24.Fujii K, Ikai Y, Mayumi T, Oka H, Suzuki M, Harada K-i. Anal Chem. 1997;69:3346. [Google Scholar]
- 25.Fujii K, Ikai Y, Oka H, Suzuki M, Harada K-i. Anal Chem. 1997;69:5146. [Google Scholar]
- 26.Kaneko I, Minekura H, Takeuchi Y, Kodama K, Nakamura T, Haruyama H, Sakaida Y. Organization, W. I. P, editor. European Patent Office. Sankyo Co., Ltd; Japan: 1994. p. 94. [Google Scholar]
- 27.Leloup L, Shao H, Bae YH, Deasy B, Stoltz D, Roy P, Wells A. J Biol Chem. 2010;285:33549. doi: 10.1074/jbc.M110.123604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanovic A, Samel SA, Essen LO, Marahiel MA. Science. 2008;321:659. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- 29.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. Proc Natl Acad Sci U S A. 2000;97:2509. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw-Reid CA, Kelleher NL, Losey HC, Gehring AM, Berg C, Walsh CT. Chem Biol. 1999;6:385. doi: 10.1016/S1074-5521(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 31.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Breitling R, Takano E. Nucleic Acids Res. 2011;39:W339. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. Nucleic Acids Res. 2011;39:W362. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:12756. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorrestein PC, Blackhall J, Straight PD, Fischbach MA, Garneau-Tsodikova S, Edwards DJ, McLoughlin SM, Lin M, Gerwick WH, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:1537. doi: 10.1021/bi052333k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chem Biol. 2004;11:195. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Keating TA, Suo Z, Ehmann DE, Walsh CT. Biochemistry. 2000;39:2297. doi: 10.1021/bi992341z. [DOI] [PubMed] [Google Scholar]
- 37.Suo Z, Tseng CC, Walsh CT. Proc Natl Acad Sci U S A. 2001;98:99. doi: 10.1073/pnas.021537498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLoughlin SM, Kelleher NL. J Am Chem Soc. 2005;127:14984. doi: 10.1021/ja0555264. [DOI] [PubMed] [Google Scholar]
- 39.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. Nucleic Acids Res. 2010;38:D211. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. PLoS One. 2012;7:e34064. doi: 10.1371/journal.pone.0034064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Biochemistry. 2000;39:890. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- 42.Meehan MJ, Xie X, Zhao X, Xu W, Tang Y, Dorrestein PC. Biochemistry. 2011;50:287. doi: 10.1021/bi1014776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Bioorg Med Chem Lett. 2008;18:3107. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.