

SUMMARY
Acetylation is increasingly recognized as an important metabolic regulatory post-translational protein modification, yet the metabolic consequence of mitochondrial protein hyperacetylation is unknown. We find that high-fat diet (HFD) feeding induces hepatic mitochondrial protein hyperacetylation in mice and downregulation of the major mitochondrial protein deacetylase SIRT3. Mice lacking SIRT3 (SIRT3KO) placed on a HFD show accelerated obesity, insulin resistance, hyperlipidemia, and steatohepatitis compared to wild-type (wt) mice. The lipogenic enzyme stearoyl-CoA desaturase 1 is highly induced in SIRT3KO mice, and its deletion rescues both wt and SIRT3KO mice from HFD-induced hepatic steatosis and insulin resistance. We further identify a single nucleotide polymorphism in the human SIRT3 gene that is suggestive of a genetic association with the metabolic syndrome. This polymorphism encodes a point-mutation in the SIRT3 protein, which reduces its overall enzymatic efficiency. Our findings show loss of SIRT3 and dysregulation of mitochondrial protein acetylation contribute to the metabolic syndrome.
INTRODUCTION
The metabolic syndrome is defined by metabolic abnormalities, including central obesity, insulin resistance, hyperlipidemia, hyperglycemia, and hypertension (Reaven, 1988). Prevalence of the metabolic syndrome is rising in the Western world and will lead to future increases in diabetes and cardiovascular disease (Ford et al., 2008). Sedentary lifestyles (Ardern et al., 2004) and high-fat “Western” diets (Feldeisen and Tucker, 2007) have been implicated in the increase in metabolic syndrome. In addition to lifestyle and diet, multiple metabolic pathways are implicated in the pathogenesis of metabolic disease, including aberrant lipogenesis (Roden et al., 1996; Samuel et al., 2004), increased inflammation (Hotamisligil et al., 1993; Uysal et al., 1997), and reduced fatty acid oxidation (Ji and Friedman, 2008; Ji and Friedman, 2007). Identifying the molecular mechanisms underlying the metabolic syndrome has been described as one of the most critical endeavors in modern medicine (Taubes, 2009).
The sirtuins (SIRT1-SIRT7) are a family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases (Imai et al., 2000), and regulate multiple cellular processes, including metabolic homeostasis [for a review, see (Finkel et al., 2009)]. SIRT3 is a mitochondrial sirtuin (Onyango et al., 2002; Schwer et al., 2002) and SIRT3 regulates fatty acid oxidation during fasting and ATP production (Ahn et al., 2008; Hirschey et al., 2010). SIRT3KO mice have hyperacetylated mitochondrial proteins (Lombard et al., 2007). Acetylation controls the enzymatic activity of mitochondrial metabolic enzymes such as malate dehydrogenase in the TCA cycle (Zhao et al., 2010), enoyl–coA hydratase/3-hydroxyacyl–coA dehydrogenase (Zhao et al., 2010) and long-chain acyl–CoA dehydrogenase (LCAD) (Hirschey et al., 2010) in the fatty acid oxidation pathway, 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2) in the ketone body synthesis pathway (Shimazu et al., 2010), carbamoyl phosphate synthetase 1 (Nakagawa et al., 2009) and ornithine transcarbamoylase (Hallows et al., 2011) in the urea cycle, and manganese superoxide dismutase (Qiu et al., 2010; Tao et al., 2010) in the antioxidant system.
Recent large-scale proteomics analyses revealed that every major metabolic pathway contains acetylated proteins, including glycolysis, the tricarboxylic acid (TCA) cycle, the urea cycle, fatty acid metabolism and glycogen metabolism (Choudhary et al., 2009; Kim et al., 2006; Wang et al., 2010; Zhao et al., 2010). Global mitochondrial protein acetylation is regulated by the nutritional status of the cell, and is sensitive to fasting (Kim et al., 2006), calorie restriction (Schwer et al., 2009), and ethanol metabolism (Picklo, 2008). However, the metabolic consequence of mitochondrial protein hyperacetylation is not known. Here, we show decreased SIRT3 expression, reduced SIRT3 activity, or complete ablation of SIRT3 induces mitochondrial protein hyperacetylation and accelerates the development of the metabolic syndrome in mice and possibly in humans.
RESULTS
Chronic high-fat diet feeding induces global mitochondrial protein hyperacetylation and loss of SIRT3
Western blot analysis of hepatic mitochondrial extracts with an anti-acetyllysine antibody revealed that chronic high-fat diet (HFD) feeding (13 weeks) but not acute HFD feeding (1 week) induced global mitochondrial protein acetylation (Fig. 1A). Because SIRT3KO mice have hyperacetylated mitochondrial proteins (Lombard et al., 2007), and HFD feeding leads to mitochondrial protein hyperacetylation, we tested the possibility that the expression of SIRT3, the primary mitochondrial protein deacetylase, might be suppressed in the liver of wild-type (wt) mice fed a HFD.
Fig. 1. Chronic HFD feeding results in global mitochondrial hyperacetylation and reduces hepatic SIRT3.
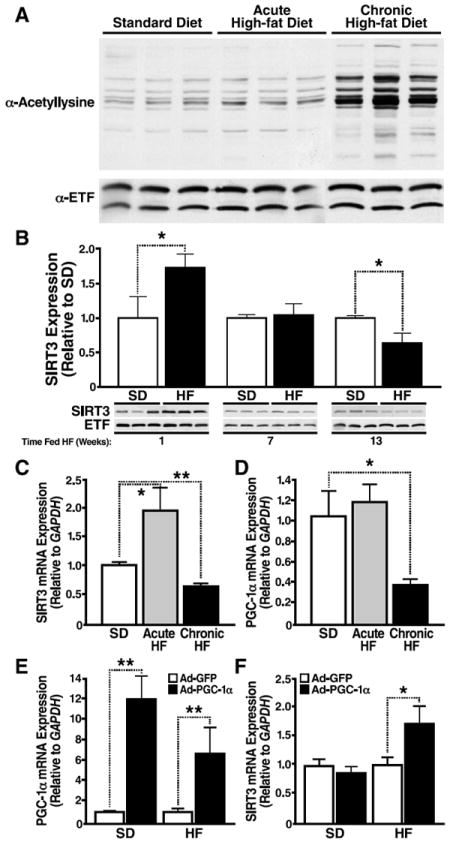
(A) Mitochondria were isolated from livers of wt mice fed a standard or HFD for 1 week or 13 weeks (Jackson Laboratory) and analyzed for mitochondrial protein acetylation by western blot analysis with an antiserum specific anti-acetyllysine; n=3 mice/condition. (B) Mitochondria were isolated from livers of wt mice fed a standard or HFD for 1 week, 5 weeks or 13 weeks (Jackson Laboratory) and analyzed for SIRT3 expression by western blot analysis with an antiserum specific for SIRT3. Integrated density values were calculated for SD and HFD fed wt mice; data represented in arbitrary units (AU) ±SEM, n=3 mice/condition, *p<0.05; (C, D) mRNA transcript levels were quantified by qPCR from wt mice, (from panel B, *p<0.05, n=3/genotype, standard or HFD, ±SEM). (E, F) mRNA transcript levels were quantified by qPCR from wt mice fed a standard (SD) or high-fat (HF) diet overexpressing adenoviral PGC-1α or GFP as a control, (*p<0.05, **p<0.01, n=5/condition, ±SEM).
Hepatic SIRT3 expression was initially increased in response to a one-week HFD feeding in wt mice (Fig. 1B). In contrast, hepatic SIRT3 was suppressed with chronic HFD feeding (13 weeks on a HFD) compared to a standard diet (SD) (Fig. 1B). Suppression of SIRT3 with chronic HFD feeding resulted in elevated global mitochondrial protein acetylation (Fig. 1A).
To determine if reduced Sirt3 transcription underlies the suppression of hepatic SIRT3 in HFD fed mice, we measured SIRT3 mRNA. We found significantly reduced hepatic SIRT3 mRNA in mice chronically fed a HFD (Fig. 1C). Because Sirt3 gene expression is regulated by peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) (Kong et al., 2010), we measured hepatic PGC-1α mRNA in mice chronically fed a HFD and found significantly reduced levels, consistent with the reduction in Sirt3 gene expression (Fig. 1D). To confirm PGC-1α regulates Sirt3 gene expression in HFD fed mice, adenoviral constructs overexpressing PGC-1α, or GFP as a control, were injected into wt mice fed a standard or HFD. After intravenous administration, hepatic tissue homogenates were collected and assessed for PGC-1α and SIRT3 mRNA expression. We observed a 6.8- to 12.1-fold increase in hepatic PGC-1α expression in mice overexpressing adenoviral PGC-1α fed a HF or SD, respectively, compared to mice overexpressing adenoviral GFP (Fig. 1E). Furthermore, we found a 1.7-fold increase in SIRT3 expression in mice overexpressing adenoviral PGC-1α fed a HFD, compared to mice overexpressing adenoviral GFP (Fig. 1F). In contrast, no differences were observed in SIRT3 expression between mice overexpressing PGC-1α or GFP fed a SD (Fig. 1F). These data demonstrate that the reduction in SIRT3 observed in mice fed a HFD is a direct result of the reduction in hepatic PGC-1α, and can be rescued by exogenous PGC-1α overexpression.
HFD feeding induces LCAD hyperacetylation and reduces enzymatic activity
Previous studies show SIRT3 regulates fatty acid oxidation by reversibly deacetylating proteins in the fatty acid oxidation pathway, such as LCAD, and ablation of SIRT3 increases LCAD acetylation and reduces LCAD activity (Hirschey et al., 2010). To test the possibility that HFD-induced loss of SIRT3 could influence LCAD activity, we measured LCAD acetylation and activity in SD and HFD fed mice, both in the presence and absence of SIRT3. As shown previously (Hirschey et al., 2010; Lombard et al., 2007), SIRT3KO mice fed a SD showed a significant increase in global hyperacetylated mitochondrial proteins compared to wt mice (159% increase; Fig. 2A). Wt mice fed a HFD also showed an increase in global hyperacetylation of mitochondrial proteins compared to SD fed mice (51% increase; Fig. 2A). While SIRT3KO mice fed a HFD show increased acetylation compared to wt mice, no further increase in protein acetylation was observed when comparing the global mitochondrial protein acetylation levels between SD fed and HFD fed SIRT3KO mice (Fig. 2A).
Fig. 2. Chronic HFD feeding induces LCAD hyperacetylation and reduces enzymatic activity.
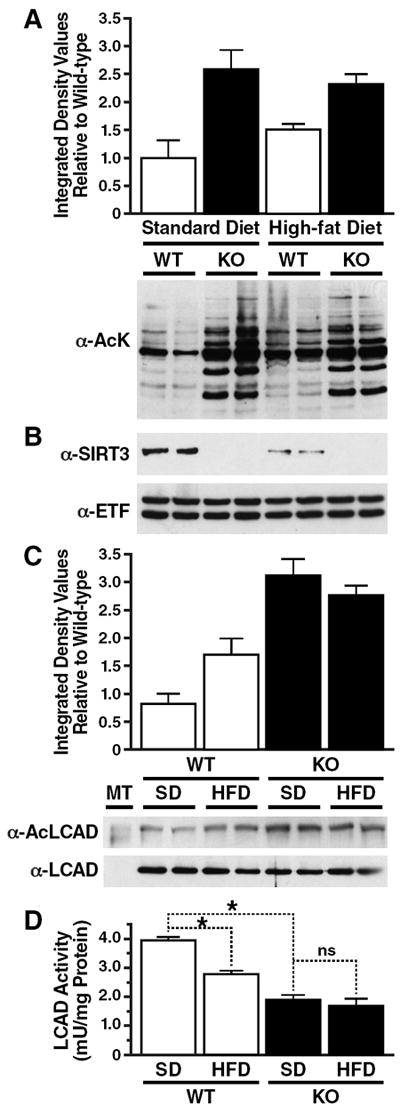
(A) Mitochondria were isolated from livers of 6-month old wt and SIRT3KO mice fed a standard or HFD and analyzed for mitochondrial protein acetylation with an antiserum specific anti-acetyllysine; average integrated densitometry values (IDV) were calculated relative to wt mice fed a standard diet, ±SEM. (B) Mitochondria were isolated from livers of wt and SIRT3KO mice fed a standard or HFD and analyzed for SIRT3 expression by western blot analysis with an antiserum specific for SIRT3. (C) Liver extracts from wt and SIRT3KO mice fed a standard or HFD were immunoprecipitated with an anti-acetyllysine antiserum and analyzed with anti-LCAD antiserum; average integrated densitometry values (IDV) were calculated relative to one wt mouse fed a standard diet and LCAD input was used as a reference, ±SEM. (D) Liver extracts from wt and SIRT3KO mice fed a standard or HFD were assessed for enzymatic activity ex vivo using 2, 6 dimethylheptanoyl-CoA as a substrate; *p<0.05, all samples from same mice, n=3/condition, representative samples shown, ±SEM.
To determine if SIRT3 protein levels, and not diet composition, could regulate mitochondrial protein acetylation, we measured SIRT3 expression in mice fed a SD and HFD fed mice. We observed a significant reduction in SIRT3 protein in wt mice fed a HFD (67% reduction compared to SD; Fig. 2B). The reduction in SIRT3 in 6-month old HFD fed mice (Fig. 2B) was stronger than in 4-month old HFD fed mice (Fig. 1B), and demonstrates SIRT3 continues to be suppressed by HFD feeding over time. Furthermore, we observed a direct correlation between the level of SIRT3 and the levels of mitochondrial protein acetylation. HFD feeding suppresses SIRT3 and induces moderate mitochondrial protein hyperacetylation, whereas ablation of SIRT3 in standard or HFD fed conditions induces severe mitochondrial protein acetylation. These data support the finding that SIRT3 levels are regulated by diet, and directly influence global mitochondrial protein acetylation levels.
Furthermore, we observed low levels of LCAD acetylation in wt mice fed a SD and a 134% increase in LCAD acetylation in SIRT3KO mice fed a SD (Fig. 2C), as reported previously (Hirschey et al., 2010). Wt mice fed a HFD showed a significant increase in LCAD acetylation (55% increase compared to wt mice fed a SD; Fig. 2C). HFD fed SIRT3KO mice also showed an increase in LCAD acetylation compared to HFD fed wt mice (Fig. 2C). However the level of LCAD acetylation was similar in SIRT3KO mice fed a standard or HFD (Fig. 2C).
In the absence of SIRT3, hyperacetylation of LCAD causes reduced enzymatic activity (Hirschey et al., 2010). Thus, we tested the possibility that diet-induced LCAD hyperacetylation might also influence enzyme activity by measuring endogenous LCAD activity levels from wt and SIRT3KO mice fed either SD or HFD. LCAD activity is reduced by 53% in SIRT3KO mice compared to wt mice fed a SD (Fig. 2D), as reported previously (Hirschey et al., 2010). Additionally, wt mice fed a HFD showed a 30% reduction in LCAD activity compared to wt mice fed a SD (Fig. 2D), demonstrating diet-induced LCAD hyperacetylation is sufficient to reduce enzymatic activity. Furthermore, SIRT3KO mice fed a HFD showed a 57% reduction in LCAD activity compared to wt mice fed a SD, but was not significantly reduced compared to SIRT3KO mice fed a SD (Fig. 2D). Thus, we find a direct correlation between SIRT3 expression, global acetylation levels, LCAD acetylation levels, and LCAD activity.
SIRT3KO mice develop diet-induced obesity and insulin resistance
To identify the metabolic consequences of chronic mitochondrial protein hyperacetylation, we placed SIRT3KO mice on a HFD, and measured several metabolic parameters. No early differences in weight were noted between wt and SIRT3KO mice (Fig. 3A). However, SIRT3KO mice developed diet-induced obesity at an accelerated rate when maintained on a HFD (Fig. 3A), but not a SD (Fig. S1). On a HFD, SIRT3KO mice weighed 7% more than wt mice (p=0.075) by 18 weeks, 10% more by 33 weeks and 15% more by 52 weeks (p-values: 0.042 and 0.018, respectively). Dual energy X-ray absorptiometry (DEXA) analyses showed that the increased weight in SIRT3KO mice was due to increased adiposity (Fig. S1).
Fig. 3. SIRT3KO develop diet-induced obesity and insulin resistance.
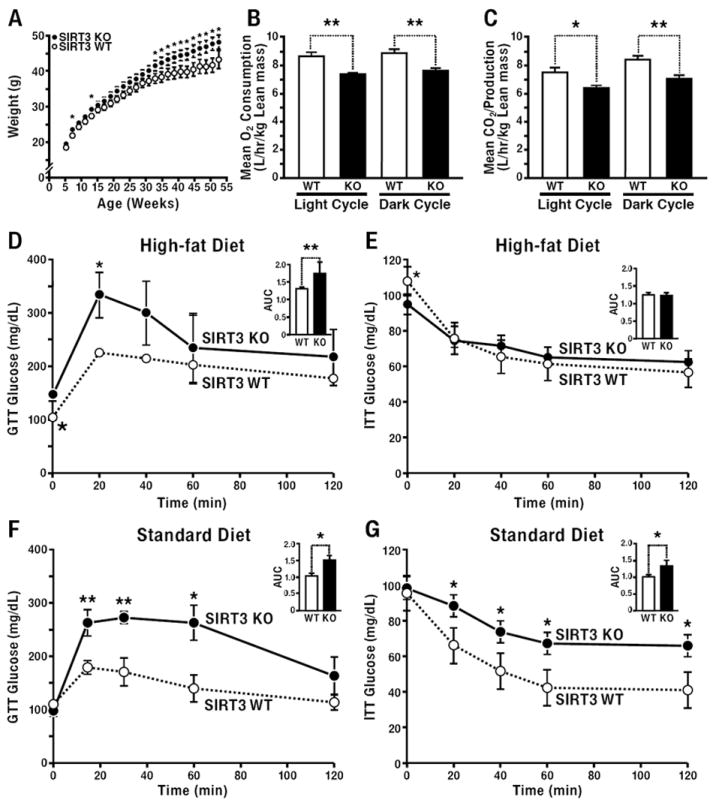
(A) Body weight measurements were recorded from wt and SIRT3KO mice weaned onto and maintained on a HFD (n=20/genotype); (B, C) 3-month old WT and SIRT3KO mice were assessed for the volume of oxygen consumption (VO2) (B) and carbon dioxide exhalation (VCO2) (C), in metabolic cages, n=10/genotype, 3 independent trials, data collected over 48 h and normalized to lean body mass, averages were totaled for dark and light cycles, ±SEM. (D, E) 12-month old SIRT3KO and wt mice fed a HFD were tested for glucose (D) and insulin tolerance (E) and measured for blood glucose levels; inset data represent area under the curve (AUC), ±SEM. (F, G) 12-month old SIRT3KO and wt mice fed a standard diet were tested for glucose (F) and insulin tolerance (G) and measured for blood glucose levels; inset data represent AUC, (n=5/genotype, fasted 6 h, standard diet); *p<0.05, **p<0.01, ±SEM.
To assess the relative contributions of energy intake (food consumption) and energy expenditure (activity, respiration) to increased obesity, metabolic cage analyses were performed in wt and SIRT3KO mice. Oxygen consumption (VO2) was 15% lower in SIRT3KO mice during light (p=0.001) and 14% lower during dark (p=0.003) cycles (Fig. 3B). Additionally, SIRT3KO mice had lower CO2 exhalation (VCO2) during both light and dark cycles (15% lower during the light cycle and 16% lower during the dark cycle, p=0.01 and 0.003, respectively) (Fig. 3C), however no differences in the respiratory exchange ratio (RER) were observed (Fig. S1). Because no significant differences were observed in food intake or spontaneous activity (Fig. S1), we concluded lower energy expenditure in SIRT3KO mice preceded the onset of diet-induced obesity and is the primary driver for the development of increased adiposity and weight gain.
Insulin resistance is a hallmark of obesity and the metabolic syndrome (Biddinger and Kahn, 2006; Kahn et al., 2006; Reaven, 1988). We therefore measured glucose tolerance and insulin sensitivity in wt and SIRT3KO mice. Obese 12-month-old SIRT3KO mice fed a HFD exhibited hyperglycemia during glucose-tolerance testing and were insulin resistant by insulin tolerance testing (Figs. 3D and 3E). To assess the role of SIRT3 on insulin resistance in the absence of obesity, we measured glucose and insulin tolerance in non-obese 12-month old SD fed wt and SIRT3KO mice (Fig. S1). We observed marked hyperglycemia in SIRT3KO mice upon intraperitoneal glucose injection [52% increase in area under the curve (AUC)] and marked insulin resistance upon intraperitoneal insulin injection (34% increase in AUC; Fig. 3F and 3G, respectively).
To measure the progression of the observed insulin resistance in 12-month old SD and HFD fed SIRT3KO mice, we measured glucose and insulin sensitivity in 3-month old SD and HFD fed wt and SIRT3KO mice. No differences were observed in glucose or insulin tolerance in 3-month old wt and SIRT3KO mice fed a SD (Fig. S2). However, glucose and insulin tolerance tests in 3-month old wt and SIRT3KO mice maintained on a HFD revealed key differences. Namely, SIRT3KO mice displayed equal ability to regulate glucose levels as wt mice in response to intraperitoneal glucose injection, but generated higher insulin levels to maintain glucose homeostasis (Fig. S2). HFD fed SIRT3KO mice were also insulin resistant upon insulin tolerance challenge by intraperitoneal insulin injection (Fig. S2). Together, these data demonstrate lack of SIRT3 and mitochondrial protein hyperacetylation leads to disrupted insulin signaling and insulin resistance with age. Furthermore, HFD feeding accelerates the development of insulin resistance and glucose intolerance in SIRT3KO mice at 3 months of age in the absence of obesity, which is significantly exacerbated in obese 12-month old mice.
Hepatic steatosis and non-alcoholic steatohepatitis in SIRT3KO mice
Abnormal hepatic lipid accumulation has been proposed as possible mechanism for the development of insulin resistance in the metabolic syndrome (Reaven, 1988). We therefore measured lipids in wt and SIRT3KO mice fed a HFD. Staining of liver sections for total lipids with Oil Red O showed high lipid levels in wt mice fed a HFD but even higher levels in SIRT3KO mice fed the same diet (Figs. 4A, 4B and S3). Direct measurement of lipids in liver tissue homogenates showed that 3-month old SIRT3KO mice had 38% more hepatic triglycerides than wt mice, and 41% more hepatic cholesterol esters (Fig. 4B). Metabolomic analyses of 3-month old wt and SIRT3KO mice fed a HFD revealed increased accumulation of hepatic long-chain acylcarnitine species in SIRT3KO mice, but not organic acids or amino acids (Fig. S4). These results are consistent with the previously demonstrated reduced fatty acid oxidation in SIRT3KO mouse livers (Hirschey et al., 2010).
Fig. 4. SIRT3KO mice fed a HFD develop hepatic steatosis and inflammation.
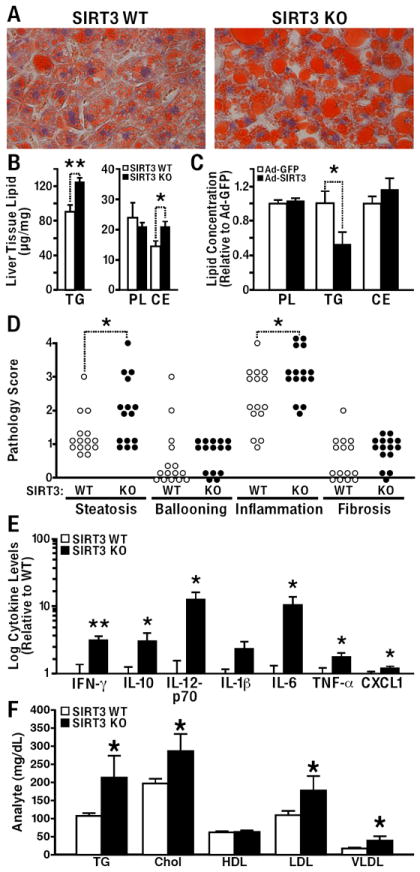
(A) Histological analysis of livers from HFD fed wt and SIRT3KO mice with Oil Red O Stain (fed or fasted 24 h). (B) Livers extracts from wt and SIRT3KO mice fed a HFD were analyzed for total phospholipids, triglycerides and cholesterol esters (n=5/genotype, fasted 24 h, ±SEM). (C) Hepatic lipids were measured in wt mice 1 week after injection with adenovirus expression vectors for GFP or SIRT3. (D) Hepatic sections from 12-month-old wt and SIRT3KO mice fed a HFD were fixed, stained with hematoxylin & eosin (H&E), Masson’s trichrome, or reticulin, and scored for inflammation, steatosis, ballooning, and fibrosis, (n=15/genotype, Wilcoxon rank-sum test, *p<0.05). (E) Cytokine analyses were conducted on serum obtained from 12-month-old wt and SIRT3KO mice maintained on a HFD (n=5/genotype, ±SEM). (F) Serum triglyceride (TG), cholesterol (Chol), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and very-low-density lipoprotein (VLDL) were measured in 12-month-old wt and SIRT3KO mice fed a HFD ad libitum (fasted 24 h, n=5 mice/genotype, ±SEM).
Because SIRT3 deficiency reduces fatty acid oxidation and results in accumulation of hepatic lipids, we sought to determine if SIRT3 overexpression is protective against hepatic lipid accumulation. Recombinant adenoviruses containing the cDNA encoding Sirt3, or green fluorescent protein (GFP) as a control, were injected into the tail veins of wt mice, and hepatic tissue homogenates were assessed for total lipid levels. Hepatic triglyceride levels were 50% lower in wt mice injected with SIRT3-expressing adenovirus than in mice injected with the GFP-expressing virus (Fig. 4C). These data demonstrate that SIRT3 overexpression reduces hepatic lipids and suggest a possible therapeutic role for enhanced SIRT3 expression in the management of the metabolic syndrome.
Histological analysis of liver sections from 12-month old mice fed a HFD showed higher lipid levels in SIRT3KO mice than in wt mice with features consistent with steatohepatitis. Aged SIRT3KO mice had more macrovesicular steatosis, which was evaluated based on the percentage of fat in a histologic section of the liver parenchyma. Strikingly, 12-month-old SIRT3KO mice also had more lobular lymphoplasmacytic inflammation (Figs. 4D and S3), as determined by histological scoring of the average number of lymphoplasmacytic aggregates in five 100X fields. Additionally, a trend toward more hepatocyte ballooning degeneration and hepatic fibrosis was observed in the SIRT3KO mice compared to wt mice (Figs. 4D and S3).
Because we observed signs of hepatic inflammation, which has been linked to obesity and metabolic dysfunction (Hotamisligil et al., 1993), we measured serum inflammatory cytokines from wt and SIRT3KO mice fed a HFD. We found aged SIRT3KO mice had markedly higher levels of inflammatory cytokines than wt mice (Fig. 4E). In particular, interferon-γ (3-fold), IL-10 (3-fold), IL-12p70 (12-fold), IL-6 (10-fold), TNF-α (1.7-fold), and CXCL1 (1.2-fold) were all significantly higher in obese 12-month-old SIRT3KO mice fed a HFD compared to wt mice, but were unchanged in non-obese 3-month-old SIRT3KO mice (Fig. 4E). These data show that lack of SIRT3 and the accompanying mitochondrial protein hyperacetylation is linked to increased cytokines and the development of steatohepatitis in aged, obese mice.
SIRT3KO mice develop hyperlipidemias with HFD feeding
Obesity, insulin resistance, and the metabolic syndrome often coincide with lipid abnormalities, including hypertriglyceridemia, hypercholesterolemia, and other dyslipidemias (Reaven, 1988). Serum lipid measurements from 12-month-old SIRT3KO mice fed a HFD revealed higher levels of triglyceride (97% increase) and cholesterol (141% increase) than in wt mice (Fig. 4F). We also found higher levels of low-density lipoproteins (LDL, 60% increase) and very-low-density lipoproteins (VLDL, 100% increase) in SIRT3KO mice (Fig. 4F). No differences in HDL levels were detected (Fig. 4F). Consistent with the insulin resistance data discussed above, fasting insulin levels were increased in 12 month-old SIRT3KO mice (285% increase), but levels of leptin, adiponectin, and resistin were unchanged (Table S1).
Increased hepatic saturated lipids induce SCD1 expression in SIRT3KO mice
To characterize possible changes in nuclear gene expression that occur as a consequence of SIRT3 deficiency, microarray analyses were performed on livers of wt and SIRT3KO mice. Because HFD feeding is associated with large changes in gene transcription (Kim et al., 2004), and to identify gene expression changes unique to SIRT3 deficiency, we compared gene expression in wt and SIRT3KO mice fed a SD. Of 28,800 genes tested, 18 were significantly upregulated (increased greater than 1.2-fold, p<0.001), and nine were significantly downregulated (decreased greater than 1.2-fold, p<0.001) in liver from SIRT3KO mice (Fig. S5). Interestingly, the most highly induced gene (1.7-fold increase) was stearoyl-CoA desaturase 1 (SCD1) (Fig. S5). SCD1 is a fatty acid synthesis enzyme that catalyzes the biosynthesis of monounsaturated long-chain acyl CoAs from saturated long-chain acyl CoAs (Miyazaki et al., 2001). Mice with hyperlipidemias or humans with obesity and type II diabetes have increased SCD1 expression (Attie et al., 2002; Hulver et al., 2005). Increased SCD1 mRNA abundance was independently validated using quantitative RT-PCR and a five-fold increase in mRNA was detected in SIRT3KO mice compared to wt mice (Fig. 5A). Surprisingly, SCD1 was the only mRNA for lipogenic genes whose expression was increased (Fig. 5A and S5).
Fig. 5. SIRT3KO mice have high expression and activity of hepatic SCD1.
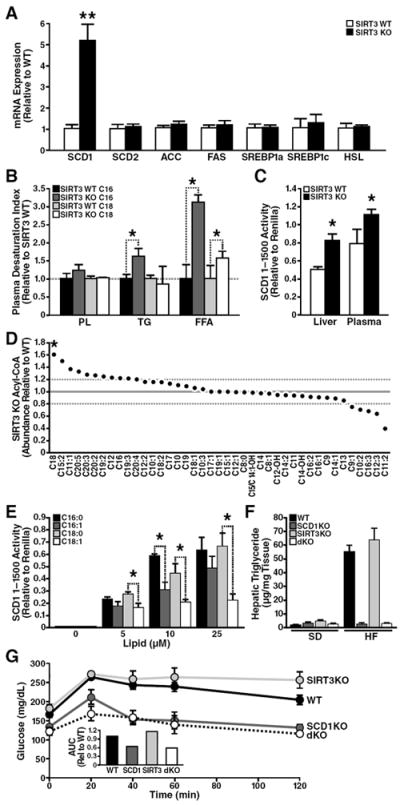
(A) mRNA transcript levels were quantified by qPCR from wt and SIRT3KO mice, (*p<0.05, n=3/genotype, 3-month old mice, standard diet, ±SEM). (B) Plasma samples from 3-month old SIRT3KO and wt mice were analyzed for desaturation indices in triglyceride (TG) phospholipids (PL) and free fatty acids (FFA) by measuring palmitate (C16), palmitoleate (C16:1), stearate (C18) and oleate (C18:1) (*p<0.05, n=5/genotype, 3-month-old mice, standard diet, ±SEM). (C) Adenoviral SCD1 promoter activity in Huh7 cells after treatment with lipids extracted from plasma or liver tissue in wt or SIRT3KO (*p<0.05, n=3/genotype, 12-week-old mice, standard diet). (D) Hepatic extracts from SIRT3KO and wt mice were analyzed for acyl-CoA lipid species (*p<0.05, n=5/genotype, 12-week-old mice, standard diet). (E) Adenoviral SCD1 promoter activity in Huh7 cells after treatment with palmitate, palmitoleate, stearate, oleate (n=3/condition, ±SEM, *p<0.05) (F) Hepatic triglycerides from wt, SCD1KO, SIRT3KO, and SCD1KO/SIRT3KO (dKO) mice fed a standard (SD) or high-fat (HF) diet were measured (n=5/genotype, fed or fasted 24 h, ±SEM). (G) 2-month old wt, SIRT3KO, SCD1KO, and dKO mice fed a HFD were tested for glucose tolerance and measured for blood glucose levels; inset data represent area under the curve (AUC), ±SEM.
To determine if increased SCD1 expression correlated with increased SCD1 activity, we measured the plasma desaturation index. This index represents the ratio of serum palmitoleate:palmitate (16:1/16:0) or oleate:stearate (18:1/18:0) and is a well-documented marker for SCD1 activity (Attie et al., 2002). In the plasma of SIRT3KO mice fed a SD, the free fatty acid desaturation indexes for C16:1/C16:0 and C18:1/C18:0 were increased (213% and 62%, respectively) (Fig. 5B). Triglyceride desaturation indices were also increased (66% for triglyceride C16:1/C16:0) (Fig. 5B).
Scd1 gene transcription is sensitive to hormones (e.g., insulin) and fatty acids, (e.g., polyunsaturated fatty acids) (Ntambi et al., 2004) and regulated by multiple transcription factors [e.g., sterol regulatory element-binding protein 1c (SREBP-1c)], as part of a coordinated fatty acid synthetic effort (Shimano et al., 1999). However, because no differences were observed in the levels of SREBP-1c or other lipogenic enzymes in SIRT3KO mice (Fig. 5A and S5), we sought to determine the molecular mechanism underlying the specific increase in SCD1.
Multiple lipids are implicated in regulating Scd1 gene transcription [for a review, see (Paton and Ntambi, 2009)], and palmitoleate (C16:1) suppresses Scd1 gene transcription and enhances insulin sensitivity in mice (Cao et al., 2008). Accordingly, we tested the possibility that accumulated lipids in SIRT3KO mice increased Scd1 gene transcription. The Scd1 promoter region (1–1500) was cloned in front of a luciferase gene and packaged into an adenoviral reporter (Cao et al., 2008). Huh7 hepatoma cells were infected with this adenovirus and incubated with murine lipid extracts, and Scd1 promoter activity was measured. As a control, cells were co-infected with another adenovirus encoding renilla luciferase under control of the CMV promoter. We found higher levels of Scd1 promoter activity upon treatment with hepatic lipids extracted from SIRT3KO mouse (64% higher than from wt mice) (Fig. 5C). The activity of the Scd1 reporter was also sensitive to lipids extracted from SIRT3KO mouse plasma (40% higher than from wt mice) (Fig. 5C).
To identify lipid species in SIRT3KO mice responsible for increased SCD1 transcriptional activity, we measured levels of hepatic lipids from SIRT3KO mice. Lipids are activated by attaching Coenzyme A to an acyl chain before storage. Long-chain acyl CoAs acts as signal transduction intermediate and directly regulates gene expression via transcription factor binding (Black et al., 2000). Thus, we measured the abundance of acyl-CoAs in wt and SIRT3KO mouse livers. Of 47 hepatic acyl CoAs measured ranging from acetyl-CoA (C2) to eicospentanoyl-CoA (EPA, C20:05), the acyl-CoA moiety in the greatest abundance (62% increase for SIRT3KO) was stearoyl-CoA (Fig. 5D). The abundance of saturated stearoyl-CoA suggested that saturated lipids might regulate Scd1 promoter activity.
We then measured Scd1 promoter activity in response to long-chain saturated (palmitate, stearate) fatty acids with the adenoviral luciferase reporter described above. We infected Huh7 hepatoma cells with an adenovirus reporter encoding the Scd1 promoter and measured its sensitivity to saturated fatty acids. Palmitate and stearate increased Scd1 promoter activity in a dose-dependent manner (Fig. 5E). Thus, these data show accumulation of saturated lipids directly increased Scd1 gene transcription, and suggest high hepatic stearate levels in SIRT3KO mice cause increased Scd1 gene expression and contribute to the pathogenesis of the metabolic syndrome in SIRT3KO mice.
Because Scd1 is elevated in mice and in humans with elevated lipids (Attie et al., 2002; Hulver et al., 2005) and mice lacking Scd1 (SCD1KO) have reduced hepatic lipids (Cohen et al., 2002), we sought to identify the physiological significance of Scd1 up-regulation in SIRT3KO mice and generated mice lacking both Sirt3 and Scd1 (dKO). Under SD fed conditions, no differences in hepatic lipids were observed between wt, SIRT3KO, SCD1KO or dKO mice (Fig. 5F). However, HFD-induced hepatic steatosis was observed in wt mice and was exacerbated in SIRT3KO mice as described above (Fig. 5F). Hepatic steatosis was absent in SCD1KO mice fed a HFD, as previously reported (Ntambi et al., 2002). Furthermore, dKO mice showed markedly reduced hepatic triglycerides with HFD feeding (Fig. 5F) and improved insulin sensitivity (Fig. 5G), consistent with the results that loss of the lipogenic gene Scd1 protects from HFD-induced metabolic dysfunction.
Genetic association of a single nucleotide polymorphism (SNP) in SIRT3 with the metabolic syndrome in humans
The metabolic syndrome is a complex disease with several contributing etiologies, and while no single gene can explain the full collection of metabolic disorders in humans, several genes have been shown to influence susceptibility to the metabolic syndrome (Lusis et al., 2008). Because SIRT3KO mice develop several metabolic disorders at an accelerated rate compared to wt mice fed a HFD, we tested if variability in the SIRT3 gene was correlated to increased susceptibility for developing metabolic dysfunction in humans. We took a gene candidate approach and interrogated the SIRT3 gene in 834 DNA samples from a cross-sectional cohort of patients with nonalcoholic fatty liver disease, and initially tested if a SNP in SIRT3 increased a patient’s susceptibility for accelerated progression from fatty liver disease to more complex metabolic diseases including the metabolic syndrome [obtained from the Non-alcoholic Steatohepatitis Clinical Research Network (NASH CRN), Table S2]. Thirteen SNPs spanning SIRT3 were measured, nine of which were found in the patient cohort and passed all quality control criteria (Table S3A). Two SNPs in near-perfect linkage disequilibrium [rs7934919 and rs11246020; D’: 1.0, r2 = 0.98, (Fig. 6A)] were associated with the metabolic syndrome (MetSyn). The strength of these associations was enhanced following exclusion of individuals with type 2 diabetes.
Fig. 6. A functional SNP in the human SIRT3 gene is associated with human metabolic syndrome and encodes a point-mutation.
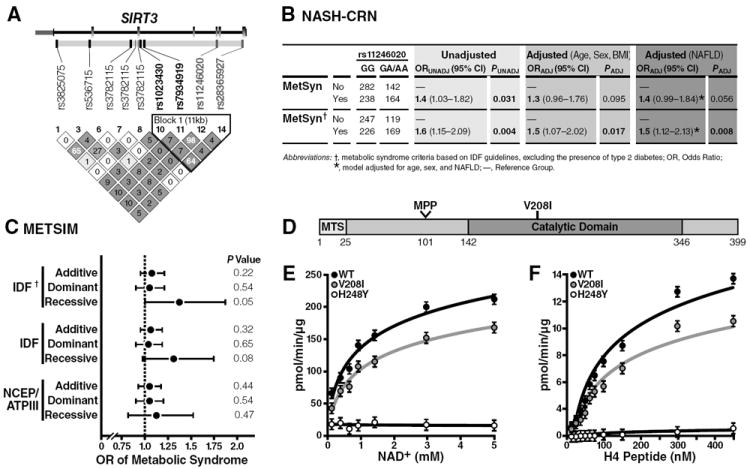
(A) Heat map detailing the pairwise LD among the 7 SNPs spanning the SIRT3 coding region; the pairwise correlations (D’) are rendered within each diamond with greater LD reflected by darker shades of gray; SIRT3 gene structure is depicted above the LD heat map: exons are depicted in gray boxes, introns as connecting black lines, and untranslated regions as smaller boxes shades in pink; approximately 10 kbp of flanking DNA sequence was included in the tagSNP selection procedure; tagSNPs the promoter region is included in the diagram to the right of the transcription start site indicated by the arrow found directly above the SIRT3 gene structure diagram; SNPs rendered in bold are included in the haploblock outlined in the black triangle; the two SNPs that encode for nonsynonymous polymorphisms (rs11246020 and rs28365927; green). (B) Association of SIRT3 rs11246020 with the metabolic syndrome in the NASH-CRN cohort. Abbreviations: P, p-value for the test statistic; OR (95% CI), the odds ratio and 95% confidence interval for the test statistic; VNTR, variable number tandem repeat; UNADJ, unadjusted; ADJ, adjusted for age, sex, and BMI unless otherwise indicated. (C) Association of SIRT3 rs11246020 with the metabolic syndrome in the METSIM cohort, adjusted for age and BMI. †Metabolic syndrome criteria based on the IDF guidelines, excluding the presence of type 2 diabetes. *p-value for the overall model. (D) Schematic of SIRT3 protein; mitochondrial targeting sequence (MTS), mitochondrial processing peptidase (MPP) site. (E, F) Steady-state kinetic analyses of SIRT3 activity; rates of activity were measured as a function of [NAD+] (E) or [3H-histone H4 peptide] (F), as measured by organic-soluble radioactive signal, n=3 independent measurements/sample, ±SEM.
Next, haplotype analysis of the haplotype block was performed to refine the association signal and to determine if either SNP (rs7934919 and rs11246020) was a surrogate of the other. The four-SNP haplotypes did not improve the association signal (Table S3B). The rs7934919 SNP appears to be a surrogate of rs11246020 given the associations detected were strongest for rs11246020, and adjusting for rs7934919 in multivariable analyses completely attenuated the association signal. Regression analysis of the inferred diplotypes for the 4-SNP haplotype provided additional evidence that rs11246020 underlies the association signal between SIRT3 gene variation and the metabolic syndrome (p=0.007), based on the observation that the association signal did not improve substantially in comparison with analysis of rs11246020 alone (Table S3B, shaded row). Assuming a recessive mode of inheritance, the rs11246020 “A” minor allele was associated with increased odds for meeting the criteria for the metabolic syndrome (odds ratio [OR]: 1.5, 95% Confidence Interval [CI]: 1.07—2.02, p=0.017), after adjustment for age, sex, and BMI (Fig. 6B, middle column). Additionally, the precision of the point estimate improved when NAFLD was controlled for in the model examining the relationship between rs11246020 and metabolic syndrome (Fig. 6B, right column, OR: 1.5, 95% CI: 1.12—2.13, p=0.008).
To further investigate the correlation between rs11246020 and the metabolic syndrome in humans, we evaluated rs11246020 in the Metabolic Syndrome in Men (METSIM) cohort containing approximately 8000 Finnish men (Stancáková et al., 2009). We estimated the association between the rs11246020 minor allele and the metabolic syndrome using additive, dominant, and recessive genetic models, and found the “A” minor allele again showed a positive association with increased odds for meeting the criteria for diagnosis of the metabolic syndrome (OR: 1.30, 95% CI: 1.00—1.71, p=0.053), after adjustment for type 2 diabetes (Fig. 6C). Assumption of either the additive or dominant modes of inheritance impacted the association signals (Fig. 6C and Table S4).
Different health agencies use different criteria for the definition of the metabolic syndrome (Reaven, 2006), such as the International Diabetes Federation (IDF), the World Health Organization (WHO), the European Group for the Study of Insulin Resistance (EGIR), the US National Cholesterol Education Program Adult Treatment Panel III (NCEP/ATPIII), and the American Heart Association/National Heart, Lung, and Blood Institute (AHA/NHLBI). These definitions contain different qualifying criteria and apply different clinically-defined thresholds for several criteria (i.e., fasting glucose, blood pressure). Thus, we sought to measure the impact of these varying definitions on the association between rs11246020 in SIRT3 and the metabolic syndrome used by the IDF with diabetics excluded, IDF with diabetics included, or the NCEP/ATPIII definition. We found different definitions impacted both the odds ratios and the precision of the estimates (i.e., 95% CI) (Fig. 6C and Table S4). Namely, the IDF definition of the metabolic syndrome which includes individuals with diabetes was associated with rs11246020, which strengthened when individuals with diabetes were excluded. However, when using the NCEP/ATPIII definition, the association was attenuated.
Functional SNP in SIRT3 causes a point mutation and reduces enzymatic activity
The non-synonymous point mutation encoded by rs11246020 results in a change of valine to isoleucine at residue 208 of the SIRT3 polypeptide. The V208I polymorphism lies within the conserved catalytic deacetylase domain of SIRT3 [Fig. 6D and (Frye, 2000)] and could therefore affect its enzymatic activity. To test this possibility, we expressed recombinant wt SIRT3, SIRT3-V208I and catalytically-inactive SIRT3-H248Y in E. coli and tested their deacetylase activity in vitro. A steady-state kinetic analysis of SIRT3 activity was performed, and the initial rates of radioactive release were measured as a function of NAD+ concentration. The resulting saturation curves were fitted to the Michaelis-Menten equation and the Vmax and KM kinetic parameters were compared among wt SIRT3, SIRT3-V208I and catalytically-inactive SIRT3-H248Y. We observed an 18% increase in the KM for NAD+ in SIRT3-V208I, compared to wt SIRT3 (Figs. 6E and S6), indicating more NAD+ is required in SIRT3-V208I deacetylation reaction. Coincident with the increase in KM, we observed a 19% reduction Vmax for NAD+ by SIRT3-V208I, compared to wt SIRT3 (Figs. 6E and S6). Additionally, we observed a 28% reduction in SIRT3-V208I Vmax for the peptide substrate, compared to wt SIRT3, but no change in the KM for the peptide substrate (Figs. 6F and S6). These results indicate that the SIRT3-V208I mutation reduces the catalytic efficiency by 34% compared to wt SIRT3, in three independent enzyme preparations (Fig. S6). These observations demonstrate the mutant SIRT3-V208I has reduced enzyme efficiency and could partially explain how human patients with rs11246020 have increased susceptibility to developing the metabolic syndrome.
DISCUSSION
In this study, we identify SIRT3 and mitochondrial protein acetylation as crucial regulators of metabolic homeostasis and demonstrate that deficiency of SIRT3 leads to accelerated development of the diseases of the metabolic syndrome. While some genes influence several traits associated with the metabolic syndrome, thus far no single gene influences the entire spectrum (Lusis et al., 2008), and mouse models encompassing a single or several traits of the metabolic syndrome continue to provide a better understanding of the mechanisms underlying the metabolic syndrome in humans. Our observations support the model that SIRT3 deficiency and the associated mitochondrial protein hyperacetylation results in mitochondrial dysfunction, and we identify three distinct conditions associated with decreased SIRT3 expression or activity that all lead to metabolic dysfunction.
First, wt mice fed a HFD develop obesity, hyperlipidemia, type 2 diabetes mellitus, insulin resistance, and non-alcoholic steatohepatitis (Collins et al., 2004; Petro et al., 2004; Rossmeisl et al., 2003; Surwit et al., 1995), and these deleterious effects of HFD feeding are further exacerbated in mice with a genetic deletion of Sirt3. We previously reported that SIRT3 deacetylates mitochondrial proteins and that ablation of SIRT3 is associated with LCAD hyperacetylation, reduced LCAD enzymatic activity, and decreased fatty acid oxidation (Hirschey et al., 2010). Interestingly, LCAD deficiency is also associated with accelerated development of a insulin resistance in mice (Zhang et al., 2007). Ablation of LCAD in mice results in hepatic steatosis and hepatic insulin resistance, primarily attributed to lipid accumulation from reduced fatty acid oxidation (Kurtz et al., 1998). Additionally, ablation of malonyl-CoA decarboxylase (MCD), an enzyme that regulates mitochondrial fatty acid oxidation, leads to reduced fatty acid oxidation and insulin resistance (Koves et al., 2008). We speculate that increased SCD1 expression also contributes to the metabolic syndrome phenotype in SIRT3KO mice. Lipids are implicated in regulating Scd1 gene transcription [for a review, see (Paton and Ntambi, 2009)], and elevated hepatic lipids in SIRT3KO mice lead to increased transcriptional signals that further activate Scd1 expression. Notably, increased Scd1 expression in SIRT3KO mice precedes the development of the metabolic syndrome phenotype, and ablation of Scd1 in SIRT3KO mice rescues the hepatic steatosis phenotype induced by HFD feeding. Thus, primary lesions in fatty acid oxidation upon ablation of SIRT3, LCAD, or MCD all result in elevated lipids and insulin resistance, and strongly support a role for mitochondrial lipid oxidation in the maintenance of insulin signaling and metabolic homeostasis.
Second, prolonged exposure to HFD feeding in wt mice results in a reduction of hepatic SIRT3 expression and increased mitochondrial protein acetylation, as reported previously (Bao et al., 2010). Furthermore, HFD feeding results in LCAD hyperacetylation and reduces LCAD activity, demonstrating that HFD feeding partially mimics the phenotype of the SIRT3KO mice. The HFD-induced suppression of Sirt3 occurs at the transcriptional level, and is primarily driven by the HFD-induced suppression of PGC-1α (Crunkhorn et al., 2007; Li et al., 2007), which regulates the expression of SIRT3 (Kong et al., 2010). Overexpression of exogenous PGC-1α was sufficient to rescue the loss of SIRT3 in HFD fed mice. However, overexpression of exogenous PGC-1α did not result in overexpression of SIRT3 in SD fed mice, which displayed basal levels of SIRT3. These data show that SIRT3 levels are tightly regulated at the transcriptional level under SD and HFD feeding. Because fatty acid oxidation is also suppressed by HFD feeding (Ji and Friedman, 2008; Ji and Friedman, 2007), we hypothesize that reduced SIRT3 and increased mitochondrial protein acetylation could be a new mechanism to reduce fatty acid oxidation in a HFD fed state.
Third, we find a correlation between a genetic polymorphism in SIRT3 in humans and the development of the metabolic syndrome. Although SNPs in SIRT3 have not been identified in large-scale genome wide association studies in obesity (Heid et al., 2010; Lindgren et al., 2009; Speliotes et al., 2010), diabetes (Dupuis et al., 2010; Prokopenko et al., 2009), or cholesterol and lipid metabolism (Musunuru et al., 2010; Teslovich et al., 2010), we tested the possibility that SNPs in SIRT3 were associated with metabolic dysfunction in a Caucasian cohort diagnosed with fatty liver disease (The NASH CRN). This cohort was chosen because SIRT3KO mice have clear signs of fatty liver disease, and we hypothesized these patients might be enriched for one of 12 SNPs in SIRT3. Our unbiased approach identified a single SNP that correlated with increased risk of the metabolic syndrome from over 30 clinical parameters tested.
In a subsequent study focusing specifically on the rs11246020 “A” minor allele, we sought to validate the SNP association in a population of approximately 8000 Finnish men (Stancáková et al., 2009). We observed an additional correlation between the frequency of this allele and meeting the criteria for diagnosis of the metabolic syndrome. While these data are suggestive and support the findings in the NASH-CRN study, these data are not definitive and further human genetic studies will fully elucidate the association between rs11246020 and the metabolic syndrome. Although, the complexity of different experimental designs and measures introduce heterogeneity in the results that are more difficult to interpret together, the both studies provide evidence of an association between a single SNP in SIRT3 and the metabolic syndrome.
To further define this relationship, we examined the functional impact of the non-synonymous point mutation (V208I) in the SIRT3 protein. Indeed, the V208 lies within the conserved sirtuin catalytic deacetylase domain, and mutation from valine to isoleucine reduces SIRT3 enzyme efficiency, by both increasing the KM for NAD+ and reducing the Vmax. These data are consistent with the model that reduction in SIRT3 enzymatic activity associated with the V208I mutation plays a pathogenic role in humans, as in mice, and increases susceptibility to the metabolic syndrome. Taken together, our observations highlight the importance of using primary cellular and mouse data to direct human genetic studies and the power of integrating these data to glean insights into the relationships between human SNPs and the underlying biology.
We propose that reduction or loss of SIRT3 and the resulting mitochondrial protein hyperacetylation lead to global mitochondrial dysfunction (Fig. 7). Since, every major metabolic pathway in human liver contains acetylated proteins (Zhao et al., 2010), the function of other critical mitochondrial proteins is likely to be dysregulated in the absence of SIRT3. A number of abnormalities in mitochondria have been identified in prior studies as key pathogenic mechanisms in the development of the metabolic syndrome, including reduced mass (Kelley et al., 2002), altered morphology (Civitarese et al., 2010), reduced fatty acid oxidation (Zhang et al., 2007), lower oxidative phosphorylation (Befroy et al., 2007; Petersen et al., 2005), and increased reactive oxygen species (Civitarese et al., 2006; Patti et al., 2003; Petersen et al., 2004; Ukropcova et al., 2007). SIRT3KO show an overlapping group of mitochondrial abnormalities including reduced fatty acid oxidation (Hallows et al., 2011; Hirschey et al., 2010), lower oxidative phosphorylation (Ahn et al., 2008), and increased reactive oxygen species (Kim et al., 2010; Qiu et al., 2010; Someya et al., 2010; Tao et al., 2010). Thus, these data support the hypothesis that a primary mitochondrial lesion results in global metabolic dysfunction, and can progress to metabolic disease.
Fig. 7. Working model.
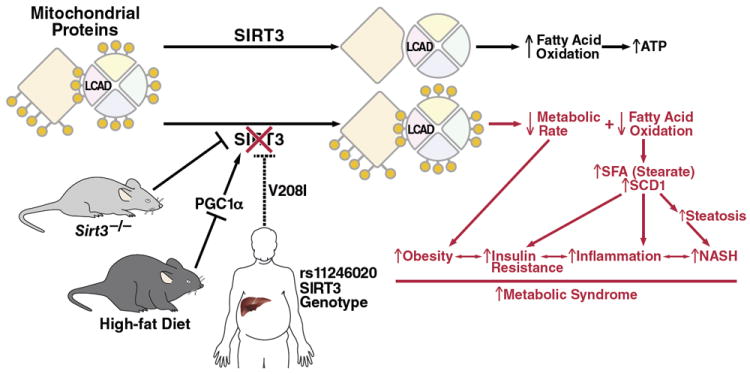
SIRT3 functions to deacetylate mitochondrial proteins, and increase fatty acid oxidation and energy production. In SIRT3KO mice or mice fed a HFD, mitochondrial proteins are hyperacetylated, resulting in reduced energy expenditure and less fatty acid oxidation, which contributes to insulin resistance, obesity, and increased inflammation. Similarly, humans with a unique SNP in the SIRT3 gene have reduced SIRT3 enzymatic efficiency and increased risk to develop the metabolic syndrome.
We conclude that mitochondrial protein acetylation is a critical post-translational modification, whose regulation by SIRT3 is necessary to maintain metabolic health in mice and possibly in humans. Future studies will examine the therapeutic potential of manipulating SIRT3 expression or activity in the liver or other tissues to ameliorate manifestations of the metabolic syndrome.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies used were specific for monoclonal and polyclonal acetyllysine (Cell Signaling Technology, Danvers, MA), SIRT3 [as described (Lombard et al., 2007)], ETF and LCAD (Jerry Vockley, University of Pittsburgh, PA).
Animal Studies
All animal studies were performed using IACUC-approved protocols. Studies used male wt and SIRT3KO 129Sv mice (Lombard et al., 2007), at either 3-months or 12-months of age, maintained on a standard chow diet (5053 PicoLab diet, Ralston Purina Company, St. Louis, MO) or a high-fat ‘Western diet’ (TD.88137; Harlan Teklad, Indianapolis, IN), unless otherwise indicated. Mice were sacrificed at 7:00 h for fed mouse studies, or transferred to a new cage without food for 24 h from 7:00 h to 7:00 h, and then sacrificed for fasted mouse studies. For SCD1 mouse studies, wt (C57Bl/6), SIRT3KO (C57Bl/6-Sirt3-/-), SCD1KO (B6.129-Scd1tm1Ntam/J, The Jackson Laboratory, Bar Harbor, ME), or dKO (C57Bl/6-Scd1-/-Sirt3-/-), mice were used. Mice were sacrificed at 7:00 h for fed mouse studies, or transferred to a new cage without food for 24 h from 7:00 h to 7:00 h, and then sacrificed for fasted mouse studies.
DEXA and Metabolic Measurements
For metabolic measurements, body weight and composition were measured in wt and SIRT3KO mice at 3-4 months or 12 months of age by dual-energy X-ray absorptiometry (DEXA) scanning. The Comprehensive Lab Animal Monitoring System (CLAMS) method was used to measure activity level, food intake, volume of O2 consumption, volume of CO2 production over a 48 h period (Oxymax OPTO-M3 system, Columbus Instruments, Colombus, OH). Mean VO2 and VCO2 were calculated for dark and light cycles and normalized to lean body mass. Mean activity and respiratory exchange ratio were calculated for dark and light cycles. Glucose and insulin tolerance tests were performed according to The Jackson Laboratory protocol.
Cell Biology and Immunoprecipitation
Wt and recombinant human SIRT3 was cloned into pTrcHis2 expression vectors, generated by standard PCR-based cloning strategies and verified by DNA sequencing. For immunoprecipitation experiments, murine liver mitochondria were prepared and purified as described (Graham, 2001a, b; Hirschey et al., 2009). Briefly, mitochondria were lysed by sonication and resuspended in a low-stringency IP buffer [0.05% NP-40, 50 mM NaCl, 0.5 mM EDTA, 50 mM Tris-HCl, pH 7.4, 10 mM nicotinamide, 1 μM trichostatin A, protease inhibitor cocktail (Roche)].
Statistical Analyses
Results are given as the mean ± standard error. Statistical analyses represent a one-tailed Students t-test or a Wilcoxon rank-sum test and null hypotheses were rejected at 0.05.
Supplementary Material
Acknowledgments
We thank C. Miller and J.D. Fish for histology preparation, C. Harris, L. Swift, and the MMPC (DK59637) for plasma and tissue lipid analysis; T. Tran for metabolic experiments; A. Wilson, G. Maki and J. Carroll for Fig. preparation; G. Howard and S. Ordway for editorial review. The Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grants U01DK061718, U01DK061728, U01DK061731, U01DK061732, U01DK061734, U01DK061737, U01DK061738, U01DK061730, U01DK061713), and the National Institute of Child Health and Human Development (NICHD). Several NASH CRN clinical centers use support from General Clinical Research Centers or Clinical and Translational Science Awards in conduct of NASH CRN Studies (grants UL1RR024989, M01RR000750, M01RR00188, UL1RR02413101, M01RR000827, UL1RR02501401, M01RR000065, M01RR020359, UL1RR025741). Funding for this work was supported in part by a UCSF Postdoctoral Research Fellowship Award from the Sandler Foundation (MDH and BS), from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research (KL2 RR024130) (BA), grants from the Academy of Finland (contract 124243), The Finnish Heart Foundation, The Finnish Diabetes Research Foundation, TEKES (contract 1510/31/06), Kuopio University Hospital (EVO grants 5232 and 5263), a Senior Scholarship in Aging from the Elison Medical Foundation (FWA and EV), FWA is an Investigator of the Howard Hughes Medical Institute, the UCSF Liver Center though the NIDDK (P30 DK026743) (EV), an R24 grant from NIDDK (DK085610) (CBN, CRK, and EV), and institutional support from the J. David Gladstone Institutes (EV).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, Supplemental References, six Fig.s, and four tables and can be found with this article online
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardern CI, Katzmarzyk PT, Janssen I, Leon AS, Wilmore JH, Skinner JS, Rao DC, Després JP, Rankinen T, Bouchard C. Race and sex similarities in exercise-induced changes in blood lipids and fatness. Medicine and science in sports and exercise. 2004;36:1610–1615. doi: 10.1249/01.mss.0000139798.54405.af. [DOI] [PubMed] [Google Scholar]
- Attie AD, Krauss RM, Gray-Keller MP, Brownlie A, Miyazaki M, Kastelein JJ, Lusis AJ, Stalenhoef AF, Stoehr JP, Hayden MR, et al. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J Lipid Res. 2002;43:1899–1907. doi: 10.1194/jlr.m200189-jlr200. [DOI] [PubMed] [Google Scholar]
- Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befroy D, Petersen K, Dufour S, Mason G, de Graff R, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- Black PN, Færgeman NJ, DiRusso CC. Long-Chain Acyl-CoA–Dependent Regulation of Gene Expression in Bacteria, Yeast and Mammals. J Nutr. 2000;130:305S–309S. doi: 10.1093/jn/130.2.305S. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen M, Rehman M, Walther T, Olsen J, Mann M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Civitarese A, Ukropcova B, Carling S, Hulver M, DeFronzo RA, Mandarino L, Ravussin E, Smith SR. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metabolism. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civitarese AE, Maclean PS, Carling S, Kerr-Bayles L, Mcmillan RP, Pierce A, Becker TC, Moro C, Finlayson J, Lefort N, et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metabolism. 2010;11:412–426. doi: 10.1016/j.cmet.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439–15450. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldeisen SE, Tucker KL. Nutritional strategies in the prevention and treatment of metabolic syndrome. Applied physiology, nutrition, and metabolism = Physiologie appliquée, nutrition et métabolisme. 2007;32:46–60. doi: 10.1139/h06-101. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ES, Li C, Zhao G, Pearson WS, Mokdad AH. Prevalence of the metabolic syndrome among U.S. adolescents using the definition from the International Diabetes Federation. Diabetes Care. 2008;31:587–589. doi: 10.2337/dc07-1030. [DOI] [PubMed] [Google Scholar]
- Frye R. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- Graham JM, et al. Isolation of mitochondria from tissues and cells by differential centrifugation. In: Bonifacino Juan S, et al., editors. Current protocols in cell biology / editorial board. Unit 3.3. Chapter 3. 2001a. [DOI] [PubMed] [Google Scholar]
- Graham JM, et al. Purification of a crude mitochondrial fraction by density-gradient centrifugation. In: Bonifacino Juan S, et al., editors. Current protocols in cell biology / editorial board. Unit 3.4. Chapter 3. 2001b. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Yu W, Smith BC, Devires MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, et al. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Molecular cell. 2011;41:139–149. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Mägi R, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nature Genetics. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey M, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard D, Grueter C, Harris C, Biddinger S, Ilkayeva O, et al. SIRT3 regulates mitochondrial fatty acid oxidation via reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Huang J, Verdin E. Acetylation of mitochondrial proteins. Meth Enzymol. 2009;457:137–147. doi: 10.1016/S0076-6879(09)05008-3. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL, Houmard JA, et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metabolism. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Ji H, Friedman M. Reduced hepatocyte fatty acid oxidation in outbred rats prescreened for susceptibility to diet-induced obesity. International journal of obesity (2005) 2008;32:1331–1334. doi: 10.1038/ijo.2008.71. [DOI] [PubMed] [Google Scholar]
- Ji H, Friedman MI. Reduced capacity for fatty acid oxidation in rats with inherited susceptibility to diet-induced obesity. Metab Clin Exp. 2007;56:1124–1130. doi: 10.1016/j.metabol.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kelley D, He J, Menshikova E, Ritov V. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kim H-S, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Sohn I, Ahn J-I, Lee K-H, Lee YS, Lee YS. Hepatic gene expression profiles in a long-term high-fat diet-induced obesity mouse model. Gene. 2004;340:99–109. doi: 10.1016/j.gene.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. Sirtuin 3, a New Target of PGC-1alpha, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE. 2010;5:e11707. doi: 10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JRB, Newgard CB, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, Hamm DA, Brix AE, Lindsey JR, Pinkert CA, et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci USA. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, Qi L, Speliotes EK, Thorleifsson G, Willer CJ, Herrera BM, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Kim YC, Ntambi JM. A lipogenic diet in mice with a disruption of the stearoyl-CoA desaturase 1 gene reveals a stringent requirement of endogenous monounsaturated fatty acids for triglyceride synthesis. J Lipid Res. 2001;42:1018–1024. [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntambi JM, Miyazaki M, Dobrzyn A. Regulation of stearoyl-CoA desaturase expression. Lipids. 2004;39:1061–1065. doi: 10.1007/s11745-004-1331-2. [DOI] [PubMed] [Google Scholar]
- Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, Attie AD. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci USA. 2002;99:11482–11486. doi: 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery J, Boeke J, Feinberg A. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton CM, Ntambi JM. Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab. 2009;297:E28–37. doi: 10.1152/ajpendo.90897.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti M-E, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro AE, Cotter J, Cooper DA, Peters JC, Surwit SJ, Surwit RS. Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metabolism. 2004;53:454–457. doi: 10.1016/j.metabol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Picklo MJ. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem Biophys Res Commun. 2008;376:615–619. doi: 10.1016/j.bbrc.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, Loos RJF, Manning AK, Jackson AU, Aulchenko Y, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metabolism. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Reaven GM. The metabolic syndrome: is this diagnosis necessary? Am J Clin Nutr. 2006;83:1237–1247. doi: 10.1093/ajcn/83.6.1237. [DOI] [PubMed] [Google Scholar]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmeisl M, Rim JS, Koza LP. Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes. 2003;52:1958–1966. doi: 10.2337/diabetes.52.8.1958. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Schwer B, Eckersdorff M, Li Y, Silva J, Fermin D, Kurtev M, Giallourakis C, Comb M, Alt F, Lombard D. Calorie Restriction Alters Mitochondrial Protein Acetylation. Aging Cell. 2009 doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey M, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard D, Li Y, Bunkenborg J, Alt FW, Denu JM, et al. SIRT3 Deacetylates Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase 2, Increases its Enzymatic Activity and Regulates Ketone Body Production Cell Metab. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan Ja, Mägi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nature Genetics. 2010 doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancáková A, Javorský M, Kuulasmaa T, Haffner SM, Kuusisto J, Laakso M. Changes in insulin sensitivity and insulin release in relation to glycemia and glucose tolerance in 6,414 Finnish men. Diabetes. 2009;58:1212–1221. doi: 10.2337/db08-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffe-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park S-H, Jiang H, Kim H-S, Flynn CR, Hill S, Hayes McDonald W, et al. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Molecular cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubes G. Insulin resistance. Prosperity’s plague. Science. 2009;325:256–260. doi: 10.1126/science.325_256. [DOI] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukropcova B, Sereda O, de Jonge L, Bogacka I, Nguyen T, Xie H, Bray GA, Smith SR. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes. 2007;56:720–727. doi: 10.2337/db06-0521. [DOI] [PubMed] [Google Scholar]
- Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci USA. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.