

Abstract
Current antiretroviral therapy for HIV-1 infection effectively suppresses but does not eradicate HIV-1. Patients on Highly Active Anti-Retroviral Therapy (HAART) maintain a persistent low-level viremia requiring lifelong adherence to antiretroviral therapies. This viremia may arise from latently infected reservoirs such as resting memory CD4+ T-cells or sanctuary sites where drug penetration is suboptimal. Understanding the mechanisms of HIV latency will help efforts to eradicate the infection. This review examines the dynamics of persistent viremia, viral reservoirs, the mechanisms behind viral latency, and methods to purge the viral reservoirs. This article forms part of a special issue of Antiviral Research marking the 25th anniversary of antiretroviral drug discovery and development.
Keywords: Heterochromatin, repression, latency, transcription, transcriptional interference, chromatin, primary latency models, virus, epigenetics, histone
Introduction
Highly active anti-retroviral therapy (HAART) was introduced for the treatment of HIV infection 15 years ago after development of effective anti-HIV drugs targeting the viral reverse transcriptase and the viral protease (Lassen, et al., 2004). HAART reduces plasma HIV RNA levels below the detection limit of viral RNA and greatly decreases the morbidity of HIV-1-infected patients. However, HAART interruption results in rapid rebound of replicating viruses within a few weeks in most patients. The inability of HAART to cure HIV has led to renewed interest in understanding the site of HIV persistence during HAART and to novel therapeutic strategies aiming at eliminating persistent HIV.
Plasma viral loads decay in two phases during HAART (Perelson, et al., 1996). In the first phase, infected activated CD4+ T cells are rapidly cleared and the virus levels decrease with a half-life of days. During the second decay phase, characterized by a half-life of weeks, other infected cells, including macrophages, and partially activated T cells can be detected in many patients on HAART using ultrasensitive techniques around 1–5 copies of viral RNA/ml, indicating the persistence of HIV production under HAART.
Different models have been proposed for this low level viremia: latency with episodic reactivation, persistent infection in replicating cells or in long-lived cells that are not eliminated by the immune system and the existence of anatomical sanctuaries that are not reached by the HAART drugs, for example in the gut-associated lymphatic tissues and the central nervous system. This review focuses on HIV latency in resting memory CD4+ T-cells and its mechanisms.
Pre- and post-integration latency
Following entry into its target cell, HIV integrates its own genome into the cellular genome. Most of these infections are productive: the viral replication cycle is completed within days (Perelson, et al., 1997, Perelson, et al., 1997). However, a small fraction of incoming HIV enters a latent mode of infection. This reservoir of infected cells produces infectious virus under appropriate stimulation. Although relatively small in number, the latent population is of great clinical importance because it serves as a source of virus for reinfection after interruption of HAART. The frequency of latently infected cells is very low, estimated to be on the order of 0.03–3 infectious units per million resting CD4+ T cells (Siliciano, et al., 2003).
Viral latency exists in two forms: pre- and post-integration latency. Pre-integration latency occurs when virions fail to undergo integration and remain in the cytoplasm of the infected cell for days as a labile pre-integration complex (PIC). This can occur when HIV infects a resting CD4+ T cell, in which viral nuclear import and integration are inhibited by the low metabolic state of the cell. If host cell activation occurs before decay of the PIC, integration and replication occur and the viral lifecycle is completed.
This review focuses on post-integration latency, which occurs when a provirus integrates successfully within the cellular genome but fails to effectively express its genome and thus is maintained in a transcriptionally silent state. Such a latent state with provirus blocked at the transcriptional level has been detected in patients infected with HIV and on HAART predominantly in memory CD4+ T lymphocytes (Chomont, et al., 2009). Viral production in latently infected resting CD4+ T cells can be induced by antigenic stimulation, cytokines, mitogens, or phorbol esters. Low level spontaneous reactivation from this latent pool has been proposed as a source of rebounding virus when HAART is interrupted but this has not been proven.
Molecular mechanisms of HIV latency
The study of HIV latency has benefited from the early development of in vitro models of HIV latency using transformed lymphoid cell lines. A critical and unanswered question is what distinguishes productive vs. latent infections at the molecular level. A number of molecular mechanisms have been advanced that can explain HIV latency. Since HIV integrates in a large variety of chromatin environments, it could integrate within chromatin environments that are incompatible with HIV transcription. Such integration events would lead to a reversible state of transcriptional inactivation of the HIV provirus, i.e. latency. Studies using an in vitro model of HIV latency, the J-LAT cells, which is a Jurkat-based cell line model, has shown that 1–2% of infection events lead to latency (Jordan, et al., 2003). These studies also showed that cis-acting effects (i.e. the site of integration of the provirus) and not transacting effects (i.e. the cellular factors that regulate HIV transcription) is responsible for latency in this system. Analysis of latent integration sites within the J-Lat system further showed that latent HIV preferentially integrates within the most highly transcribed genes (transcriptional interference), within gene deserts and within heterochromatin (Lewinski, et al., 2005). In agreement with previous observations, Lewinski and coworkers found that integration near alphoid repeats was enriched 8.6 fold in the latent population (4.3% of latent integration sites), suggesting that integration within or near heterochromatin can lead to latency. Furthermore, latent viral integration sites that were outside of known genes had a tendency to be located in “gene deserts”, while stably expressed proviruses were more likely to be found in short intergenic regions. Gene deserts, or long stretches between active genes, are thought to be compacted into heterochromatin and therefore less conducive to transcription than gene-rich regions.
Much of the early research efforts on HIV latency have been focused on studying the mechanism by which chromatin and associated factors can regulate HIV transcription and latency. In particular, we review the chromatin modifications that are known so far to act on the HIV promoter both to repress and activate transcription, respectively. Additionally, we also review a list of the transcription factors and co-factors that participate in the repression and activation of the HIV promoter.
Epigenetic Regulation of HIV latency
The DNA of eukaryotes is densely packed into a structure called chromatin in which DNA is tightly wrapped around nucleosomes. Each nucleosomes is composed of octamers of histone proteins containing two molecules of histone H2A, H2B, H3 and H4. The N-terminal domains of each histones projects outside of the nucleosomal core and are subject to numerous types of post-translational modifications including phosphorylation, acetylation, methylation and ubiquitination. These histone modifications are thought to contribute to transcriptional regulation by defining an “open” or “closed” state of chromatin (Robison & Nestler, 2011). The “open” configuration of chromatin facilitates transcription whereas the “closed” state is more often associated with a transcriptionally repressed state. Below, we discuss histone acetylation and methylation and the factors/complexes responsible in conducting these modifications on the HIV promoter.
1. Histone Acetylation/Deacetylation
We first reported 15 years ago that latent HIV could be transcriptionally reactivated by treatment of latently-infected cell lines by two HDAC inhibitors, trichostatin and trapoxin (Van Lint, et al., 1996). These results indicated that one or several HDACs were bound to the HIV promoter under latency conditions and that deacetylation of the HIV promoter chromatin by these enzymes played a role in the establishment and maintenance of HIV latency. This observation also indicated that treatment of HIV-infected patients with HDAC inhibitors might be of therapeutic value by forcing the reactivation of HIV expression and the elimination of the latent HIV reservoir (“flushing hypothesis”)(Van Lint, et al., 1996).
Later experiments revealed that transcriptional repressors recruit HDACs to the HIV promoter. Ying-Yan 1 (YY-1) and late SV40 factor (LSF) recruit histone deacetylase 1 (HDAC1) to the 5′LTR (Coull, et al., 2000) (Fig 1). The transcription factor NFκB, containing two subunits p50 and p65, plays a dual role on the HIV promoter. While the p50/p65 heterodimer is a potent activator of HIV transcription, the p50 homodimer represses HIV transcription in latently infected cell lines by recruiting HDAC1 to the HIV promoter (Williams, et al., 2006) (Fig 1 and Table 1)). Knockdown of p50 expression reduces HDAC1 binding to the 5′LTR and induces RNA polymerase II recruitment (Williams, et al., 2006). The transcriptional repressor c-promoter binding factor (CBF-1) also recruits HDAC1 by binding to the NFκB element in the enhancer region of the HIV-1 LTR (Tyagi & Karn, 2007). Knockdown of CBF-1 resulted in the partial reactivation of latent HIV-1, reduced HDAC1 binding, and recruitment of RNA polymerase II to the 5′LTR. In addition to all these factors that recruit HDAC1 to the HIV promoter, the corepressor COUP TF interacting protein (CTIP2), which interacts with Sp1, is also required in recruiting HDAC1 and as well as HDAC2 (Marban, et al., 2005, Marban, et al., 2007, Keedy, et al., 2009). Transcription factors AP-4 (Imai & Okamoto, 2006), c-Myc and Sp1(Jiang, et al., 2007) also recruit HDAC1. Another HDAC, HDAC3, is recruited to the HIV-1 5′LTR region in HIV-1 latently infected cell lines and is involved in the transcriptional repression of the HIV promoter (Marban, et al., 2007) (Fig.1).
Figure 1. The HIV promoter contains binding sites for transcriptional repressors and activators.

(A) Transcriptional repressors of the HIV promoter that maintain the provirus in the silent state consist of transcription factors such as CBF1, SP1, CTIP2, LSF and YY1. These transcription factors mediate the recruitment of effectors such as chromatin remodeling complexes (BAF), histone deacetylases (HDACs) and histone methyltransferases (SUV39H1, EZH2 and G9A). HDACs deacetylate histones and keep the HIV promoter inactive. The histone methyl transferases methylate histones and create a repressive mark. Nucleosome 1 (nuc-1) is stabilized by the BAF complex downstream of the HIV transcriptional start site on the silent HIV LTR; (B) Reactivation of latency is achieved by the concerted activities of histone acetyltransferases (HATs), such as p300, which acetylate the histones. p300 also acetylates the HIV transactivator protein Tat and the cellular NFκB subunit p65. The PBAF complex remodels nuc-1 in an ATP-dependent manner to facilitate transcriptional elongation.
Table 1.
Table 1 summarizes the different factors that play a role in HIV latency and their regulation by small molecules. The factors are grouped into boxes based on the type of function they are involved in i.e. interaction with histone deacetylases or histone acetyl transferases, whether they are histone methyl transferases, whether they bind to methylated DNA or whether they function on stabilizing nuc-1.
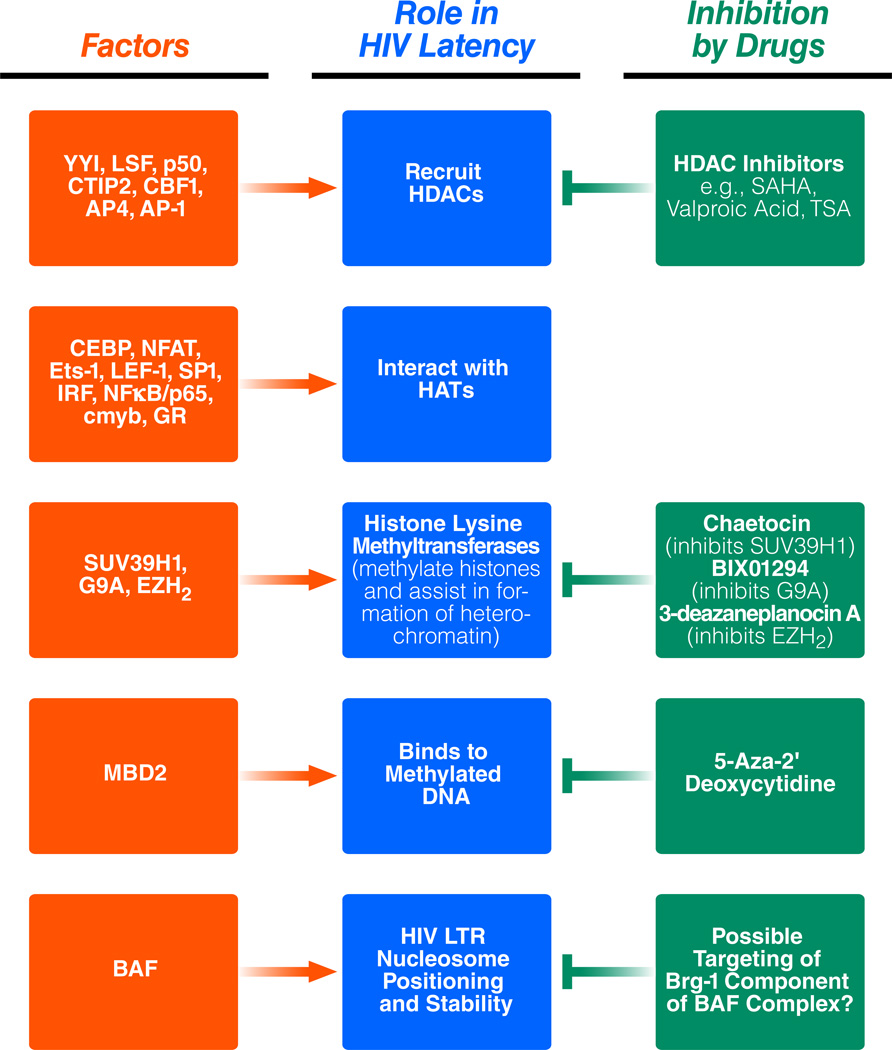 |
Upon activation of the HIV promoter by various stimuli, the NFκB p65 subunit accumulates in the nucleus, followed by the recruitment of cellular histone acetyltransferases (HATs), such as p300/CBP, PCAF, and GCN5, to 5′LTR promoter/enhancer region. These HATs are critical in the acetylation of the histone tails. Other transcription factors, such as AP-1, cMyb, GR, C/EBP, NFAT, Ets-1, LEF-1, Sp1, and IRF are recruited to the HIV LTR and interact with HATs to activate the HIV promoter (Lusic, et al., 2003, Marsili, et al., 2004, Colin & Van Lint, 2009) (Table 1). In addition, the HIV transactivator Tat also interacts with HATs, such as p300 and PCAF, and recruits them to the viral promoter, thus leading to acetylation of histones (Benkirane, et al., 1998). Acetylation of histones subsequently results in the recruitment of the SWI/SNF remodeling complexes (reviewed later) and the assembly of the transcriptional machinery to the viral promoter.
2. Histone Methylation
Histone methylation also plays a role in regulating the latent HIV promoter. The histone methyltransferase SUV39H1 is recruited to the HIV promoter and methylates histones. SUV39H1 is an H3Lys9 methyltransferase that initiates the formation of heterochromatin in Drosophila via recruiting the HP1 proteins (Benkirane, et al., 1998) (Cheutin et. al 2003). HP1 proteins exist in three isoforms (i.e., α, β and γ) and mostly establish a heterochromatic environment. However, HP1γ in particular, and some portion of HP1β, also localizes to euchromatic sites (Maison & Almouzni, 2004)(Maison and Almouzni 2004).
In the context of HIV latency, three studies have analyzed the molecular mechanism of maintenance of H3K9 methylation and HP1 and SUV39H1 recruitment to the 5′LTR. In HeLa cells, latently infected U-1 cells, Jurkat cells as well as peripheral blood mononuclear cells (PBMCs) from healthy donors, SUV39H1 is required for the deposition of trimethylated histones and that the trimethylated H3 histones provide a platform for HP1γ recruitment and subsequent repression of the HIV LTR (du Chene, et al., 2007). Upon HP-1γ knockdown, SUV39H1 was displaced from the LTR and levels of the H3K9 trimethylation mark were reduced. At the same time, the promoters were occupied by two well-known activators of HIV transcription: PCAF and pTEF-B. This resulted in the derepression of the HIV promoter (du Chene, et al., 2007).
Similarly, Coup-TF interacting protein 2 (CTIP2) associates with and recruits the chromatin-modifying complex of HDAC1, HDAC2, and SUV39H1 in microglial cells (Marban, et al., 2005) (Fig. 1 and Table 1). CTIP-2 recruitment of SUV39H1 allows trimethylation of H3K9 in the 5′LTR region, which in turn recruits all three isoforms of HP1 and creates a repressive chromatin environment. In luciferase assays with an integrated and episomal LTR, in TZM-bl and microglial cells, when SUV39H1 and CTIP2 were overexpressed together, there was functional cooperation in decreasing HIV LTR activity (Marban, et al., 2005). Thus, CTIP2 and SUV39H1 seem to functionally cooperate to maintain a repressed HIV LTR. Upon CTIP2 knockdown, levels of SUV39H1 recruitment and H3K9 trimethylation were decreased, and HP1β and HP1γ were displaced (Marban, et al., 2005) (Fig. 1). The exact interplay of HP1 isoforms in HIV-1 transcription remains controversial. HP1β and HP1λ are required to both maintain repression and to reactivate the promoter, respectively. (Mateescu, et al., 2008). HP1β recruitment results in H3K9 trimethylation of the promoter. H3K14 and H3S10 double modifications have been found at the promoter upon activation and upon the occupancy of HP1γ onto the promoter (Mateescu, et al., 2008). The exact mechanism still needs to be further elucidated.
In addition to SUV39H1, another methyltransferase, named G9a, also regulates HIV latency (Imai, et al.) (Fig. 1 and Table 1). Following G9a knockdown, the following was observed: increased basal LTR transcription, increased TNF-α-stimulated LTR transcription, increased viral replication and reduced H3K9me2 activity. Treatment with a specific inhibitor of G9a, BIX01924, resulted in a decrease in H3K9me2 activity and, in fact, reactivated latent LTR expression (Imai, et al.).
Work done by the Karn laboratory has more recently shown that H3K27 tri-methylation of the HIV promoter plays a role in the establishment of latency. The complex responsible for the establishment of the H3K27 trimethylation chromatin mark is Polycomb repressive complex 2 (PRC2) (Fig. 1). Knockdown of EZH2, a critical enzymatically active component of the Polycomb Repressive 2 (PRC2) complex, induced a loss of H3K27tri-methylation and reactivation of HIV latency by up to 40%. In fact, the authors showed that knocking down EZH2 had a much higher effect on reactivating latent provirus than did the methyltransferase SUV39H1 which only had a 5% effect of reactivating the provirus. Furthermore, 3-deazaneplanocin A (DZNep), a broad histone lysine methyltransferase inhibitor and a selective inhibitor of EZH2 significantly reactivated latent HIV (Table 1). On the other hand, chaetocin and BIX 01924, selective inhibitors of SUV39H1 and G9A, respectively, had a very modest effect on latent HIV (Table 1).
3. DNA methylation and Effect of HDAC Inhibition
DNA modification via methylation also participates in the regulation of HIV transcription during latency. DNA methylation is a common epigenetic repressive mark on many promoters. DNA methylation occurs on cytosine residues and is preferentially observed in CpG rich sequences. The HIV-1 LTR promoter is methylated in latently infected cells generated in vitro in Jurkat cells and in primary lymphoid cells (Kauder, et al., 2009). The methyl-CpG-binding domain protein, MBD2, recognizes the methylated promoter region of HIV-1 and subsequently mediates the recruitment of HDAC2 (Fig.1 and Table 1)). Two CpG islands flank the HIV-1 transcription start site and are methylated in latently infected Jurkat cells and primary CD4+ T cells. MBD2 and histone deacetylase 2 (HDAC2) are found at one of these CpG islands during latency. Inhibition of cytosine methylation with 5-aza-2′deoxycytidine (aza-CdR) abrogates recruitment of MBD2 and HDAC2. Furthermore, aza-CdR potently synergizes with the NF-κB activators prostratin or TNF-α to reactivate latent HIV-1 (Kauder, et al., 2009).
Viral transcription can be reactivated with HDAC inhibitors. Reactivation was measured in a latently infected Jurkat cell line that contained high levels of promoter methylation (Blazkova, et al., 2009). This clonal cell line, containing hypermethylated provirus, was most efficiently reactivated using HDAC inhibitors, such as trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA) (Table 1). In addition, levels of methylation of H3K27, a repressive chromatin mark, were much higher in this cell line than in cell lines that contained latent provirus with little or no promoter methylation.
The significance of these findings was shown in vivo. Some aviremic HIV patients were found to have provirus with heavily methylated promoter regions, suggesting that proviral methylation contributes to a more restrictive chromatin state during latency (Blazkova, et al., 2009).
4. Chromatin remodeling
In addition to the posttranslational modifications discussed above, the HIV promoter shows distinct chromatin organizations under latent and transcriptionally active conditions (Van Lint, et al., 1996). A single nucleosome, called nuc-1, is positioned ~50 bp downstream of the HIV transcriptional start site and is removed rapidly during the transcriptional activation of the HIV promoter. Subsequent work showed that nuc-1 remodeling is mediated by the SWI/SNF proteins (Fig. 1). SWI/SNF complexes are composed of several subunits and remodel nucleosomes in an ATP-dependent manner. The Brg-1 and Brm subunits of the complex contain the enzymatic activity. SWI/SNF complexes exist in two different complexes, BAF and PBAF. The defining subunit of the BAF complex is BAF250a/ARID1a, and those of the PBAF complex are BAF180, BAF 200 and Brd7. BAF and PBAF complexes can either repress or activate transcription in a context-dependent manner.
SWI/SNF complexes participate in the activation of HIV-1 transcription. Agbottah et al. showed that Tat activates transcription in the G1/S phase of the cell cycle. The modified form of Tat (acetylated by HATs such as p300) shows increased binding to Brg-1, the enzymatically active subunit of SWI/SNF at the G1/S phase of the cell cycle (Agbottah, et al., 2006). Acetylated Tat and Brg-1 are recruited to the 5′ LTR. Furthermore, using latently infected cell lines, such as U-1 and ACH-2, stimulated with TNF, the authors showed that, upon knockdown of Brg-1, viral expression decreased significantly (Agbottah, et al., 2006). Treand et al. also showed that Brm, another catalytic subunit of the SWI/SNF complex, is required for proper Tat-mediated activation of the HIV LTR (Treand, et al., 2006). siRNA knockdown of Brm failed to transactivate the integrated LTR. Furthermore, Tat is required to recruit Brm to the promoter. Only the acetylated form of Tat interacts with Brm. These studies were all conducted in Hela cells.
Our laboratory reported that SWI/SNF is required for Tat-mediated transactivation of the HIV LTR (Mahmoudi, et al., 2006). We showed that Brg-1 and Ini-1 cooperate with p300 to synergistically activate the 5′LTR (Mahmoudi, et al., 2006). Furthermore, the acetylated form of Tat showed increased interaction with Brg-1. Recently, we reported that establishment and maintenance of HIV latency requires BAF, which helps to position the repressive nucleosome-1 immediately downstream of the HIV transcriptional start site. Depletion of BAF specific subunits resulted in de-repression of HIV latency concomitant with loss of nuc-1 on the HIV promoter. Upon transcriptional activation, BAF was lost from the HIV promoter, while PBAF was selectively recruited by acetylated Tat to facilitate LTR transcription. Thus BAF and PBAF, recruited during different stages of the HIV life cycle, display opposing functions on the HIV promoter. (Rafati, et al., 2011)
Tat acetylation is required for its interaction with the SWI/SNF complex and hence transactivation of the HIV promoter. Treand et al showed that Tat acetylation at lysine 50 is required for Brm-Tat interaction and HIV transcription. A mutant of Tat at lysine 50 abrogated the interaction. We found that Tat lysine 50 is required for interaction with BAF180, a subunit of the PBAF complex ((Rafati, et al., 2011). In another report, Easley et al. showed that the PBAF complex (BAF200) of SWI/SNF is required for Tat-mediated activation of the viral LTR, transcriptional elongation and viral production (Easley, et al.). These reports highlight SWI/SNF as a critical factor regulating HIV latency and its reactivation from latency and indicate that the ATP-dependent BRG1 component of BAF might represent a novel therapeutic target to deplete the latent reservoir in patients (Table 1).
Experimental models for HIV latency
Both in vivo and in vitro models have been used to study the mechanisms behind HIV latency. In vivo models developed in non-human primates have provided valuable information (see (Deere, et al., 2011) for review). In addition, in vitro models of HIV latency in transformed cell lines, such as the J-Lat cell line, have also led to a better understanding of the mechanisms governing latency (Han, et al., 2007). While much has been learned from the study of HIV latency in transformed cell lines, concerns have been raised on the relevance of such models to latency occurring in primary lymphoid cells (Jordan, et al., 2003). Here, we will focus solely on recent developments in the area of in vitro models of HIV latency in primary CD4+ T-cells.
As discussed above, latent HIV is primarily found in central memory and transitional memory CD4+ T-cells (Chomont, et al., 2009). Latency is thought to be established by infection of activated CD4+ T-cells that escape the cytotoxic effects of circulating CD8 T cells and revert back to a quiescent state (Jordan, et al., 2003, Lassen, et al., 2004). Since HIV transcription is tightly coupled to the state of activation of T cells, this return to quiescence is thought to lead to a suppression of HIV transcription and to latency. Alternatively, resting CD4+ T-cells can also be directly infected by HIV, resulting in integration but no viral transcription (Swiggard, et al., 2005, Agosto, et al., 2007, Dai, et al., 2009). The latter was noted in both naïve and memory CD4+ T-cells, but infection of such quiescent cells is far less efficient than infection of activated CD4+ T-cells. Nonetheless, we will discuss below primary latency models that make use of infection of both activated and resting CD4+ T-cells.
1. Infection of activated CD4+ T cells
Several groups have attempted to mimic the infection of activated CD4+ T-cells in vitro, with HIV or HIV-derived vectors, followed by the transition of these infected cells to a resting state to establish latency in memory CD4+ T-cells. This is a difficult process due the complicated nature of generating and maintaining memory CD4+ T-cells in vitro (MacLeod, et al., Marrack & Kappler, 2004). The major problem is that most CD4+ T-cells die soon after activation if not continuously cultured in the presence of specific cytokines. This results in too few cells that have transitioned to a quiescent state and contain integrated HIV that can be used for further study. Culturing of CD4+ T-cells in the presence of cytokines, such as IL-2 or IL-7, after activation increases their survival and allows time (several weeks) for more cells to transition to a memory state with integrated HIV. However, IL-2 and IL-7 have been implicated in the reactivation of latent HIV (Chun, et al., 1998, Brooks, et al., 2003, Wang, et al., 2005). Therefore, these cytokines must be used at a concentration that can maintain CD4+ T-cells in culture without reactivating latent HIV. Alternatively, activated and HIV-infected CD4+ T-cells can be maintained in culture and allowed to transition back to a resting state in the absence of cytokines by utilizing strategies, such as transduction with anti-apoptotic proteins or co-culture with feeder cell lines (Tyagi, et al., Sahu, et al., 2006, Yang, et al., 2009) (Fig.2 and Table 2). All these strategies lead to more viable cells that have been infected and returned to a resting state, allowing for HIV latency to be studied in a physiologically relevant setting.
Figure 2. Models for HIV latency in primary lymphocytes.

HIV latency can be established in primary human CD4 +T cells using the procedures illustrated on this diagram. The different models are defined by whether activated CD4+ T cells or resting CD4+ T cells are used. If the cells are activated, the mechanism of activation is shown and the type of HIV vector (full length viral vector or not). The procedure used to return cells to a resting state, the reagent used to reactivate the latent pool (i.e. the stimulating agent) and the type of assay used to assess viral reactivation are also shown. Each primary latency model system is named by the first author of the publication.
Table 2.
Table 2 summarizes the features of all primary latency cell models published thus far. The table summarizes the studies based on authors, the activation status of the CD4+ T cells they used (activated or resting), and the subset of T cells used (central memory (TCM), effector memory (TEM). The table also further categorizes the primary latency cell model studies based on multiplicity of infection (MOI) used in each of the studies , the HIV viral vector employed in each of the models, the percentage of productive and latent infections and the time the experiment takes from start to finish in each of the models.
| Phenotype of Resting CD4+ T Cells |
MOI |
HIV Virus/Viral Vector |
% of Cells Productively Infected |
% of Cells Latently Infected |
Length of Time to Complete Model |
|
|---|---|---|---|---|---|---|
|
Activated CD4+ T Cells |
||||||
| Sahu |
TCM |
1–10 |
Replication-competent |
~80% |
>5% |
2 Months |
| Tyagi |
TCM |
N/A |
Δgag/Δtat |
~85% |
20% |
2 Months |
| Marini |
TCM |
0.002 |
Replication-competent |
5–10% |
2% |
2 Months |
| Bosque |
TCM/TEM |
50 |
Δenv |
3%NP/14.5%Th1/ 3%Th2 |
45%NP/11%Th1/ 40%Th2 |
1 Month |
| Yang |
TEM |
<0.01 |
Δgag/Δvif/Δvpr/Δvpu/Δenv |
5–10% |
1–3% |
~4 Months |
|
Resting CD4+ T Cells |
||||||
| Swiggard |
Naïve/TCM/TEM |
22–150 |
Replication-competent |
0.3% |
4.5% |
Several Days |
| Saleh |
Naïve/TCM/TEM |
1 |
Replication-competent |
N/A |
N/A |
Several Days |
The first model of HIV latency in primary lymphocytes was developed by Sahu et al. in 2006 (Sahu, et al., 2006) (Fig.2 and Table 2). They isolated CD4+ T-cells from blood of uninfected donors and activated them with plate-bound α-CD3 antibody in the presence of IL-2. Activated cells were then infected with a replication-competent virus at a multiplicity of infection (moi) of 1–10 and allowed to transition to a quiescent, memory state. Cells were allowed to transition to a resting state by being co-cultured with a brain tumor-derived cell line, H80. This feeder cell line promoted cell survival in the absence of any cytokines. The exact mechanism by which the H80 feeder cell line works is still unknown, but it allowed the production of a long-lived, mostly central memory CD4+ T-cell population. However, despite being phenotypically similar to resting cells in many respects, a significant fraction of this population continued to express the early activation marker, CD69, suggesting that these cells were not completely resting. Interestingly, a small fraction of quiescent cells also continuously expressed low levels of p24, suggesting that low-level viral replication is ongoing in resting cells. Notably, this low-level virus production came from resting cells that were CD69− and CD69+. Reactivation of this population of cells by the protein kinase C-activator prostratin, led to ~twofold increase in p24-positive cells, showing that latently infected cells were also present within the resting CD4+ T-cell population, accounting for >5% of the cell population.
Tyagi et al. published a study in which they used the H80 feeder cell line to transition activated CD4+ T-cells that had been infected with a HIV vector back to a resting state (Tyagi, et al.) (Fig. 2 and Table 2). Freshly isolated CD4+ T-cells were isolated from both uninfected donor blood or uninfected tonsils and activated and expanded with αCD3/αCD28 antibodies in the presence of IL-2. Activated cells where then infected with a Δgag HIV vector that contained a mutated tat and encoded a fluorescent marker in place of nef. Infected cells were enriched by fluorescence-activated cell sorting (FACS) and expanded via the αCD3/αCD28/IL-2 cocktail. These cells where then co-cultured with the H80 feeder cells and allowed to return to a resting state, producing a mostly central memory CD4+ T-cell population that continued to express significant levels of the late activation marker, CD25 (CD69 expression was not measured). However, a decrease in viral production, as measured by fluorescence, coincided with the cells returning to a mostly resting state. Still, low-level GFP expression was observed within the resting cell population showing, again, that low-level viral expression is ongoing in this primary latency model. Latently infected cells were also present within the central memory CD4+ T-cell population as demonstrated by the significant increase in GFP expression upon CD3/CD28 stimulation as compared to infection with a wild-type Tat. This model of HIV latency produced enough cells for further biochemical characterization that implicates epigenetic silencing and low levels of the transcription factor, P-TEFb, in HIV latency.
A primary latency model in CD4+ T-cells utilizing more physiologically relevant parameters has been developed by Marini and coworkers (Marini, et al., 2008) (Fig. 2 and Table 2). First, freshly isolated CD4+ T-cells from peripheral blood of uninfected donors were activated by co-culture with antigen-loaded monocyte-derived dendritic cells (Ag-MDDCs). Activated cells where then infected with a replication competent HIV-1 at an moi of 0.002. Ongoing infection, as measured by p24, increased over the course of 15 days within the activated CD4+ T-cell population. After the 15-day infection period, CD4+ T-cells that remained activated were enriched for a cell population that was activated and infected by HIV, and eliminating uninfected cells as well as resting cells that may be infected. These activated cells were then allowed to transition to a resting state in the presence of IL-7. After 4 weeks, ~20% of the cells remained alive with the majority exhibiting a central memory phenotype. These quiescent cells were negative for CD25 (CD69 expression not shown) but were larger and more granular than freshly isolated CD4+ T-cells suggesting that they may not be completely resting. Reactivation of this resting central memory CD4+ T-cell population indicated that ~2% of the cells were latently infected.
In 2009, Bosque et al. developed a primary latency model in activated CD4+ T-cells that allowed for the study of both central memory and effector memory populations (Bosque & Planelles, 2009) (Fig. 2 and Table 2). Their system also shortened the time period from activation of freshly isolated CD4+ T-cells to reactivation of latent virus down to less than a month. Isolated CD4+ T-cells were first activated with αCD3/αCD28 antibodies in the presence of IL-2 and then were cultured for several days in three different conditions that produced Th1-helper, Th2-helper and non-polarized CD4+ T-cells (Messi, et al., 2003). Biochemical analysis indicated that the Th1 and Th2 populations closely resembled both effector memory and central memory CD4+ T-cells, in vivo, while the non-polarized population more closely resembled central memory CD4+ T-cells. These three different populations of CD4+ T-cells were then infected with an envelope-deficient HIV virus that was pseudotyped with HIV env, thus limiting infection to a single round. After infection these cells were allowed to transition back to a resting state over the course of 7 days in the presence of IL-2. Three days post-infection cells were analyzed for productive infection by intracellular staining for the viral p24gag protein. Cells expressing high levels of p24gag were considered to be productively infected and die in culture within 3–5 days after infection due to virus-induced apoptosis, leaving both uninfected and latently infected cells alive in culture. Seven days post-infection, the remaining cells were stimulated with αCD3/αCD28 antibodies, and p24gag levels were determined to assess the latently infected population. Cells expressing p24gag after reactivation were considered the latently infected population. However, activation markers such as CD25 and CD69 were not assessed before reactivation, giving no indication if these cells were resting or active. Additionally, it is unclear if reactivated virus arose completely from post-integration latency. Since integration was not assessed, it is possible that a fraction of the reactivated virus came from pre-integration latency. Also of note, mechanistic studies performed using this primary latency model showed that NFκB was not critical for latent reactivation.
A system allowing the screening of drugs that reactivate latent provirus in CD4+ T-cells has been described by Yang and colleagues (Yang, et al., 2009) (Fig. 2 and Table 2). Freshly isolated CD4+ T-cells from uninfected donor blood were first activated and expanded using a αCD3/αCD28/IL-2 cocktail and then transduced with a lentiviral vector carrying the Bcl-2 gene. In this system, Bcl-2, a downstream target of IL-7, acts as an anti-apoptotic protein that allows CD4+ T-cells to remain long-lived in culture without the need for feeder cells or cytokine stimulation. Bcl-2-transduced cells were carried in culture for several weeks, allowing them to return to a resting state. During this time, non-transduced cells eventually died off in the absence of any cytokine signaling leaving only Bcl-2-transduced cells alive in culture. Characterization of these cells showed that they more closely resembled effector memory cells. Bcl-2-transduced cells were reactivated and expanded with a αCD3/αCD28/IL-2 cocktail and then infected with a HIV-derived vector (moi<0.1). This HIV-vector lacked several genes, including gag, vif, vpr, vpu and env, and encoded GFP in place of nef. Infected cells were allowed to transition back to a resting state over the course of several weeks in the absence of cytokines. To enrich for latently infected cells, GFP-negative cells were isolated via FACS and then used to test more than 4000 compounds for their ability to reactivate latent provirus. Before reactivation, however, cells were first assessed for activation markers to prove that they were truly in a resting state. A small fraction of cells still expressed CD69 and CD25. To look at only latent infection in resting cells, CD69− and CD25− cells were purified and used for the reactivation studies. In this primary latency model, 1–3% of GFP-negative cells could be induced to express latent provirus.
2. Infection of resting CD4+ T-cells
Direct infection of resting CD4+ T-cells is an inefficient process due to several blocks imposed by the cellular environment of resting T cells (Zack, et al., 1990, Pierson, et al., 2002, Yoder, et al., 2008). Lack of dNTPs needed for reverse transcription, and a lack of ATP needed for nuclear import of the viral DNA, as well as a restrictive cortical actin barrier, all make infection of resting CD4+ T-cells difficult but not impossible.
The direct infection of resting CD4+ T-cells with HIV in the absence of any activating stimuli has been described by Swiggard and coworkers (Swiggard, et al., 2005) (Fig. 2 and Table 2). Freshly isolated CD4+ T-cells that included a mixture of naive, TCM and TEM were infected with a replication-competent HIV (moi~22–150) by centrifugation of the virus with the target cells in a minimal volume. Integration occurred in resting cells, albeit at a much lower frequency than activated CD4+ T-cells. Integration of the virus took up to 3 days to complete and produced a productive infection in which only 0.3% of cells expressed HIV-Gag. However, reactivation of the cells 3 days post-infection with αCD3/αCD28 antibodies induced 4.5% of the cells to express HIV-Gag showing that a fraction of these cells contained latent provirus.
Saleh et al. developed a primary latency model in resting CD4+ T-cells based on the observation that the majority of HIV-infected resting CD4+ T-cells express CCR7, a lymphoid organ homing receptor (Saleh, et al., 2007) (Fig.2 and Table 2). Freshly isolated resting CD4+ T-cells from uninfected donor blood were first stimulated with the CCR7 ligands, CCL19 and CCL21. These chemokines did not induce either CD69 or CD25 expression, but did increase the susceptibility of resting CD4+ T-cells to infection by a replication competent HIV virus (moi=1). Both reverse transcripts and integrated provirus were detected in these cells, and the production of reverse transcripts after αCD3/αCD28 antibody stimulation increased, showing that latent virus was present in these cells.
Future Directions
Current long-term antiretroviral treatment suppresses new cycles of HIV replication but has no effect on the residual viremia that is responsible for the rebound observed upon the cessation of treatment. Intensification of HAART aimed at complete suppression of viral replication has failed in most studies to suppress residual viremia. This suggests that residual viremia is not due to ongoing viral replication but rather secondary to the persistence of long lived productively infected cells although this is a subject of intense debate. This residual viremia is probably secondary, at least in part, to the existence of latently infected CD4+ T-cells, but likely includes other cell types such as hematopoietic stem cells. Identification of all latent reservoirs is imperative for eradication of the virus from infected individuals. Once identified these latent reservoirs could be stimulated to induce HIV reactivation. Such reactivation, conducted under the cover of HAART, could lead to the selective killing of latently infected cells and the “purging” of the latent reservoir. However, the use of therapeutics that reactivate latent HIV has shown little success in patients thus far or has proven to be too toxic. This is in part due to our lack of understanding of the mechanisms that lead to the establishment and maintenance of HIV latency. The study of HIV latency has been hindered by the relatively low number of latently infected cells within patients and by our lack of relevant in vitro latency models. New technical advances in both fields should allow the study of infected CD4+ T-cells at the single cell level in patients currently on HAART, and the study of latency, in vitro, using primary CD4+ T-cells.
Acknowledgements
We would like to thank Gary Howard for editorial assistance. We would also like to thank John Carroll and Teresa Roberts for graphics and Veronica Fonseca for administrative assistance. SH was funded by a post doctoral fellowship from California HIV/AIDS Research Program of the University of California, Grant Number F08-GI-205, LC is funded by a fellowship from National Institute of General Medical Sciences and EV is supported by NIDA Avant-Garde-1DP1 DA031126, 1R01 DA030216-01 and UNC/NIH - Federal- 5-31532. We apologize to colleagues whose work we could not cite due to space constraints.
References
- Agbottah E, Deng L, Dannenberg LO, Pumfery A, Kashanchi F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology. 2006;3:48. doi: 10.1186/1742-4690-3-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O'Doherty U. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology. 2007;368:60–72. doi: 10.1016/j.virol.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkirane M, Chun RF, Xiao H, Ogryzko VV, Howard BH, Nakatani Y, Jeang KT. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J Biol Chem. 1998;273:24898–24905. doi: 10.1074/jbc.273.38.24898. [DOI] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois-Rey F, et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc Natl Acad Sci U S A. 2003;100:12955–12960. doi: 10.1073/pnas.2233345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Engel D, Mizell SB, Ehler LA, Fauci AS. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J Exp Med. 1998;188:83–91. doi: 10.1084/jem.188.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin L, Van Lint C. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology. 2009;6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, et al. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, O'Doherty U. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol. 2009;83:4528–4537. doi: 10.1128/JVI.01910-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deere JD, Schinazi RF, North TW. Simian immunodeficiency virus macaque models of HIV latency. Curr Opin HIV AIDS. 2011;6:57–61. doi: 10.1097/COH.0b013e32834086ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Chene I, Basyuk E, Lin YL, et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. Embo J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley R, Carpio L, Dannenberg L, et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology. 2010 doi: 10.1016/j.virol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol. 2007;5:95–106. doi: 10.1038/nrmicro1580. [DOI] [PubMed] [Google Scholar]
- Imai K, Okamoto T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J Biol Chem. 2006;281:12495–12505. doi: 10.1074/jbc.M511773200. [DOI] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007;81:10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol. 2009;83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. The multifactorial nature of HIV-1 latency. Trends Mol Med. 2004;10:525–531. doi: 10.1016/j.molmed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Lewinski MK, Bisgrove D, Shinn P, et al. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol. 2005;79:6610–6619. doi: 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusic M, Marcello A, Cereseto A, Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. Embo J. 2003;22:6550–6561. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod MK, Kappler JW, Marrack P. Memory CD4 T cells: generation, reactivation and re-assignment. Immunology. 2010;130:10–15. doi: 10.1111/j.1365-2567.2010.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP, Ott M, Verdin E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Biol Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. Embo J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Redel L, Suzanne S, et al. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 2005;33:2318–2331. doi: 10.1093/nar/gki529. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol. 2008;181:7713–7720. doi: 10.4049/jimmunol.181.11.7713. [DOI] [PubMed] [Google Scholar]
- Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- Marsili G, Remoli AL, Sgarbanti M, Battistini A. Role of acetylases and deacetylase inhibitors in IRF-1-mediated HIV-1 long terminal repeat transcription. Ann N Y Acad Sci. 2004;1030:636–643. doi: 10.1196/annals.1329.074. [DOI] [PubMed] [Google Scholar]
- Mateescu B, Bourachot B, Rachez C, Ogryzko V, Muchardt C. Regulation of an inducible promoter by an HP1beta-HP1gamma switch. EMBO Rep. 2008;9:267–272. doi: 10.1038/embor.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messi M, Giacchetto I, Nagata K, Lanzavecchia A, Natoli G, Sallusto F. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat Immunol. 2003;4:78–86. doi: 10.1038/ni872. [DOI] [PubMed] [Google Scholar]
- Perelson AS, Essunger P, Ho DD. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS. 1997;11(Suppl A):S17–S24. [PubMed] [Google Scholar]
- Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- Pierson TC, Zhou Y, Kieffer TL, Ruff CT, Buck C, Siliciano RF. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J Virol. 2002;76:8518–8531. doi: 10.1128/JVI.76.17.8518-8531.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. PLoS Biol. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu GK, Lee K, Ji J, Braciale V, Baron S, Cloyd MW. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology. 2006;355:127–137. doi: 10.1016/j.virol.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood. 2007;110:4161–4164. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Swiggard WJ, Baytop C, Yu JJ, et al. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol. 2005;79:14179–14188. doi: 10.1128/JVI.79.22.14179-14188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treand C, du Chene I, Bres V, Kiernan R, Benarous R, Benkirane M, Emiliani S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. Embo J. 2006;25:1690–1699. doi: 10.1038/sj.emboj.7601074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. Embo J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Wang FX, Xu Y, Sullivan J, et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest. 2005;115:128–137. doi: 10.1172/JCI22574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. Embo J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC, Xing S, Shan L, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest. 2009;119:3473–3486. doi: 10.1172/JCI39199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A, Yu D, Dong L, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]