

Abstract
Receptor tyrosine kinases (RTKs) are proteins that upon ligand stimulation undergo dimerization and autophosphorylation. Eph receptors (EphRs) are RTKs that are found in different cell types, from both tissues that are developing and from mature tissues and play important roles in the development of the central nervous system and peripheral nervous system. EphRs also play roles in synapse formation, neural crest formation, angiogenesis and in remodeling the vascular system. Interaction of EphRs with their ephrin ligands leads to activation of signal transduction pathways and to formation of many transient protein-protein interactions that ultimately leads to cytoskeletal remodeling. However, the sequence of events at the molecular level is not well-understood.
We used Blue Native PAGE (BN-PAGE) and mass spectrometry (MS) to analyze the transient protein-protein interactions that resulted from stimulation of EphB2 receptors by their ephrinB1-Fc ligands. We analyzed the phosphotyrosine-containing protein complexes immunoprecipitated (pY-IPs) from the cell lysates of both unstimulated (−) and ephrinB1-Fc-stimulated (+) NG108 cells. Our experiments allowed us to identify many signaling proteins, either known to be part of EphB2 signaling or new for this pathway, which are involved in transient protein-protein interactions upon ephrinB1-Fc stimulation. These data led us to investigate the roles in EphB2 signaling of proteins such as FAK, WAVEs, and Nischarin.
Keywords: mass spectrometry, BN-PAGE, proteomics, signal transduction, protein-protein interactions
Introduction
Receptor tyrosine kinases (RTKs) are proteins that undergo dimerization and/or multimerization and autophosphorylation upon ligand stimulation. Eph receptors (EphRs) are RTKs that are found in different cell types, from tissues that are developing and from mature tissues and play important roles in the development of the central nervous system and peripheral nervous system (1–3), including synapse formation (4), neural crest formation (5), as well as angiogenesis and vascular system remodeling (6–9). Unlike other RTKs, EphRs are not known to promote cell growth and differentiation; however, they play important roles in attractive and repellent interactions between adjacent cells. Furthermore, activation of Eph receptors is affected by membrane-anchored ephrins that are not soluble, like the other RTKs. Therefore, during stimulation, EphRs may act as both receptors that are activated by ephrin ligands and trigger downstream signaling or may act as ligands that activate upstream signaling in ephrin-bearing cells.
Currently at least 14 different EphRs have been identified. They have been divided into two classes, depending on the type of ligand with which they interact: EphAs and EphBs (10). EphA receptors (EphA1-A8 and EphA10) interact with ephrinAs (ephrinA1-A5). EphB receptors (EphB1-B4 and EphB6) interact with ephrinBs (ephrinB1-B3) (10). All Eph receptors and ephrinBs are transmembrane proteins that contain extracellular domains. However, unlike ephrinBs, which contain cytosolic domains, ephrinAs are connected to the cell surface by glycosylphosphatidylinositol anchors, and therefore do not contain a cytosolic domain.
Interaction of ephrins with Eph receptors leads to formation of ephrin-Eph receptors; dimers and multimers that triggers remodeling of the cytoskeleton, which in turn underlies cell adhesion and motility in cells that contain the ligand and/or the receptor (11–13). However, intracellular molecular mechanisms leading to activation of signal transduction pathways, ultimately remodeling the cytoskeleton in cells that contain ephrin ligands and/or EphRs, are not well understood. Understanding these pathways is further complicated by the bidirectional signaling that occurs between Eph receptors and their ephrin ligands (1, 2, 14, 15).
Upon activation of signal transduction pathways, many proteins transiently interact with other proteins (transient protein-protein interactions), leading to the formation of different protein complexes. These protein complexes are constantly formed and destroyed during transition of cells from an unstimulated to stimulated state and back to the unstimulated state. However, the transient protein-protein interactions that result upon ephrin stimulation are not well understood and information about the interaction of these protein complexes at both the structural and functional level would lead to a better understanding of ephrin signaling.
Blue Native PAGE (BN-PAGE) is a variant of electrophoresis that has been used for a long time to analyze protein-protein interactions and protein complexes (16–21). This method separates intact protein complexes according to their molecular mass (mass), due to the external charge induced by Coomassie dye that binds to these complexes. If the BN-PAGE gel lane containing the intact protein complexes is further separated under reducing and denaturating conditions in a second dimension (2D) by SDS-PAGE, the subunit composition of a particular protein complex, and/or the interacting partners of a particular protein may be revealed. Mass spectrometry (MS) (22–24) is also a method used to dissect signal transduction pathways through identification of potential interacting partners of a protein or of protein complexes involved in signal transduction pathways.
Here, we used BN-PAGE and MS to analyze the transient protein-protein interactions that resulted from stimulation of EphB2 receptors by their ephrinB1-Fc ligands. We analyzed the phosphotyrosine-containing protein complexes immunoprecipitated (pY-IPs) from the cell lysates of (−) and (+) NG108 cells, a model for neurons in cell culture. These experiments allowed us to identify many signaling proteins, either known to be part of EphB2 signaling or new for this pathway, which are involved in transient protein-protein interactions upon ephrinB1-Fc stimulation. These data also led us to further investigate the role of proteins like FAK, WAVEs, and Nischarin in EphB2 signaling. This approach also should be easily applicable for analysis of protein-protein interactions in other growth factor-dependent systems.
Experimental Design
Cell culture, ephrin stimulation, and cell lysis
The NG108 cells (mouse neuroblastoma x rat glioma hybrid) that were stably transfected with ephB2 gene and that express EphB2 receptors (NG108-EphB2) were grown in Dubelcco’s modified Eagle’s medium (DMEM) supplemented with 100 units/mL penicillin/streptomycin, 1x HAT, 10% (v/v) fetal bovine serum and 400 μg gemtamycin/mL (all from GIBCO, Invitrogen Corporation, Carlsbad, CA). NG108 cells were stimulated with soluble ephrinB1-Fc (Sigma, St. Luis MO) preclustered with anti-Fc antibodies (Jackson Laboratory, Bar Harbor, ME) for 5, 10, 15, 20 and 45 minutes at 37°C, according to published procedures (25, 26). The control (−) cells were treated with soluble IgG-Fc (Jackson Laboratory, Bar Harbor, ME) pre-clustered with anti-Fc antibodies (Jackson Laboratory, Bar Harbor, ME). Upon stimulation, the cells were lysed using either NP40 buffer [150 mM NaCl, 20 mM Tris-HCl (pH 8.0), 0.2 mM EDTA, 2 mM NaF, 2 mM Na3VO4, 1% (v/v) NP40, and Complete protease inhibitor (Roche, Mannheim, Germany; 1 tablet/10 mL buffer)) or DDM buffer [50 mM NaCl, 500 mM 6-amino caproic acid (Sigma, St. Luis, MO) 20 mM Bistris (pH 7.0), 0.2 mM EDTA, 2 mM NaF, 2 mM Na3VO4, 1% (w/v) n-dodecyl β-D-maltoside (DDM, Sigma, St. Luis, MO), and Complete protease inhibitor (Roche, Mannheim, Germany; 1 tablet/10 mL buffer)). The lysates were then incubated on ice for 20 minutes and the unsolubilized material was removed by centrifugation in an Epifuge at 14000 rpm/20 min/4°C. The supernates of (−) and (+) NG108 cells were stored at −80°C and used as starting material for SDS-PAGE, BN-PAGE, or for antibody-based enrichment (anti-phosphotyrosine immunopurification: pY-IP).
Phosphotyrosine immunoprecipitation (pY-IP)
The cell lysates were incubated overnight at 4°C with 0.1 mg/10 mL lysate agarose-bound pY99 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and then washed five times with NP40 or three times with DDM buffer. Phosphotyrosine-containing protein complexes were eluted with 5 mM phenyl phosphate (Sigma, St. Luis, MO) in NP40 (small scale experiment) or DDM (larger scale experiment) buffer by incubation in ice for 30 minutes. The eluates were collected by centrifugation in an Epifuge microcentrifuge for 3 min at 5000 rpm and then stored at −80°C.
SDS-PAGE and Western blotting (WB)
The cell lysates or pY99-IPs were solubilized for 5 minutes in Laemmli sample buffer/95°C, loaded on 10% Tris-HCl gels (Bio-Rad, Hercules, CA) and separated by SDS-PAGE. The gels were then Coomassie-or silver-stained, or electroblotted on PVDF membrane (Millipore, Bedford, MA), immunodecorated with different antibodies and visualized by enhanced chemiluminiscent (ECL) reaction kit (Pierce, Rockford, IL). All antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), except antibodies against Flag (Sigma, St. Luis, MO), EphB2 (Abcam, Cambridge, MA) and Nischarin (a kind gift of Dr. S.K. Alahari).
BN-PAGE and WB
BN-PAGE was performed using a Protean Xi system (Bio-Rad, Hercules, CA) on a 4–13% self made linear acrylamide-bisacrylamide gradient, according to published procedures (16, 18, 19), with modifications (16). Immediately before the electrophoresis, the cell lysate or pY-IP samples were supplemented with 5% (v/v) of a 5% Coomassie Blue G-250 in 500 mM 6-aminocaproic acid, loaded onto the gel lane, and separated for 3–4 hours at 100 V/4°C, followed by overnight separation at 150 V and 4°C. The cathode buffer contained 15 mM Bistris, 50 mM tricine, pH 7.0 and 0.02% (w/v) Coomassie G-250. For enhanced visualization, the initial cathode buffer was replaced after 3–4 hours with a buffer containing 0.002% (w/v) Coomassie G-250. The gel lanes that resulted in the first dimension (1D) were either Coomassie- or silver-stained or electroblotted, or were reduced and denatured by shaking in 2% (w/v) SDS and 1% (v/v) β-mercapto-ethanol for 60 minutes and further separated by denaturing SDS-PAGE (2D). The 2D SDS-PAGE gels were then stained by silver staining or electroblotted onto PVDF membraqne and analyzed by WB.
Protein digestion and peptide extraction
Proteins separated by either SDS-PAGE or BN-PAGE 1D were stained by Coomassie dye and then cut in either 14 (SDS-PAGE) or 24 to 32 (BN-PAGE) gel pieces for each condition (−) and (+) cells. The gel bands were washed in HPLC grade water, cut into small pieces and destained by incubating three times in 50 mM ammonium bicarbonate, 50 mM ammonium bicarbonate/50% acetonitrile, and 100% acetonitrile under moderate shaking, followed by drying in a vacuum centrifuge (27). The gel bands were then rehydrated with 50 mM ammonium bicarbonate containing 15 ng/μL trypsin and incubated overnight at 37°C under low shaking. The resulting peptides were extracted twice with 5% formic acid/50 mM ammonium bicarbonate/50% acetonitrile and once with 100% acetonitrile under moderate shaking. Peptide mixture was then dried by vacuum centrifugation, solubilized in 20 μL of 0.1% formic acid/2% acetonitrile and cleaned with a P10 ZipTip μ-C18 (Millipore Corporation, Billerica, MA). For HPLC analysis, the samples were resuspended in 10 μL of 0.1% formic acid/2% acetonitrile.
Mass Spectrometry and protein identification
The peptide mixture that resulted upon trypsin digestion was analyzed by reverse phase liquid chromatography (LC) and MS (LC-MS/MS) using a NanoAquity UPLC coupled directly to a Q-Tof Premier MS (Waters, Milford, MA). The peptides were loaded onto a 100 μm × 10 mm nanoAquity BEH130 C18 1.7 μm UPLC column (Waters, Milford, MA) and eluted over a 120 minutes gradient of 10–85% acetonitrile in 0.1% formic acid at a flow rate of 250 nL/min. In addition, peptides were also eluted over a 180 minute gradient of 5–40% acetonitrile in 0.1% formic acid at a flow rate of 400 nL/min. The 180 minute gradient runs also had a channel for lockspray calibration delivered by the auxiliary pump at a flow rate of 1 μL GluFib peptide/min. The column was coupled to a Picotip Emitter Silicatip nano-electrospray needle (New Objective, Woburn, MA). MS data acquisition involved survey MS scans and automatic data dependent MS/MS of 2+, 3+ or 4+ ions. The MS/MS was triggered when the MS signal intensity exceeded 10 counts/second. In survey MS scans, the three (120 minutes gradient) or four (180 minutes gradient) most intense peaks were selected for CID and fragmented until the total MS/MS ion counts reached 10,000 or for up to 6 seconds each. In the 180 minutes gradient runs, the lockspray channel recorded signals for one second at every 30 seconds interval. The raw data produced by the 120 minute gradient were processed using ProteinLynx software with the following parameters: background subtraction of polynomial order 5 adaptive with a threshold of 35%, two smoothings with a window of three channels in Savitzky-Golay mode and centroid calculation of top 80% of peaks based on a minimum peak width of 4 channels at half height. The raw data produced by the 180 minute gradient were processed with the above-mentioned parameters, plus lockspray calibration of MS and MSMS using GluFib peptide (785.8426 Da; 3 scans, mass tolerance of 0.3 Da). The resulting pkl files were submitted for database searching and protein identification to an in-house Mascot server (version 2.2.1, Matrix Science, London, UK) using the following parameters: mouse-rat forward and reverse databases from NCBI as of 06/07/06 (28), parent mass error of 50 ppm, product ion error of 0.15 Da, enzyme used: trypsin, one missed cleavage, and no fixed or variable modifications. Before submission to the database search, the pkl files that resulted from 120 minute gradient runs were re-calibrated post-acquisition as previously reported (28). The Mascot search identified a list of proteins for each gel band. These lists were exported as csv files using a significance threshold with p<0.01 to eliminate the false positive results. The csv files for each individual gel band were then uploaded onto web-based ProteinCenter software (Proxeon.com). The results from the 120 and 180 minute gradient runs for a particular gel band were merged and clustered for 98% homology in ProteinCenter and then exported as csv files. The csv files were then grouped into an excel file according to their position in the initial gel. Only proteins identified by two or more peptides and with a Mascot score >50 were considered. Furthermore, to eliminate false positive results from BN-PAGE carry-over, most of the proteins identified by a Mascot score lower than 100 were eliminated, unless specified. For the proteins identified by only one peptide and included in the supplemental material, the MSMS spectra of these peptides were manually inspected to confirm the identity of the peptide. Additional MS runs were performed using an Eksigent NanoLC-2D coupled to an LTQ-Orbitrap MS (Thermo Scientific, Waltham, MA) as described in the supplementary methods.
Immunofluorescence (IF)
Cells were grown on coverslips and treated as described above. After treatment, cells were fixed in 4% paraformaldehyde/20% sucrose/phosphate-buffered saline (PBS) for 10 min at room temperature. They were then incubated in 50 mM NH4Cl/PBS followed by 0.1% TritonX-100/PBS. Cells were blocked in blocking buffer (10% normal donkey serum, 2% bovine serum albumin, 0.25% fish skin gelatin in Tris-buffered saline (TBS)) for 30 min at room temperature, incubated with the relevant primary antibodies for 30 min in blocking buffer, washed in TBS/0.25% fish skin gelatin, followed by secondary antibodies (AlexaFluor-conjugated, 1:1000, Invitrogen) for 30 min in blocking buffer, washed and mounted in Mowiol. Cells were imaged using a LSM510 laser-scanning confocal microscope equipped with a 40X Plan Neofluor NA1.3 DIC oil immersion objective (both Carl Zeiss MicroImaging, Thornweed, NY). Images were processed using ImageJ (NIH).
Western blotting and MS characterization of protein complexes
Two main approaches were used for analysis of protein-protein interactions in NG108 cells: 1) BN-PAGE (2D) and WB of the lysates from (−) and (+) cells and 2) BN-PAGE and MS of the lysates and pY-IPs from (−) and (+) cells. Separation of stable or transient protein complexes from cell lysates or pY-IPs was performed by using one-dimensional BN-PAGE (BN-PAGE; 1D) or two-dimensional BN-PAGE (BN-PAGE; 2D) electrophoresis. The workflow used for characterization of ephrin signaling is shown in Figure 1. Supplemental Figure 1 shows a schematic of characterization of the protein complexes upon ephrin stimulation.
Figure 1.
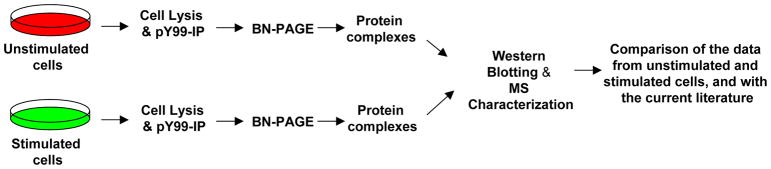
A workflow for the biochemical and MS characterization of ephrin signaling. Unstimulated (−) or stimulated (+) cells were lysed and the lysates (and/or the pY99-IPs from the lysate) were separated by BN-PAGE. The protein complexes were characterized by Western blotting and/or by MS, followed by comparison with the current literature.
Results and discussion
Investigation of the cell lysates from (−) and (+) cells by SDS-PAGE, BN-PAGE, and WB: Many proteins become phosphorylated at tyrosine residues upon ephrinB1-Fc stimulation, which leads to formation of dynamic and transient protein-protein interactions
To identify the differences between the cell lysates from (−) and (−) cells that resulted upon eprinB1-Fc stimulation, we analyzed them by SDS-PAGE and BN-PAGE.
At first, the cell lysates from (−) cells and from cells stimulated for 45 minutes were fractionated by SDS-PAGE and then Coomassie- or silver-stained (Supplemental Figure 2), or further analyzed by WB (Figure 2A). No detectable differences between the Coomassie- or silver-stained (Supplemental Figure 2) gel lanes of lysates from (−) and (+) cells were observed. However, when the SDS-PAGE gels were analyzed by WB using pY99 antibodies, many differences were observed (Figure 2A). More tyrosine phosphorylated proteins were observed in the lysate from the (+) cells than in the lysate from the (−) ones (Figure 2A).
Figure 2.

Analysis of lysates of (−) and (+) cells by SDS-PAGE and BN-PAGE. The lysates of (−) and (+) cells were separated by SDS-PAGE (A) and BN-PAGE 1D (B) and blotted to membranes and analyzed by WB using pY99 antibodies. The gel lanes from BN-PAGE 1D were excised and further reduced and denatured and separated in 2D SDS-PAGE and electroblotted by WB and analyzed with pY99 antibodies (C). In (C), the lysates were from (−) cells (0′) or cells stimulated with ephrinB1-Fc for 5, 10, 15, 20 and 45 minutes. The direction of migration in BN-PAGE is indicated by arrows followed by either 1D or 2D. The mass markers are indicated for each gel in 1D or 2D.
To confirm the previous observations and to further our findings, we analyzed the lysates from both (−) and (+) cells by BN-PAGE. BN-PAGE separates proteins and protein complexes according to their molecular mass and under native conditions, due to the external charge induced by Coomassie Blue dye that is added in the separation buffer. Therefore, in under BN-PAGE conditions, many stable and transient protein-protein interactions are preserved. To identify the differences (at the protein complex level) between the lysates from (−) cells and (+) cells that resulted upon ephrinB1-Fc stimulation, we fractionated the two cell lysates by BN-PAGE in the first dimension 1D or in the second dimension 2D and visualized them by either Coomassie- or silver staining (Supplemental Figure 2) or further analyzed them by WB using pY99 antibodies (Figures 2B and C). No difference could be observed in the Coomassie- (data not shown) or silver-stained BN-PAGE gel lanes, in either 1D or 2D (Supplemental Figure 2). However, significant differences between the phosphorylation pattern in the cell lysates of (−) and (+) cells could be observed in the WB of the BN-PAGE from 1D (Figure 2B) or 2D (Figure 2C).
To further investigate whether ephrin stimulation of NG108 cells determines formation of dynamic and transient protein-protein interactions, we separated the cell lysate from (−) cells and from cells stimulated for 5, 10, 15, 20 and 45 minutes by BN-PAGE 1D, SDS-PAGE 2D, and WB with pY99 antibodies (Figure 2C). As observed, dramatic changes happened in this time-course experiment, suggesting that subunits of stable protein complexes are either phosphorylated and/or dephosphorylated upon ephrinB1-Fc stimulation.
Investigation of the cell lysates from (−) and (+) cells by BN-PAGE and MS
Initially, we tested the combination of BN-PAGE and MS on cell lysates to analyze well known stable protein interactions. We observed that both subunit composition and the masses of both homo- or hetero-protein complexes (Valosin-containing protein or Proteasome) agree with the previous reports (29, 30) (Supplemental Figures 3 and 4). Overall, these data suggest that BN-PAGE and MS can be combined into a powerful tool and used for identification of the mass and subunit composition of a particular protein complex, and may be the method of choice for investigation of transient protein-protein interactions.
Well known transient protein-protein interactions may be detected by BN-PAGE and MS: Small scale analysis of pY99-IPs from (−) and (+) cells by SDS-PAGE, BN-PAGE and MS
To identify transient protein-protein interactions that resulted upon ephrinB1-Fc stimulation, we analyzed the phosphorylated proteins or protein complexes from the lysates of both (−) and (+) cells. Specifically, we performed pY99-IPs to enrich for tyrosine phosphorylated proteins and for proteins associated with tyrosine phosphorylated proteins. In a small scale experiment, we isolated the pY99-IPs and separated them by SDS-PAGE and MS, or BN-PAGE and MS. SDS-PAGE and MS analysis identified the differences between the proteins immunopurified by pY99 antibodies from the (−) and (+) cells, while BN-PAGE and MS identified the interactions between the proteins already identified by SDS-PAGE and MS.
The proteins that were identified by SDS-PAGE and MS of pY-IPs from (−) cells are shown in Supplemental Table 1 and from (+) cells are shown in Supplemental Table 2. In the SDS-PAGE of pY99-IP of (−) cells, we identified a 130 kDa protein, named focal adhesion kinase (FAK) or protein tyrosine kinase 2 (PTK2, Supplemental Table 1 and text below). Among the proteins identified in the pY99-IP of (+) cells analyzed by SDS-PAGE and MS and known to be involved in ephrin signaling (25, 31), in addition to EphB2 receptor, two important proteins were identified. The first protein was FAK or PTK2. The second protein was a 95 kDa protein named breast cancer anti-estrogen resistance 1 or p130Cas. FAK and p130Cas proteins are known to interact with each other via a SH3 domain (11, 32). Therefore, identification of 1) FAK and p130Cas in the pY99-IP from the (+) cells and 2) only of FAK in the (−) cells suggests that FAK and p130Cas interact with each other upon ephrinB1-Fc stimulation.
To test this hypothesis, we analyzed the pY99-IPs from both (−) and (+) cells by BN-PAGE and MS. The gel bands that were cut out are shown in Figure 3A (left). A schematic of our findings is shown in Figure 3A. In the gel bands from (−) cells, we identified FAK/PTK2 in the 130–170 kDa range (B19(−), mascot score 54) and 190–210 kDa range (B15(−), mascot score 32), and in the (+) cells in the 150–170 kDa range (B17(+), mascot score 138) and 230–250 kDa range (B13(+), mascot score 50) (Figure 3A, right). In addition, we identified the 95 kDa p130Cas protein in the 230–300 kDa range in the gel band from (+) cells (B12(+), mascot score 33 and B13(+), mascot score 63) but not (−) cells (Figure 3A, right). The outcome of this experiment is summarized in Supplemental Table 3. Since in BN-PAGE and MS experiments 1) FAK shifted from a molecular weight region consistent with a monomeric state in the pY-IP of (−) cells to a higher mass consistent with a protein complex in the pY-IPs of the (+) cells; 2) The 95 kDa p130Cas protein was identified as part of a protein complex with a mass of 230–300 kDa in the (+) cells but not in the (−) cells; 3) The calculated mass of FAK/PTK2 is 130 kDa and of p130Cas is 95 kDa, and the calculated mass of FAK/PTK2-p130Cas protein complex is 225 kDa; 4) FAK and p130Cas were identified by BN-PAGE and MS in the same BN-PAGE gel band (at about 230–300 kDa); 5) it is well established that FAK/PTK2 interacts with the p130Cas protein, it is possible to conclude that FAK/PTK2 forms a hetero-protein complex with the 95 kDa p130Cas protein upon ephrin stimulation. Overall, these data suggest that BN-PAGE and MS of the pY-IPs from both (−) and (+) cells can be used to identify both transient and stable protein-protein interactions. We also identified EphB2 receptor in the pY-IP of (+) cells (B13(+), mascot score 51; B14(+), mascot score 528 and B15(+), mascot score 447), but not in (−) cells.
Figure 3.

Representation of the proteins found by BN-PAGE and MS of the pY-IPs of (−) and (+) cells. A: Identification of well known protein interactions by analysis of small scale pY-IP in the presence of NP-40 detergent. The pY-IP from (−) and (+) cells were separated by BN-PAGE and the gel lanes were excised, cut into pieces/bands, digested by trypsin, and analyzed by MS. The approximate mass of the bands of interest was between 110–130 and 250–300 kDa. The proteins identified are labeled with different colors, shown at the bottom of the figure. B: as in A, except that identification of well known protein interactions were performed by analysis of larger scale pY-IP in the presence of DDM detergent. The mass markers are indicated for each gel.
A larger scale investigation of pY99-IPs from (−) and (+) cells by BN-PAGE and MS suggests a variety of known and potential new stable and transient protein-protein interactions
We continued our search for identification of both transient and stable protein-protein interactions that formed as a result of ephrinB1-Fc stimulation and performed a larger scale purification of pY-IPs from the (−) and (+) cells and analyzed them by BN-PAGE and MS. The outcome of this experiment is summarized in Supplemental Table 4 and in Figures 3B and 4. Most of the MS results shown in Figure 3B and Supplemental Table 4 were further confirmed using an Eksigent NanoLC-2D coupled to an LTQ-Orbitrap MS and the outcome regarding the proteins shown in Figure 3B is presented in Supplemental Table 5.
Figure 4.
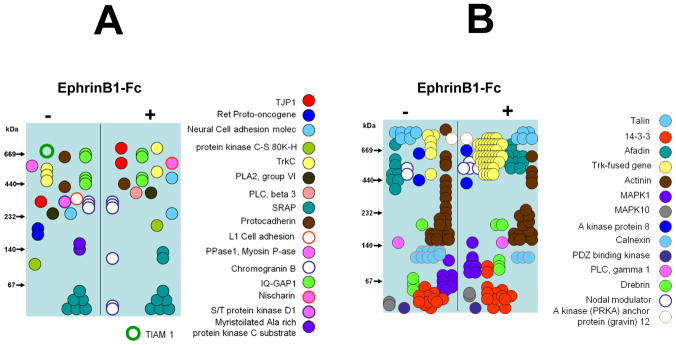
Representation of selected proteins found by BN-PAGE and MS of the larger scale pY IPs of (−) and (+) cells. A: Proteins found by BN-PAGE and MS in either (−) or (+) cells or in both and shifted their molecular mass from low to high or from high to lo w. B: Proteins found by BN-PAGE and MS in both (−) and (+) cells that did not change in measured molecular mass (did not shift in BN-PAGE) upon ephrinB1-Fc stimulation, but changed their abundance by either increase or decrease of their relative amounts in the pY IP experiment.
Three categories of proteins were identified in our experiments: 1) previously demonstrated protein interactions, but in a new context (i.e. after ephrinB1 stimulation) as well as previously described interactions. 2) proteins found in pY-IPs from either (−) or (+) cells or both cells that were identified in BN-PAGE and MS experiments, and that shifted their molecular mass in BN-PAGE from low to high or from high to low. 3) Proteins found by BN-PAGE and MS in both (−) and (−) cells that did not change in measured molecular mass (did not shift in BN-PAGE), but changed their abundance by either increase or decrease of their relative amounts.
-
Well known protein interactions, but in a new context, as well as well known events. We identified EphB2 receptor (110 kDa) in the gel bands from (+) (190–300 kDa range) but not (−) cells (Supplemental Table 4 and Figure 4B), results that confirmed our previous experiments by SDS-PAGE and MS (Supplemental Tables 1 and 2) and BN-PAGE and MS (Figure 3A), where the estimated mass was 190–250 kDa (mascot score 528 and 447).
Two proteins identified in our experiment, signal transducing adapter molecule (STAM1, mass 60 kDa) and hepatocyte growth factor-regulated tyrosine kinase substrate (HRS, mass 115 kDa), well known to interact with each other and involved in receptor down-regulation by internalization in the endosomal vesicles (28, 31, 33–35), but not in EphB2 receptor down-regulation. STAM1 was identified at about 180–200 kDa in the (−) cells and at 250–300 kDa in the (+) cells, while Hrs was identified only in the (+) cells at about 250–400 kDa (Supplemental Table 4 and Figure 3B). In addition, these two proteins overlapped with the EphB2 receptor in (+) cells. Since STAM1 and Hrs are known to interact with each other and were found in the same gel bands only in (+) cells, but not in (−) cells, this suggests that ephrinB1-Fc stimulation leads to transient interaction of these two proteins. Furthermore, since 1) STAM1 and Hrs are well known to down-regulate receptor tyrosine kinases; 2) both proteins were found to overlap with EphB2 receptor; and 3) the calculated mass of STAM1 (60 kDa), Hrs (115 kDa) and EphB2 (110 kDa) together is 285 kDa, this suggests that ephrinB1-Fc stimulation leads to a transient interaction between STAM1 and Hrs, which ultimately down-regulate the EphB2 receptor and attenuates its signaling. Based on this rationale, we also conclude that the EphB2 receptor identified in BN-PAGE and MS in the 190–300 kDa range is in the monomeric state and may form a complex with STAM1 and Hrs.
Two other proteins well-known to interact with each other and identified in our experiment were Cytoplasmic FMR1-interacting protein 1/Specifically Rac1-associated protein (Cyfip1/Sra1/Shyc; mass 145 kDa) and Nck-associated protein 1 (Nckap1/p125Nap1; mass 129 kDa). Shyc was detected at around 400 kDa and 600–700 kDa in both (−) and (−) cells, while Nckap1 was detected around 600–700 kDa in the (−) cells and around 400 kDa and 600–700 kDa in the (+) cells (Supplemental Table 4 and Figure 3B). Cyfip1/Sra1/Shyc and Nckap1/p125Nap1 are known to interact with each other and also with Abi-1 (52 kDa) and WAVE 2 (54 kDa), and all of them are components of the WAVE 2 complex (36). Additional MS experiments of the BN-PAGE gel bands from the 300–900 kDa range identified WAVE 1/2 protein at about 700 kDa in the (−) cells and at about 600 kDa in the (+) cells, and Abi-1 protein at about 600 kDa in the (+) cells (Figure 3B). The WAVE 2 complex has a calculated mass of about 380 kDa and an experimentally determined mass of 550 kDa (36). Since the intact WAVE 2 complex was identified at about 600 kDa in the (+) cells and three out of four of its subunits at about 700 kDa in the (−) cells, this suggests that the WAVE 2 complex from the (−) cells interacts with additional protein/s and loses it/them upon ephrinB1-Fc stimulation. However, the components of the WAVE2 complex may also be involved in other interactions. For example, Shyc/SRA1 and Nckap1/NAP1 interact with each other in a WAVE2 subcomplex, but also are often found together in other complexes different from WAVE2 complex (37–39). Upon fibronectin (FN) stimulation, Abi-1 promotes activation of WAVE2 and Abl-mediated tyrosine phosphorylation is required for WAVE activation. (40). Recombinant WAVE isoforms co-immunoprecipitate (41). WAVE1 is an A-kinase anchoring protein and interacts with cAMP-dependent protein kinase (PKA) and Abelson tyrosine kinase (Abl) (41), and Rac1 or Nck are thought to interact with WAVE1 complexes (37). Keeping in mind all these possible interactions and that (except for the case of WAVE1 complex which is activated by disintegration (37)) in all (−) and EGF or PDGF(+) cells the integrity of the WAVE2 complex was not altered (36), it is more likely that the WAVE2 complexes identified in our experiments are intact and the WAVE2 complex from the (−) cells contains additional subunits.
BN-PAGE and MS also allow us to identify proteins that are not involved in protein-protein interactions, but specifically phosphorylated. For example, a protein identified in our experiment was Non-catalytic region of tyrosine kinase adaptor protein 2 (Nck2; mass 43 kDa). Nck2 was detected as monomer in the BN-PAGE and MS of pY-IPs of the (+) (at about 50 kDa) but not (−) cells (Supplemental Table 4 and Figure 3B), suggesting that Nck2 is phosphorylated upon stimulation and that, at these particular conditions, it does not interact with any protein. At this point, we would like to point out that phenylphosphate elution used in our experiments, in addition to elution of the protein complexes, it may also disrupt specific phosphotyrosine-SH2 domain or phosphotyrosine- phosphotyrosine-binding domain (PDB) interactions. Before we started our study, we carefully considered the possibility that phenylphosphate may disrupt the protein-protein interactions by competitive inhibition. Therefore, we tested different concentrations of phenylphosphate (5, 10, 20, 50 and 100 mM phenylphosphate) in small scale phosphotyrosine IP experiments. Specifically, the pY-IP proteins were eluted by incubation with different concentrations of phenylphosphate and their presence in the eluates was verified by SDS-PAGE and WB using anti-pY antibodies or by silver staining. We also verified the presence of the protein complexes in the eluates by BN-PAGE and WB. We found that the best concentration that we can use is 5 mM phenylphosphate. At this concentration, phenylphosphate does not elute all proteins, but we have the best compromise possible, using the lowest concentration of phenylphosphate in the shortest incubation time and which allows us to obtain the highest amount of the proteins in our IP.
-
Proteins found in the pY-IPs of either (−) or (+) cells or in both by BN-PAGE and MS experiments and shifted their molecular mass from low Mw to high Mw or from high Mw to low Mw. In this category, many proteins were identified, whose behavior was unknown before this study was performed. For example, Nischarin (148 kDa and 210 kDa isoforms) a protein reported to be a participant in ephrin signaling (25) and in cytoskeletal remodeling by inhibition of Rac1 (42–44) was identified in the pY-IP of the (+) cells at around 600 kDa, but not in the (−) cells (Supplemental Table 4 and Figure 4A). Nisharin interacts specifically with the integrin alpha 5 subunit of the fibronectin receptor (45) and with insulin receptor substrates (IRS, IRS4; (46). Nischarin also interacts with PAK1 and active Rac, in a ternary complex, or only with active Rac and supresses PAK1-dependent and PAK1-independent Rac signaling (43, 47). However, it was not known that this protein is involved in a high Mw (600 kDa) protein complex. Our results suggest that upon ephrinB1-Fc stimulation, Nischarin either becomes phosphorylated or interacts with a phosphorylated protein and forms a 600 kDa complex upon ephrin stimulation.
Ret proto-oncogene (124 kDa) is a transmembrane receptor tyrosine kinase protein and is involved in neural crest cell migration, neuron maturation and receptor protein tyrosine kinase signaling pathway, but it is not known whether it is a component of the ephrin signaling. We found this protein in the pY-IP of (−) (at around 180–200 kDa), but not in the (+) cells (Supplemental Table 4 and Figure 4A). This suggests that, unlike the EphB2 receptors, Ret proto-oncogene is tyrosine phosphorylated in the (−) cells (or interacts with a phosphorylated protein) and is de-phosphorylated upon ephrinB1-Fc stimulation (or no longer binds to phosphoprotein upon ephrinB1-Fc stimulation). Since the difference between the theoretical mass of this protein (124 kDa) and its experimentally determined (180–200 kDa) is very small, we suspect that this protein is in monomeric form. Another protein of interest is T-lymphoma invasion and metastasis-inducing protein 1 (Tiam 1, 178 kDa), a RhoGEF protein involved in positive regulation of axonogenesis and Rac1 activation, and in connection of extracellular signals to cytoskeletal activities (48). This protein is also predicted to be involved in the ephrin signaling pathway. We found this protein in the pY-IP of (−) (at around 700 kDa), but not in the (+) cells (Supplemental Table 4 and Figure 4A). This suggests that, proto-oncogene, Tiam1 is tyrosine phosphorylated in the (−) cells (or interacts with a phosphorylated protein) and is de-phosphorylated upon ephrinB1-Fc stimulation (or no longer binds to phosphoprotein).
Proteins found in the pY-IPs of both (−) and (+) cells by BN-PAGE and MS experiments that did not change in the molecular mass (did not shift in BN-PAGE) upon ephrinB1-Fc stimulation, but changed their abundance by either increase or decrease of their relative amounts, as determined by both emPAI score (data not shown) (49) or spectral counting (50). In this category, we identified proteins that had an increased, decreased, or unchanged spectral counting score in the pY-IP of the (+) cells, compared with the (−) cells. Because spectral counting is influenced by spectral quality and number of peptided identified, it is sometimes used as an approximate measure of relative quantitation for a particular protein. An increased spectral counting score in the bands from (+) cells reflects a higher amount of a protein, suggesting either a higher degree of phosphorylation of that protein, or an association with a highly phosphorylated protein upon ephrinB1-Fc stimulation. Conversely, a decreased spectral counting score in the (+) cells reflects a reduction of the amount of a protein, suggesting either a dephosphorylation event or an association with a protein with a lower phosphorylation level that resulted upon ligand stimulation. This information, combined with the results about the mass of the protein complex that contains that protein, can suggest new hypotheses about the behavior of these proteins during signaling events.
One protein that had a spectral counting score that increased in the pY-IP of (+) cells, compared with the (−) cells is Trk-fused gene protein (43 kDa), identified in the mass range of 500–700 kDa (Figure 4B). This protein is suspected to have signal transducing activity, but so far has no demonstrated function (51–54). For this protein, the spectral counting score for this protein in the (+) cells compared with the (−) cells was 1.543. This suggests that Trk-fused gene is more phosphorylated and/or interacts with other proteins/phosphorylated proteins upon ephrinB1-Fc stimulation.
Calnexin (62 kDa) is a protein that had a spectral counting score that decreased in the pY-IP of (+) cells compared with the (−) cells. Calnexin is a calcium-binding protein that interacts with newly synthesized glycoproteins in the endoplasmic reticulum. This protein was identified in both (−) and (+) cells, at about 120 kDa (Figure 4B). The spectral counting score for this protein in the (+) cells, compared with the (−) ones was −0.695. Since Calnexin does not have a tyrosine phosphorylation motif (this protein has only been shown to be phosphorylated at serine and threonine residues), this suggests that the Calnexin’s interactor is dephosphorylated or Calnexin no longer interacts with a phosphoprotein, upon ephrinB1-Fc stimulation.
A protein that had a spectral counting score that did not change in the pY-IPs of (−) and (+) cells was actin monomer (43 kDa), a constitutive element of our pY-IPs. In our experiments, this protein had a spectral counting score very similar in both (−) and (+) cells (−0.065). This suggests that Actin, most likely a constitutive contaminant of our pY-IPs, is post-translationally unaffected by the ephrinB1-Fc stimulation.
To confirm that the three above mentioned proteins had either an increased (Trk-fused gene), decreased (Calnexin) or unchanged (Actin) level in the (−) cells compared with the (+) cells, as determined by the spectral counting score, we further compared the intensity of precursor ions for particular peptides for each protein that were found in both conditions. For each peak, the intensity scale for the spectra from both conditions was normalized to identical number of counts. Figure 5 depicts such a comparison. As observed, for the peak with m/z of 906.52 (2+) that corresponds to a Trk-fused gene peptide, a higher amount of this peptide is observed in the pY-IP of (+) cells, compared with the (−) cells (Figure 5A). Conversely, for the peak with m/z of 695.71 (3+) that corresponds to a Calnexin peptide, a lower amount of this peptide is observed in the pY-IP of (+) cells, compared with the (−) cells (Figure 5B). Finally, for the peak with m/z of 1107.90 (2+) that corresponds to an Actin peptide, almost no change in the amount of this peptide is observed in the pY-IP of (+) cells, compared with the (−) cells (Figure 5C). Taken together, these data suggests that in our BN-PAGE and MS experiments, changes in the relative amounts of proteins (due to protein phosphorylation or to interaction with a phosphorylated protein) between the (−) and (−) cells may be identified.
Figure 5.

Comparison of the intensities of MS spectra for peaks corresponding to peptides that are part of proteins found by BN-PAGE and MS. A: The peak with m/z of 906.52 (2+) that corresponds to a Trk-fused gene peptide QSTQVMAASMSAFDPLK has a higher intensity of this peptide in the pY-IP of (+) cells, compared with the (−) cells. Total ion count over one minute retention time was 4.46e3 versus 1.01e3. B: The peak with m/z of 695.71 (3+) that corresponds to a Calnexin peptide IADPDAVKPDDWDEDAPSK, has a lower intensity of this peptide in the pY-IP of (−) cells, compared with the (+) cells. Total ion count over one minute retention time was 1.25e3 versus 6.04e3. C: The peak with m/z of 1107.90 (2+) that corresponds to an Actin peptide DLYANTVLSGGTTMYPGIADR, had almost no change in the intensities of this peptide is observed in the pY-IP of both (−) and (+) cells. Total ion count over one minute retention time was 9.40e3 versus 8.62e3. The intensity scale for the spectra from both (−) and (+) cells for each individual peptide was identical.
Comparison of BN-PAGE and MS of the pY-IP with BN-PAGE and WB of the lysates from both (−) and (+) cells
By comparing the data obtained by BN-PAGE and MS of the pY-IP with the data obtained by BN-PAGE and WB of the lysates from both (−) and (+) cells, a more comprehensive picture of the behavior of a particular protein may be obtained. In Figure 6, the profiles of some proteins (previously identified in our BN-PAGE and MS experiments of the pY-IP samples) in BN-PAGE and WB of the cell lysates is shown. As observed in the BN-PAGE and WB experiments of the cell lysates, the EphB2 receptor was detected in both (−) cells (at around 120 kDa) and (+) cells (at around 120 kDa and 300 kDa) (Figure 6A). While the EphB2 receptor from 120 kDa band is in a monomeric state, the composition of the receptor-containing 300 kDa band is not known. If compared with the previous BN-PAGE and MS experiments of the pY-IPs, this band could contain the phosphorylated receptor that was identified in the small (Figure 3A) and large (Figure 3B) scale BN-PAGE and MS at around 200–300 kDa range, as well as with the phosphorylated EphB2 receptor detected by BN-PAGE and WB of the pY-IP sample from (+) cells (Figure 6A). Therefore, by comparison of the data that resulted from BN-PAGE and MS experiments of the pY-IPs, BN-PAGE and WB of the pY-IPs, and BN-PAGE and WB of the lysates, we can conclude that only a small portion of the receptor is actually phosphorylated and is involved in transient protein-protein interactions.
Figure 6.
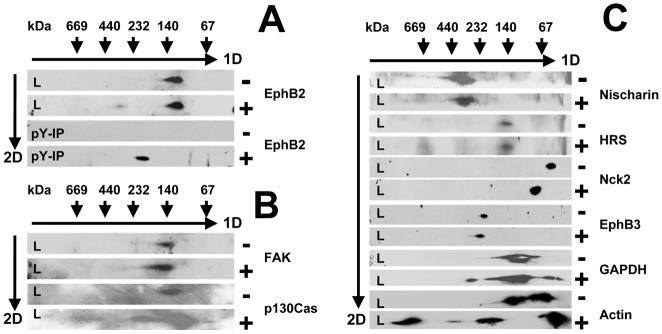
Analysis of (−) and (+) cells by BN-PAGE and WB. The lysates or pY-IPs were separated by BN-PAGE 1D and SDS-PAGE 2D and then analyzed by WB with different antibodies. A: Analysis of lysates and pY-IPs of both (−) and (+) cells by BN-PAGE and WB using EphB2 antibodies. B: Analysis of lysates of (−) and (+) cells by BN-PAGE and WB using FAK and p130Cas antibodies. C: Analysis of lysates of (−) and (+) cells by BN-PAGE and WB using Nischarin, HRS, Nck2, EphB3, GAPDH, and actin antibodies. The direction of migration is shown by arrows. The mass markers are shown for the first 1D and second 2D dimension. L stands for lysate and pY-IP for phosphotyrosine immunoprecipitate.
We also compared the data that resulted from the experiments using BN-PAGE and MS of the pY-IPs and BN-PAGE and WB of the lysates from both (−) and (+) cells for other proteins. For example, most FAK was detected at a molecular weight region in the gels consistent with a monomer in both pY-IPs and lysates from both (−) and (+) cells (Figures 3A and 6B) and only a small portion of it appears to interact with other proteins (in our case most likely with p130Cas at around 250 kDa) (Figure 3A). Another protein, p130Cas, was also mostly detected as a monomer in lysates from both (−) and (+) cells (at around 140 kDa) and apparently associated with another protein (perhaps FAK) in the lysate of the (+) cells (at around 200–250 kDa; Figure 6B). In BN-PAGE and MS of the pY-IPs, p130Cas was detected only in the (+) cells, at around 250–300 kDa, at similar apparent molecular weight as FAK (Figure 3A). This suggests that, although in the lysates and pY-IPs of both (−) and (+) cells, most of FAK and p130Cas are in monomeric form, and only a small portion of the pool of these proteins interact with each other as a result of ephrinB1-Fc stimulation. However, these results were obtained after IP in the presence of NP40 buffer, which solubilizes only a small portion of the membrane fraction, and a different buffer with a different detergent may give us additional information. For example, buffer containing DDM, used in the large scale pY-IP, solubilizes mostly membrane fraction. In our experiments, we observed that FAK was identified mostly in apparent protein complex(es) with masses between 200–500 kDa in the samples from both (−) and (+) cells, with high abundance in a complex from the (+) cells, with a mass of about 500 kDa (Supplemental Table 4). Alternatively, these complexes are stable in DDM buffer, but not NP-40 buffer.
In our previous experiments, Nischarin was identified in the BN-PAGE and MS of the pY-IP from (+) cells at around 600–650 kDa (Figure 4A). The experiments using BN-PAGE and WB of the lysate from (−) and (+) cells revealed that Nischarin runs in both conditions at about 300–350 kDa (Figure 6C). At this moment, we were unable to determine whether Nischarin is permanently in interaction with a different protein, or is in a monomeric state, but runs anomalously in BN-PAGE, as reported for some proteins with pI below 5 or above 8.5 (18, 19). However, comparison of the BN-PAGE and WB of the lysate and BN-PAGE and MS of the pY-IP from both (−) and (+) cells suggests that Nischarin is either 1) phosphorylated and interacts with a protein(s) upon ephrin stimulation and shifts from 300–350 kDa in the lysate to 600–650 kDa in the pY-IP of the (+) cells; 2) not phosphorylated but it interacts with a phosphorylated protein(s) upon ephrin stimulation and shifts from 300–350 kDa in the lysate to 600–650 kDa in the pY-IP of the (+) cells.
Nck2 is a protein that was detected only as an apparent monomer in BN-PAGE and WB of the lysates of both (−) and (+) cells (Figure 6C). Nck2 was also detected as an apparent monomer in the BN-PAGE and MS of pY-IPs of the (+) but not (−) cells (Figure 3B). By comparing these data, we may conclude that Nck2 is most likely phosphorylated upon stimulation and that, under these experimental conditions, it does not stably interact with any other protein(s). As mentioned previously, it is also possible that during elution, phenylphosphate disrupted the phosphotyrosine-SH2 domain or phosphotyrosine-PTB domain interactions of Nck2. As observed in Figure 6C, the size difference of Nck2 as detected by WB of the lysate of (−) and (+) cells is about 20 kDa. Although it is possible that Nck2 may indeed interact with a different protein upon stimulation, the size difference is so small that we believe that it is experimental error.
In BN-PAGE and WB of the lysates, Hrs appeared to be in a monomeric state in the lysates of both (−) and (+) cells (Figure 6C). In BN-PAGE and MS of the pY-IPs, Hrs was found to interact with STAM1 and possibly with EphB2 receptor (Figure 3B). Comparison of these data suggests that, while most of the Hrs may be in a monomeric state, some of it interacts with STAM1 upon stimulation and minimum one of these two proteins is tyrosine phosphorylated. Taken together, this suggests that comparison of the data obtained by BN-PAGE and MS of pY-IPs and BN-PAGE and WB of the lysates from both (−) and (+) cells may provide more information about the unphosphorylated and phosphorylated populations of a particular protein.
WAVE1 and WAVE2 co-localize in IF studies and change their intracellular distribution upon ephrinB1-Fc stimulation
It has been reported that WAVE proteins coordinate actin reorganization (41) and that lamellipodium is formed when cells are (+) by growth factors (55) or when Rac is activated (41). For example, in melanoma cells and fibroblasts, WAVE complexes interact with activated Rac1 and translocate to the plasma membrane (56). Rac may regulate the WAVE2 complex by binding to it and controlling its localization (36). In addition, Abi-1 directs translocation of WAVE2 to the membrane after FN stimulation and Rac, Abl, Abi-1 and WAVE2 colocalize at the actin-rich lamellipodial leading edge (40). Furthermore, in fibroblasts, WAVE1 assembles in a WAVE1-PKA-Abl complex and migrates from the focal adhesions to the sites of actin reorganization upon PDGF stimulation (41). In our BN-PAGE and MS of pY-IPs from both (−) and (+) cells, we identified the intact WAVE2 complex that shifted from about 700 kDa in the (−) cells to about 600 kDa in the (+) cells (Figure 3B). In order to investigate whether WAVE proteins and their interactors are also regulated by ephrin signaling, we employed IF to analyze co-localization of the WAVE1, WAVE2 and actin in the (−) cells and in the cells (+) with ephrinB1-Fc for 5, 10, 20 and 45 minutes. The outcome of this experiment is presented in Figure 7.
Figure 7.

WAVE1 and WAVE2 co-localize and dramatically change their distribution upon ephrinB1-Fc stimulation. Immunofluorescence analysis of the (−) cells (0 minutes) and cells (+) for 5, 10, 20 and 45 minutes with EphB1-Fc. The cells were analyzed for the presence of Actin (blue), WAVE1 (green) and WAVE2 (red) proteins. Scale bar, 20 μm.
In the (−) cells, WAVE1, WAVE2 and actin co-localized mainly at the tips of the neurite extensions where intense staining was observed, while the stain in the cell body for WAVE proteins was very weak. Upon stimulation, the distribution of WAVE1, WAVE2 and actin proteins was very dynamic, with the most intense changes 10 and 20 minutes after ephrinB1-Fc stimulation. If we compare these IF experiments with the BN-PAGE and WB of the lysates of (−) and time-course (+) cells immunodecorated with pY99 antibodies (Figure 2C), we observe that the most dramatic changes in the IF experiments (5 and 10 minutes after stimulation) correspond to the highest phosphorylation levels in the BN-PAGE and WB experiments, suggesting that the most important changes that may be observed in our system are 5 and 10 minutes, and not 45 minutes, after stimulation.
We also analyzed other proteins known to interact with WAVE proteins and regulate them such as Abi-1 and Abl. Abl was detected in the cytoplasm and nucleus (57, 58), but also in the focal adhesions (59). Abl was also found in a complex with WAVE1 and PKA (41) upon Rac activation (41), and co-localize with Rac, Abi-1 and WAVE2 (40). A similar pattern as previously described in our IF experiments (Figure 7) was observed when WAVE1, Abl and actin were analyzed by IF (data not shown), suggesting that Abl is also a player in regulation of translocation of WAVE proteins at the site of actin re-modeling. Taken together, these data suggests that dramatic changes happen during ephrin stimulation, detected by both IF and BN-PAGE and WB of the lysates of both (−) and time-course stimulated cells and that the most intense changes happen 5 and 10 minutes after stimulation. These data also suggest that WAVE proteins are activated through a similar mechanism as shown for the PDGF pathway (they form the same ring-like structure upon ephrinB1-Fc stimulation) (41).
A possible mechanism of ephrin-dependent cytoskeletal remodeling
It is well known that activation of Rac by either growth factors or fibronectin leads to cytoskeletal remodeling and membrane ruffle formation (36, 41, 55, 60). Among many other proteins, some important downstream effectors of Rac are WAVE proteins, Arp2/3 complex and PAK family of kinases. WAVE proteins coordinate actin reorganization by coupling Rho-related small molecular weight GTP-ases to the mobilization of Arp2/3 complex, involved in actin polymerization and facilitate dendritic branching of filaments (61). In the formation of filopodia and lamellipodia, Arp2/3 complex acts downstream of WASP family of proteins, which include WASP, N-WASP and WAVEs proteins. WASP and N-WASP lead to formation of filopodia via small GTP-ase CDC42, while WAVEs are responsible for lamellipodia formation via small GTP-ase Rac (62–65). It was reported that WAVE1 was found in a complex with Abl and PKA and migrated to the sites of cytoskeletal remodeling upon Rac activation (41). WAVE1 complex, composed of WAVE1, Nap1, Cyfip2 and Abi-2, is kept intact in inactive form and activated by dissociation upon Rac activation (37). It was also reported that WAVE2, together with SRA1, Nap1, and Abi-1 (WAVE2 complex) interacted and co-localized with activated Rac1 and translocated to the plasma membrane upon Rac1 activation (36, 56) and that WAVE2 colocalized with Rac, Abl, and Abi-1 at the actin-rich lamellipodial leading edge (40). The PAK family of kinases has been implicated in control of actin filaments and in cell motility and is activated by direct interaction with the active Rac. One target for PAK involved in migration is myosin light chain kinase (MLCK), which is phosphorylated and inhibited by PAK. Therefore, MLCK is not able to phosphorylate MLC anymore, leading to reduction of contractility. Another PAK target is LIM kinase, that phosphorylates cofilin, an actin severing protein, and thus promoting filament assembly (42).
In our experiments the WAVE2 complex appears to be constitutive with an apparent molecular weight of at least 500 kDa, and that both WAVE1 and WAVE2 co-localized at the tips of the neurite extensions in the (−) cells. In the first 10 minutes after stimulation, these proteins moved to the leading edge of lamellipodia and over the next 35 minutes migrated back at the tips of the neurite extensions, into a pattern observed in the (−) cells. The ring-like structure is most likely a result of Rac activation by ephrinB1-Fc stimulation, as observed for PDGF stimulation (41), suggesting that ephrinB1-Fc activates Rac in a manner similar to other growth factors. In the simplest scenario ephrinB1-Fc activates Rac that binds to WAVE complexes and directs them from the tips of the neurite extensions to the sites of cytoskeletal re-organization, in our case the ring-like structures. Once translocated and activated, WAVE complexes activate Arp2/3 complexes, involved in actin polymerization and branching of filaments (61). However, disappearance of the ring-like structure in the cells (+) for 20 and 45 minutes and migration of the WAVE proteins/complexes back to the tips of the neurite extensions suggests that either Rac or one of its downstream components is inactivated.
It has been reported that Nischarin, a protein highly expressed in neuronal cells, reverses the effect of Rac on lamellipodia formation in fibroblasts (45). This protein interacts with PAK1 (a downstream effector of Rac) and suppresses it, and this interaction is enhanced by active Rac, in a ternary complex (43). Nischarin also inhibits LIM kinase, a PAK downstream effector (66). In addition to supressing PAK1-dependent signaling, Nischarin also inhibits PAK1-independent Rac signaling by direct interaction with active Rac (42, 47). Furthermore, Nischarin is heavily phosphorylated upon ephrinB1-Fc stimulation (25), and co-localize with PAK1 in the membrane ruffles (43). In our experiments, Nisharin was detected in monomeric form in BN-PAGE and WB of lysates from both (−) and (+) cells (Figure 6C) and as a 600 kDa complex only in BN-PAGE and MS of pY-IP of (+) cells (Figure 4A), suggesting that Nischarin is phosphorylated and activated upon ephrinB1-Fc stimulation and perhaps inhibits formation of Rac-dependent lamellipodia by inhibiting Rac, PAK or both. Since 1) in the IF experiments, WAVE proteins moved from the lamellipodia to the tips of the neurite extensions 20 and 45 minutes after stimulation and perhaps Rac was heavily activated in the ring-like-structures 10 minutes after stimulation, but was inhibited after 20 and 45 minutes (Figure 7); 2) Nisharin is heavily phosphorylated upon ephrinB1-Fc stimulation (25) and 3) in our experiments it was found in the pY-IP of (+) cells, this suggests that in our system Nischarin becomes activated about 10 minutes after stimulation, most likely by inhibition of Rac-PAK-dependent and Rac-dependent-PAK-independent pathways. Once Rac is inactivated, WAVE complexes are not controlled by Rac anymore and may be re-directed at the tips of the neurite extensions, in agreement with our IF experiments (Figure 7). This is a possible scenario of ephrin-dependent Rac activation and Nischarin-dependent Rac inactivation, a scenario that takes into account both activation and inactivation of Rac and previously unreported, but does not take into account other players from ephrin signaling like Abl, which was shown to interact with WAVE1 (41) and to co-localize with it (data not shown) or involvement of other pathways not described here, which we do not rule out.
Concluding remarks
In conclusion, we used BN-PAGE and MS of the pY-IPs, BN-PAGE and WB of the lysates of both (−) and (+) cells to analyze protein-protein interactions from signal transduction pathways involved in ephrinB1-Fc stimulation. We also used IF to explore the time-dependent localization of proteins in (−) and (+) cells. Initially, we tested the combination of BN-PAGE and MS to analyze well known stable protein interactions from the cell lysate and observed that both subunit composition and apparent mass of the protein complexes (Valosin-containing protein or Proteasome) are in agreement with the published literature (29, 30). We then applied BN-PAGE and MS to analyze transient protein interactions in the pY-IPs of the (−) and (+) cells. We concluded that FAK and p130Cas, two proteins well documented to interact with each other in other systems, also interact with each other upon ephrinB1-Fc stimulation, which has not yet reported for this system. In the large scale analysis by BN-PAGE and MS of the pY-IPs of the (−) and (+) cells, we identified many well known (Hrs-STAM1, WAVE2 complex) protein-protein interactions but new to ephrin signaling, many new protein-protein interactions (Figures 4A and B), as well as information about particular proteins (e.g. Tiam1, Nischarin) in terms of size, protein interaction or phosphorylation. In addition, many proteins identified in our experiments and possibly involved in ephrin signaling are still waiting to be investigated. For example, Nodal modulator or Trk-fused gene proteins, found in our experiments in much higher amounts in pY IPs from lysates of the (+) cells have no demonstrated function. Involvement of some kinases such as the Ret proto-oncogene or TrkC receptors in ephrin signaling is also unknown. The combination of BN-PAGE and MS of pY-IPs, BN-PAGE and WB of the lysates of both (−) and (+) cells also can be applied to the analysis of stable and transient protein-protein interactions in other ligand-stimulated RTK-dependent signal transduction pathways.
Supplementary Material
Supplemental Figure 1. Hypothetical biochemical characterization of ephrin signaling. Possible theoretical scenarios for analysis of protein-protein interactions within pY99-IPs of (−) and (+) cells. A: One protein that is either tyrosine-phosphorylated or associated with a protein phosphorylated at the tyrosine residue in the pY-IP of (−) cells may interact with other proteins upon stimulation and shifts its molecular mass towards a higher one. B: One protein complex is disassembled or dephosphorylated upon stimulation; this complex will be detected at a lower molecular mass range upon stimulation. C: One protein complex becomes over-phosphorylated upon stimulation, regardless of which subunit within the protein complex is phosphorylated; alternatively, the same subunit from the protein complex may be phosphorylated at different phosphorylation sites. D: As in C, but the protein complex is over-dephosphorylated upon stimulation (one of more subunits, and one of more phosphorylation sites per subunit.
Supplemental Figure 2. Analysis of lysates of (−) and (+) cells by SDS-PAGE and BN-PAGE. The lysates of (−) and (+) cells were separated by SDS-PAGE (A) and BN-PAGE 1D and silver-stained. The gel lanes from BN-PAGE 1D were also excised, reduced and denatured, and further separated in 2D SDS-PAGE and silver stained. The direction of migration in BN-PAGE is indicated by arrows followed by either 1D or 2D. The mass markers are indicated for each gel in 1D or 2D.
Supplemental Figure 3. BN-PAGE of lysates of (−) NG108-EphB2 cells. The gel was Coomassie stained and the gel bands of interest (bands B1 to B8) were excised and subjected to MS analysis. The approximate masses of the bands were between 700–750 (band B1) and 350–400 kDa (band B8), as shown on the right side of the gel. The molecular mass markers are shown on the left side of the gel. Mk: molecular mass markers; L: NG108-EphB2 cell lysate.
Supplemental Figure 4. MS analysis of the BN-PAGE gel bands B1 (700–750 kDa) and B4 (550–600 kDa). (A) MS/MS of the m/z (2+) 643.33 (calculated 643.35) peak that corresponds to peptide AINQGGLTSVAVR, which is part of the proteosome alpha subunit 6 (mass 27 kDa), a 700 kDa alpha-beta heterocomplex. (B) MS/MS of the m/z (2+) 586.79 (calculated 586.81) peak that corresponds to peptide LAAIQESGVER, which is part of the proteosome beta subunit 6 (mass 29 kDa), a 700 kDa alpha-beta heterocomplex. (C) MS/MS of the m/z (2+) 1093.49 (calculated 1093.43) peak that corresponds to peptide EDEEESLNEVGYDDIGGCR, which is part of the valosin-containing protein (mass 89 kDa), that forms a 540 kDa homohexamer.
Supplemental Table 1: Proteins identified by SDS-PAGE and MS of the pY99-IP of (−) cells. The searching parameters are described in the methods section. The protein name, NCBI accession number, and Mascot score are shown for each identified protein.
Supplemental Table 2: Proteins identified by SDS-PAGE and MS of the pY99-IP of (+) cells. The searching parameters are described in the methods section. The protein name, NCBI accession number, and Mascot score are shown for each identified protein.
Supplemental Table 3: Proteins identified by small scale BN-PAGE and MS analysis of the pY-IPs of (−) and (+) cells. The searching parameters are written in the materials section. There were 24 gel bands for each condition and the band name is written on the left of the table. We did not identify any other proteins except the ones in the table, plus contaminant proteins (e.g. keratin) and trypsin (this is why we did not show the data for all bands). The molecular mass markers are as described in Figure 3A. For example, band 12 corresponds to about 250–300 kDa, band 15 to about 200 kDa, and band 19 to about 130–150 kDa. The protein name, NCBI accession number, Mascot score and molecular mass and protein coverage are shown for each identified protein.
Supplemental Table 4: Proteins identified by large scale BN-PAGE and MS analysis of the pY-IPs of (−) and (+) cells. The searching parameters are written in the materials section. There were 32 gel bands for each condition and the band name is written on the left of the table. For each gel band, two technical replicates were run by MS (128 runs), uploaded to ProteinCenter, merged and clustered, and then exported as csv files. The molecular mass markers are as described in Figure 4. For example, band 3 corresponds to about 700 kDa, band 11 to about 400–450 kDa, band 17 to about 180 kDa and band 30 to about 45–50 kDa. The protein name, NCBI accession number, molecular mass and protein coverage, and Mascot score are shown for each identified protein.
Supplemental Table 5: BN-PAGE peptide samples that were analyzed by LC-MS/MS (and for which the results are shown in Supplemental Table 4) were re-run using an Eksigent NanoLC-2D coupled to an LTQ-Orbitrap MS (Thermo Scientific, Waltham, MA). The outcome regarding the subunits of WAVE protein complexes are shown. The method used is presented in the supplemental method.
Acknowledgments
We thank Drs. Vivekananda Shetty, Chongfeng Xu, David Fenyo, Daniel Spellman, Alisa G. Woods and Mr. Steven Blais for advice and discussions. We also thank Dr. S.K. Alahari for the kind gift of anti-Nischarin antibodies. Our research was supported in part by NIH NINDS grant P30 NS050276 and NIH NCRR shared instrumentation grant S10 RR017990 to TAN and Clarkson start-up to CCD.
Abbreviations
- RTK
Receptor tyrosine kinase
- BN-PAGE
Blue Native PAGE
- WB
Western blotting
- MS
mass spectrometry
- pY-IP
phosphotyrosine immunoprecipitate
- IF
immunofluorescence microscopy
- 1D
first dimension
- 2D
second dimension
- (−)
unstimulated NG108 cells
- (+)
ephrinB1-Fc-stimulated NG108 cells
References
- 1.Holland SJ, Gale NW, Mbamalu G, Yancopoulos GD, Henkemeyer M, Pawson T. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- 2.Holland SJ, Peles E, Pawson T, Schlessinger J. Cell-contact-dependent signalling in axon growth and guidance: Eph receptor tyrosine kinases and receptor protein tyrosine phosphatase beta. Curr Opin Neurobiol. 1998;8:117–127. doi: 10.1016/s0959-4388(98)80015-9. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nat Rev Neurosci. 2001;2:155–164. doi: 10.1038/35058515. [DOI] [PubMed] [Google Scholar]
- 4.Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- 5.Robinson V, Smith A, Flenniken AM, Wilkinson DG. Roles of Eph receptors and ephrins in neural crest pathfinding. Cell Tissue Res. 1997;290:265–274. doi: 10.1007/s004410050931. [DOI] [PubMed] [Google Scholar]
- 6.Adams RH. Molecular control of arterial-venous blood vessel identity. J Anat. 2003;202:105–112. doi: 10.1046/j.1469-7580.2003.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002;13:75–85. doi: 10.1016/s1359-6101(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Li R, Du D, Zhang C, Yuan H, Zeng R, Chen Z. Proteomic analysis reveals novel molecules involved in insulin signaling pathway. J Proteome Res. 2006;5:846–855. doi: 10.1021/pr050391m. [DOI] [PubMed] [Google Scholar]
- 9.Yancopoulos GD, Klagsbrun M, Folkman J. Vasculogenesis, angiogenesis, and growth factors: ephrins enter the fray at the border. Cell. 1998;93:661–664. doi: 10.1016/s0092-8674(00)81426-9. [DOI] [PubMed] [Google Scholar]
- 10.Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson DG, Pawson T, Davis S, Yancopoulos GD. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron. 1996;17:9–19. doi: 10.1016/s0896-6273(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 11.Carter N, Nakamoto T, Hirai H, Hunter T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas) Nat Cell Biol. 2002;4:565–573. doi: 10.1038/ncb823. [DOI] [PubMed] [Google Scholar]
- 12.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 13.Vearing CJ, Lackmann M. Eph receptor signalling; dimerisation just isn’t enough. Growth Factors. 2005;23:67–76. doi: 10.1080/08977190500055869. [DOI] [PubMed] [Google Scholar]
- 14.Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275:1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- 15.Cowan CA, Henkemeyer M. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature. 2001;413:174–179. doi: 10.1038/35093123. [DOI] [PubMed] [Google Scholar]
- 16.Darie CC, Biniossek ML, Winter V, Mutschler B, Haehnel W. Isolation and structural characterization of the Ndh complex from mesophyll and bundle sheath chloroplasts of Zea mays. Febs J. 2005;272:2705–2716. doi: 10.1111/j.1742-4658.2005.04685.x. [DOI] [PubMed] [Google Scholar]
- 17.Darie CC, Janssen WG, Litscher ES, Wassarman PM. Purified trout egg vitelline envelope proteins VEbeta and VEgamma polymerize into homomeric fibrils from dimers in vitro. Biochim Biophys Acta. 2008;1784:385–392. doi: 10.1016/j.bbapap.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217:220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- 19.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 20.Camacho-Carvajal MM, Wollscheid B, Aebersold R, Steimle V, Schamel WW. Two-dimensional Blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates: a proteomics approach. Mol Cell Proteomics. 2004;3:176–182. doi: 10.1074/mcp.T300010-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Litscher ES, Janssen WG, Darie CC, Wassarman PM. Purified mouse egg zona pellucida glycoproteins polymerize into homomeric fibrils under non-denaturing conditions. J Cell Physiol. 2008;214:153–157. doi: 10.1002/jcp.21174. [DOI] [PubMed] [Google Scholar]
- 22.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 23.Zhang G, Annan RS, Carr SA, Neubert TA. Overview of peptide and protein analysis by mass spectrometry. Curr Protoc Protein Sci Chapter. 16(Unit16):11. doi: 10.1002/0471140864.ps1601s62. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Ueberheide BM, Waldemarson S, Myung S, Molloy K, Eriksson J, Chait BT, Neubert TA, Fenyo D. Protein quantitation using mass spectrometry. Methods Mol Biol. 673:211–222. doi: 10.1007/978-1-60761-842-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang G, Spellman DS, Skolnik EY, Neubert TA. Quantitative phosphotyrosine proteomics of EphB2 signaling by stable isotope labeling with amino acids in cell culture (SILAC) J Proteome Res. 2006;5:581–588. doi: 10.1021/pr050362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darie CC, Shetty V, Spellman DS, Zhang G, Xu C, Cardasis HL, Blais S, Fenyo D, Neubert TA. Blue Native PAGE and mass spectrometry analysis of the ephrin stimulation- dependent protein-protein interactions in NG108-EphB2 cells. Springer-Verlag; Düsseldorf, Germany: 2008. [Google Scholar]
- 27.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 28.Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics. 2008;7:1067–1076. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 30.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Fenyo D, Neubert TA. Screening for EphB signaling effectors using SILAC with a linear ion trap-orbitrap mass spectrometer. J Proteome Res. 2008;7:4715–4726. doi: 10.1021/pr800255a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci U S A. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Row PE, Clague MJ, Urbe S. Growth factors induce differential phosphorylation profiles of the Hrs-STAM complex: a common node in signalling networks with signal-specific properties. Biochem J. 2005;389:629–636. doi: 10.1042/BJ20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bache KG, Raiborg C, Mehlum A, Stenmark H. STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J Biol Chem. 2003;278:12513–12521. doi: 10.1074/jbc.M210843200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang G, Deinhardt K, Chao MV, Neubert TA. Study of Neurotrophin-3 Signaling in Primary Cultured Neurons using Multiplex Stable Isotope Labeling with Amino Acids in Cell Culture. J Proteome Res. doi: 10.1021/pr200016n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- 37.Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- 38.Kitamura Y, Kitamura T, Sakaue H, Maeda T, Ueno H, Nishio S, Ohno S, Osada S, Sakaue M, Ogawa W, Kasuga M. Interaction of Nck-associated protein 1 with activated GTP-binding protein Rac. Biochem J. 1997;322(Pt 3):873–878. doi: 10.1042/bj3220873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, Mello CC, Kaibuchi K. The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev. 2002;16:620–632. doi: 10.1101/gad.955702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci U S A. 2005;102:1098–1103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westphal RS, Soderling SH, Alto NM, Langeberg LK, Scott JD. Scar/WAVE-1, a Wiskott-Aldrich syndrome protein, assembles an actin-associated multi-kinase scaffold. Embo J. 2000;19:4589–4600. doi: 10.1093/emboj/19.17.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alahari SK. Nischarin inhibits Rac induced migration and invasion of epithelial cells by affecting signaling cascades involving PAK. Exp Cell Res. 2003;288:415–424. doi: 10.1016/s0014-4827(03)00233-7. [DOI] [PubMed] [Google Scholar]
- 43.Alahari SK, Reddig PJ, Juliano RL. The integrin-binding protein Nischarin regulates cell migration by inhibiting PAK. Embo J. 2004;23:2777–2788. doi: 10.1038/sj.emboj.7600291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piletz JE, Wang G, Zhu H. Cell signaling by imidazoline-1 receptor candidate, IRAS, and the nischarin homologue. Ann N Y Acad Sci. 2003;1009:392–399. doi: 10.1196/annals.1304.053. [DOI] [PubMed] [Google Scholar]
- 45.Alahari SK, Lee JW, Juliano RL. Nischarin, a novel protein that interacts with the integrin alpha5 subunit and inhibits cell migration. J Cell Biol. 2000;151:1141–1154. doi: 10.1083/jcb.151.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sano H, Liu SC, Lane WS, Piletz JE, Lienhard GE. Insulin receptor substrate 4 associates with the protein IRAS. J Biol Chem. 2002;277:19439–19447. doi: 10.1074/jbc.M111838200. [DOI] [PubMed] [Google Scholar]
- 47.Reddig PJ, Xu D, Juliano RL. Regulation of p21-activated kinase-independent Rac1 signal transduction by nischarin. J Biol Chem. 2005;280:30994–31002. doi: 10.1074/jbc.M502546200. [DOI] [PubMed] [Google Scholar]
- 48.Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408:682–688. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]
- 49.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7:2373–2385. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greco A, Fusetti L, Miranda C, Villa R, Zanotti S, Pagliardini S, Pierotti MA. Role of the TFG N-terminus and coiled-coil domain in the transforming activity of the thyroid TRK-T3 oncogene. Oncogene. 1998;16:809–816. doi: 10.1038/sj.onc.1201596. [DOI] [PubMed] [Google Scholar]
- 52.Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M, Pierotti MA. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol. 1995;15:6118–6127. doi: 10.1128/mcb.15.11.6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol. 2009 doi: 10.1016/j.mce.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Miranda C, Roccato E, Raho G, Pagliardini S, Pierotti MA, Greco A. The TFG protein, involved in oncogenic rearrangements, interacts with TANK and NEMO, two proteins involved in the NF-kappaB pathway. J Cell Physiol. 2006;208:154–160. doi: 10.1002/jcp.20644. [DOI] [PubMed] [Google Scholar]
- 55.Nakanishi O, Suetsugu S, Yamazaki D, Takenawa T. Effect of WAVE2 phosphorylation on activation of the Arp2/3 complex. J Biochem. 2007;141:319–325. doi: 10.1093/jb/mvm034. [DOI] [PubMed] [Google Scholar]
- 56.Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. Embo J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McWhirter JR, Wang JY. Activation of tyrosinase kinase and microfilament-binding functions of c-abl by bcr sequences in bcr/abl fusion proteins. Mol Cell Biol. 1991;11:1553–1565. doi: 10.1128/mcb.11.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pendergast AM. Nuclear tyrosine kinases: from Abl to WEE1. Curr Opin Cell Biol. 1996;8:174–181. doi: 10.1016/s0955-0674(96)80063-9. [DOI] [PubMed] [Google Scholar]
- 59.Lewis JM, Schwartz MA. Integrins regulate the association and phosphorylation of paxillin by c-Abl. J Biol Chem. 1998;273:14225–14230. doi: 10.1074/jbc.273.23.14225. [DOI] [PubMed] [Google Scholar]
- 60.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 61.Higgs HN, Pollard TD. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J Biol Chem. 1999;274:32531–32534. doi: 10.1074/jbc.274.46.32531. [DOI] [PubMed] [Google Scholar]
- 62.Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- 63.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. Embo J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408:732–735. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- 65.Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding Y, Milosavljevic T, Alahari SK. Nischarin inhibits LIM kinase to regulate cofilin phosphorylation and cell invasion. Mol Cell Biol. 2008;28:3742–3756. doi: 10.1128/MCB.01832-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Hypothetical biochemical characterization of ephrin signaling. Possible theoretical scenarios for analysis of protein-protein interactions within pY99-IPs of (−) and (+) cells. A: One protein that is either tyrosine-phosphorylated or associated with a protein phosphorylated at the tyrosine residue in the pY-IP of (−) cells may interact with other proteins upon stimulation and shifts its molecular mass towards a higher one. B: One protein complex is disassembled or dephosphorylated upon stimulation; this complex will be detected at a lower molecular mass range upon stimulation. C: One protein complex becomes over-phosphorylated upon stimulation, regardless of which subunit within the protein complex is phosphorylated; alternatively, the same subunit from the protein complex may be phosphorylated at different phosphorylation sites. D: As in C, but the protein complex is over-dephosphorylated upon stimulation (one of more subunits, and one of more phosphorylation sites per subunit.
Supplemental Figure 2. Analysis of lysates of (−) and (+) cells by SDS-PAGE and BN-PAGE. The lysates of (−) and (+) cells were separated by SDS-PAGE (A) and BN-PAGE 1D and silver-stained. The gel lanes from BN-PAGE 1D were also excised, reduced and denatured, and further separated in 2D SDS-PAGE and silver stained. The direction of migration in BN-PAGE is indicated by arrows followed by either 1D or 2D. The mass markers are indicated for each gel in 1D or 2D.
Supplemental Figure 3. BN-PAGE of lysates of (−) NG108-EphB2 cells. The gel was Coomassie stained and the gel bands of interest (bands B1 to B8) were excised and subjected to MS analysis. The approximate masses of the bands were between 700–750 (band B1) and 350–400 kDa (band B8), as shown on the right side of the gel. The molecular mass markers are shown on the left side of the gel. Mk: molecular mass markers; L: NG108-EphB2 cell lysate.
Supplemental Figure 4. MS analysis of the BN-PAGE gel bands B1 (700–750 kDa) and B4 (550–600 kDa). (A) MS/MS of the m/z (2+) 643.33 (calculated 643.35) peak that corresponds to peptide AINQGGLTSVAVR, which is part of the proteosome alpha subunit 6 (mass 27 kDa), a 700 kDa alpha-beta heterocomplex. (B) MS/MS of the m/z (2+) 586.79 (calculated 586.81) peak that corresponds to peptide LAAIQESGVER, which is part of the proteosome beta subunit 6 (mass 29 kDa), a 700 kDa alpha-beta heterocomplex. (C) MS/MS of the m/z (2+) 1093.49 (calculated 1093.43) peak that corresponds to peptide EDEEESLNEVGYDDIGGCR, which is part of the valosin-containing protein (mass 89 kDa), that forms a 540 kDa homohexamer.
Supplemental Table 1: Proteins identified by SDS-PAGE and MS of the pY99-IP of (−) cells. The searching parameters are described in the methods section. The protein name, NCBI accession number, and Mascot score are shown for each identified protein.
Supplemental Table 2: Proteins identified by SDS-PAGE and MS of the pY99-IP of (+) cells. The searching parameters are described in the methods section. The protein name, NCBI accession number, and Mascot score are shown for each identified protein.
Supplemental Table 3: Proteins identified by small scale BN-PAGE and MS analysis of the pY-IPs of (−) and (+) cells. The searching parameters are written in the materials section. There were 24 gel bands for each condition and the band name is written on the left of the table. We did not identify any other proteins except the ones in the table, plus contaminant proteins (e.g. keratin) and trypsin (this is why we did not show the data for all bands). The molecular mass markers are as described in Figure 3A. For example, band 12 corresponds to about 250–300 kDa, band 15 to about 200 kDa, and band 19 to about 130–150 kDa. The protein name, NCBI accession number, Mascot score and molecular mass and protein coverage are shown for each identified protein.
Supplemental Table 4: Proteins identified by large scale BN-PAGE and MS analysis of the pY-IPs of (−) and (+) cells. The searching parameters are written in the materials section. There were 32 gel bands for each condition and the band name is written on the left of the table. For each gel band, two technical replicates were run by MS (128 runs), uploaded to ProteinCenter, merged and clustered, and then exported as csv files. The molecular mass markers are as described in Figure 4. For example, band 3 corresponds to about 700 kDa, band 11 to about 400–450 kDa, band 17 to about 180 kDa and band 30 to about 45–50 kDa. The protein name, NCBI accession number, molecular mass and protein coverage, and Mascot score are shown for each identified protein.
Supplemental Table 5: BN-PAGE peptide samples that were analyzed by LC-MS/MS (and for which the results are shown in Supplemental Table 4) were re-run using an Eksigent NanoLC-2D coupled to an LTQ-Orbitrap MS (Thermo Scientific, Waltham, MA). The outcome regarding the subunits of WAVE protein complexes are shown. The method used is presented in the supplemental method.