

Abstract
Background
PCR amplification and high-throughput sequencing theoretically enable the characterization of the finest-scale diversity in natural microbial and viral populations, but each of these methods introduces random errors that are difficult to distinguish from genuine biological diversity. Several approaches have been proposed to denoise these data but lack either speed or accuracy.
Results
We introduce a new denoising algorithm that we call DADA (Divisive Amplicon Denoising Algorithm). Without training data, DADA infers both the sample genotypes and error parameters that produced a metagenome data set. We demonstrate performance on control data sequenced on Roche’s 454 platform, and compare the results to the most accurate denoising software currently available, AmpliconNoise.
Conclusions
DADA is more accurate and over an order of magnitude faster than AmpliconNoise. It eliminates the need for training data to establish error parameters, fully utilizes sequence-abundance information, and enables inclusion of context-dependent PCR error rates. It should be readily extensible to other sequencing platforms such as Illumina.
Background
The potential of high-throughput sequencing as a tool for exploring biological diversity is great, but so too are the challenges that arise in its analysis. These technologies have made possible the characterization of very rare genotypes in heterogeneous populations of DNA at low cost. But when applied to a metagenomic sample, the resulting raw data consist of an unknown mixture of genotypes that are convolved with errors introduced during amplification and sequencing.
There are two broad approaches to high-throughput sequencing of metagenomes: in amplicon sequencing (also called gene-centric or gene-targeted metagenomics) a pool of DNA for sequencing is produced by using PCR to amplify all the variant sequences in a sample that begin and end with a chosen pair of primers [1-3], frequently targeting hypervariable regions of the 16S ribosomal RNA gene [4]; in de novo genome assembly total DNA is sequenced without amplification and reads are clustered into “species bins”, each providing the material for genome assembly by shotgun methods (see Table 1 in [5] for a list of such studies).
By trading off a broad survey of gene content for greater sequencing depth at the sampled loci, amplicon sequencing has the potential to detect the rarest members of the sampled community, but errors interfere more profoundly. Unlike genome assembly projects, where one needs only to determine the consensus base at each locus or decide whether a SNP is present in a population, the space of possible distributions for the sample genotypes and frequencies is effectively infinite. As a result, ambiguities in genome projects can usually be resolved by increasing the amount of data, whereas increasing depth (as much as 106 in recent studies [6,7]) increases the number of both real and error-containing sequences and makes the challenge of distinguishing minority variants from errors only greater under amplicon sequencing. Greater depth therefore calls for progressively more sophisticated methods of analysis.
The analysis of amplicon sequence data typically begins with the construction of OTUs (operational taxonomic units), clusters of sequences that are within a cutoff in Hamming distance from one another. OTUs serve to collapse the complete set of sequences into a smaller collection of representative sequences – one for each OTU – and corresponding abundances based on the number of reads falling within each cluster. OTUs were developed as a tool for classifying microbial species, but have also been repurposed to the task of correcting errors; the sequences within an OTU are typically interpreted as a taxonomic grouping without specifying whether the variation within an OTU represents errors or real diversity on a finer scale than that chosen to define the OTU. If the scale of the noise is smaller than that of the clusters, then the construction of OTUs will appropriately group error-containing sequences together with their true genotype. However, as sequencing depth increases, low probability errors outside the OTU radius will start to appear, and will be incorrectly assigned to their own OTU. Early studies using this approach on high-throughput metagenome data sets reported large numbers of low-abundance, previously unobserved genotypes that were collectively dubbed the rare biosphere[8]. Later, analyses of control data sets indicated that the diversity estimates in such studies tends to be highly inflated [9] and that results may lack reproducibility [10]. The dual purpose of OTUs for correcting errors and for taxonomic grouping is appropriate when the diversity is being sampled at a coarse level, e.g. the frequency of different phlya. However, when probing finer-scale diversity, OTU methods have intrinsically high false positive and false negative rates: they both overestimate diversity when there exist errors larger than the OTU-defining cutoff and cannot resolve real diversity at a scale finer than that (arbitrary) cutoff.
In response, a variety of approaches to disentangling errors from actual genetic variation have been proposed recently [11-14]. These include multiple rounds of OTU clustering with different hierarchical methods [11], utilizing sequence abundance information implicitly by starting new clusters with common sequences [11,12], and replacing OTU clustering with an Expectation-Maximization (EM) approach [13,14]. Accuracy has steadily improved, but all methods still fall short of maximizing the information acquired from metagenome data sets.
We believe that the way forward is to model the error process and evaluate the validity of individual sequences in the context of the full metagenomic data set, crucially including the abundances (number of reads) corresponding to each sequence. Major progress in this direction has been made recently by Quince et al[13,14]. In the specific context of pyrosequencing, often used for metagenomics, strings of the same nucleotide (homopolymers) are problematic, and Quince et al incorporated a model of the distribution of homopolymer light intensities into an Expectation-Maximization (EM) algorithm, Pyronoise, which infers the homopolymer lengths of sequencing reads [13] (Q09). Later, Quince et al released AmpliconNoise, an extension of PyroNoise that includes rates of single-nucleotide substitution errors obtained from training data [14] (Q11). These methods were shown to more accurately infer the underlying sample genotypes than other approaches, demonstrating the worth of explicitly modeling errors. However, the methods of Quince et al. have several shortcomings that we would like to rectify: (i) as the size of sequence data sets grows, AmpliconNoise becomes too slow to use in many applications; (ii) estimation of error rates relies on the existence of training data specific to the PCR and sequencing chemistries used; (iii) differentiation of fine-scale diversity is limited because read abundances are not fully utilized when calculating the distance between sequences and clusters; (iv) the parameters that determine how conservative the algorithm is in inferring real diversity are ad hoc and cannot be tuned without experiment-specific training data.
We build on the error-modeling approach pioneered in AmpliconNoise by developing a novel algorithm, DADA, to denoise metagenomic amplicon sequence data that addresses the concerns raised above [15]. We start with a parametric statistical model of substitution errors. We incorporate this error model into a divisive hierarchical clustering algorithm that groups error-containing reads into clusters consistent with being derived from a single sample genotype. Finally we couple this clustering algorithm with the inference of the error parameters from the clustered data, and perform each step in alternation until both converge. This method is presented below, and is shown to outperform previous methods in both speed and accuracy on several control data sets.
Results
Model and algorithm
We introduce a first-order model of the error process by assuming (1) each sequence read originates from a distinct DNA molecule in the sample, and therefore that the presence of errors on different reads are statistically independent, and (2) errors on different sites of the same read are also statistically independent events. The independence of errors across different reads relies on the independence of the PCR replication histories of those reads, a condition that holds when the total number of reads is significantly smaller than the total number of DNA molecules present in the initial environmental sample and there are no strong amplification biases for sequences with errors.
Under these conditions, the numbers of reads (abundances) of the error-containing sequences derived from a sample genotype follow the multinomial distribution, and the abundance r of each particular sequence is binomially distributed (see Methods) with a probability λdetermined by the particular combination of errors in that sequence and a number of trials ρ given by the total number of reads of its sample genotype. These facts allow us to establish two statistics to evaluate the hypothesis that a collection of sequencing reads derives from a single sample genotype. The abundance p-value determines when there are too many reads of the same sequence to be consistent with the error model, and the read p-value determines when a sequencing read is too far away to be an error from an inferred sample genotype.
These statistics serve as the basis of a sequence-clustering algorithm in which (1) reads are assigned to clusters, (2) a putative sample genotype is inferred for each cluster, (3) reads are reassigned to the cluster for which they are most likely to have resulted as errors from the inferred sample genotype, (4) the two p-value statistics are computed given the inferred sample genotypes and the clustering of the sequences (5) additional clusters are created if the clustering is statistically inconsistent with the error model (as suggested by small p-values).
The full algorithm (Figure 1) combines this probabilistic sequence clustering with the estimation of substitution-error probabilities that are used to compute the p-values. The algorithm begins by assuming all reads derive from a single sample genotype and estimates initial error probabilities given this assumption. It then alternates between clustering the reads and re-estimating the error probabilities until it converges to a final set of mutually consistent clusters and error probabilities.
Figure 1.
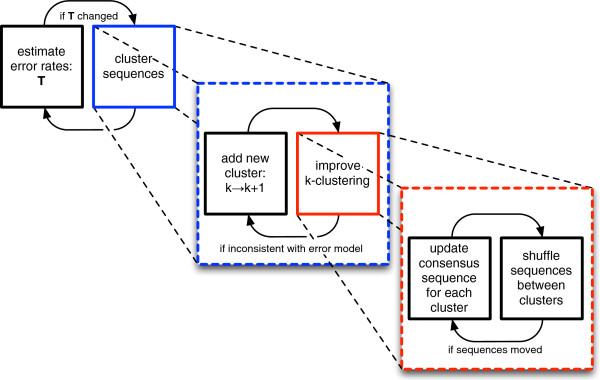
DADA schematic. The basic structure of DADA, an algorithm to denoise amplicon sequence data. See Algorithm Algorithm 1 in the Methods section for the pseudocode and a more detailed description.
The p-values
We introduce two statistics for deciding that particular sequences did not arise from errors. The read p-value is the probability of having observed at least one read of a sequence that is as improbable as the most improbable sequence amongst the observed reads. This statistic treats each read as a separate event (giving rise to its name) and therefore does not utilize sequence abundance. It results in a hard cutoff, λ∗, below which reads are decided not to be errors by DADA. This cutoff is set by the choice of a significance threshold Ωr, the probability of having observed at least one read more unlikely than λ∗. The abundance p-value, which is computed for each sequence individually, is the probability of having observed at least as many identical reads as we did of each sequence (conditioned on having observed at least one). The conservativeness of this measure is set by a significance threshold Ωa, the probability that at least one sequence should have been as overabundant as the most overabundant sequence. The abundance p-value gives DADA significantly greater sensitivity than previous methods.
Figure 2 shows simulated and real data from a typical cluster of sequences that originated from a common genotype (from the Artificial data set, introduced below). The abundance r and probability λ of each sequence is plotted, as the ability of the read and abundance p-values to discriminate between errors and non-errors is easily visualized in this parameter space. The regions where DADA will declare a sequence to be an error or a real sequence are delineated by a dashed line for each of our p-value statistics. The λ values have been log-transformed and scaled by the most common error probability, making the x-axis interpretable as an effective Hamming distance. Due to this scaling, it is also useful to interpret this plot in terms of real Hamming distances, in which case the Ωaline represents a lower bound on DADA’s resolution for any error at that distance.
Figure 2.
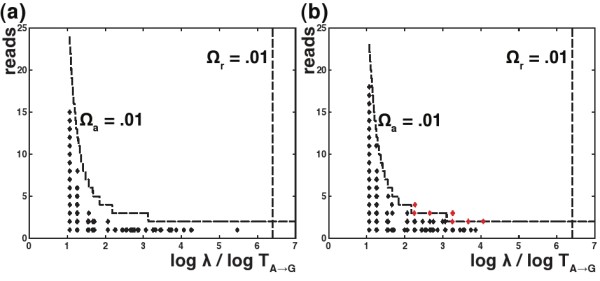
Discrimination plots for a typical cluster in the Artificial data set with 4691 reads. (a) simulated errors drawn from the error model and (b) the real errors in the cluster. Sequences (diamonds) are characterized by abundance and the probability λper read of having been produced. On the x-axis, we plot logλscaled by the most common error probability, TA→G, so that values can be interpreted as an effective Hamming distance. The dashed lines delineate the region – the lower left quadrant – where, for significance thresholds Ωaand Ωr provided by the user, DADA accepts that a sequence could have arisen via the error model. The vertical dashed lines shows the λbelow which (or the effective distance above which) the read p-value rejects sequences as being errors, and the curved dashed line shows the abundances above which the abundance p-value rejects sequences as being errors for each value of λ. There are several sequences in the real data (red diamonds) that would be rejected by the abundance p-value at the Ωa = .01 significance level; we posit that early round PCR effects are a suitable candidate to explain these departures from the error model.
For both the real and simulated data, the abundance p-value does a good job of tracking the form of the abundances of the errors, and the read p-value sits to the right of all observed data. For the real data, a small number of errors sit on or above the abundance discrimination line. Such errors were individually not expected to be observed at all, but ended up with a small number of reads larger than one. This pattern was observed across many clusters, and we believe that it reveals the presence of small violations of our assumption of the independence between reads. In particular, in a regime where the ratio of the number of error-free reads to the number of DNA molecules in the sample that act as the basis for amplification is of order one or larger, then errors during early stages of PCR may be sampled multiple times in the sequence data. As a result, the distribution for the number of reads of these errors may fall off much more slowly than what our model suggests. To deal with this effect in this paper, we lowered the Ωathreshold using an ad hoc method (discussed below) to prevent excess false positives. Doing so did not affect DADA’s ability to detect the genuine diversity in the data analyzed in this paper, which was typical of the data analyzed in many microbial metagenomics studies, but the sensitivity that is lost by using very small values of Ωa could be limiting for samples with even finer-scale diversity. Further analytics that model PCR as a branching process improve this current ad hoc threshold (unpublished work).
Treatment of insertions and deletions
DADA does not attempt to explicitly model the indel error processes, and indels do not contribute to the determination of whether sequences are related to each other via errors. Instead, sequences are aligned to each putative sample genotype, and are assigned to clusters on the basis of substitutions. During the computation of p-values, we sum together the reads of sequences within each cluster that have the same set of substitutions (forming structures that we call indel families). The number of reads of each of these indel families, rather than those of the raw sequences, are the basis of our p-values (see Methods).
Treating indels in this way does not affect the accuracy of DADA for the test data sets analyzed here, as the sample genotypes all differed from each other by at least one substitution, and these provided enough information for DADA to distinguish between them. However, DADA cannot distinguish between sequences that differ only by indels. In such cases, if the amplicons being denoised are coding regions, frame information should be used for making decisions about whether particular indels are real or errors, but in order to denoise non-coding regions with pure indel diversity, DADA is not sufficient in its current form.
Preclustering
Prior to our probabilistic sequence clustering we divided the raw data into coarse 3% single-linkage clusters (with indels not contributing to distance), subsets for which each sequence is ≤3% from at least one other sequence in its cluster and >3%from all sequences in other clusters. Due to its speed, we employed the ESPRIT algorithm for this task [16]. Single-linkage’s propensity for chaining was advantageous in this circumstance, as all error-containing sequences are very likely to be in the same cluster as their originating sample genotypes; for a sample genotype and one of its errors to end up in different clusters, the error would have to be ≥3% from the nearest error clustered with that genotype, corresponding to a large gap in the error cloud, which is unlikely under our error model.
Clustering
Each precluster is partitioned into sets of sequences that are conjectured to contain all errors arising from different sample genotypes. This partition is initialized to a single cluster containing all sequences. Two procedures then alternate. First, the indel family most unlikely to have resulted from errors is split off into a new cluster. Sequences then move between clusters based on the probability that they were generated as errors by each one, and the consensus sequence for each cluster is updated until there are no remaining reassignments that can improve the probability of the data. This second step is analogous to the assignment and update steps of standard k-means clustering. This alternation stops when the partition of the sequences fits with the current error model at the significance levels provided by the user.
Accuracy
We evaluated the accuracy of DADA by denoising three of the data sets in Q11 used to demonstrate AmpliconNoise’s accuracy relative to the earlier SLP and DeNoiser algorithms. These data are derived from mixtures of known clones that were amplified together and sequenced on the 454 platform, and consisted of different hypervariable regions of the 16S RNA subunit of bacterial ribosomes (16S rRNA), which are commonly used as a proxy for phylogenetic diversity in metagenomic studies [4]. Two of the data sets, Divergent and Artificial, with 35,190 and 31,867 reads, were sequenced with the GS-FLX chemistry and were truncated at 220 nucleotides. They were constructed by amplifying the V5 region of the 16S rRNA gene from 23 and 90 clones, respectively, isolated from lake water. The Divergent clones were mixed in equal proportions and are separated from each other by a minimum nucleotide divergence of 7%, while the Artificial clones were mixed in abundances that span several orders of magnitude, with some of the clones differing by a single SNP. The other data set, Titanium, with 25,438 reads, was sequenced with the newer Titanium chemistry and was truncated at 400 nucleotides. It contains V4-5 16S rRNA genes from 89 clones isolated from Arctic soil with varying abundance and genetic distances, similar to the Artificial set.
All data sets had undergone filtering of reads deemed to be of low quality prior to application of AmpliconNoise in Q11, so for purposes of comparison, we denoised the same set of filtered reads. The presence of a small number of low-quality reads in 454 data has been previously demonstrated [17], and as we do not expect these to be well described by our error model, we encourage the use of such quality filtering before applying DADA to non-test data. As SLP and DeNoiser were already demonstrated to be less accurate than AmpliconNoise on these data, we include here DADA’s performance only relative to that of AmpliconNoise. There were six other data sets presented in Q11 of V2 regions from a gut microbial community, but these had such an overwhelming number of chimeric sequences (reported to be as high as 85% in Q11), which neither DADA nor AmpliconNoise attempts to address, that we opted not to include these data sets in our analysis.
Tuning algorithmic sensitivity
DADA employs two tunable parameters that determine how conservative or liberal the algorithm is to be in deciding whether particular sequences could have resulted from errors: Ωa, and Ωr, the significance levels for its abundance and read p-values. Decisions about singletons, the sequences represented by a single read, depend on Ωr, whereas decisions about sequences with several reads depend on Ωa. The two values may be tuned independently to match the priority being placed on capturing the rarest and more common diversity.
Due to early-stage PCR effects discussed above, it was necessary to use Ωa significance levels lower than typical values. In order to select such values, we first performed a loose clustering of each data set with larger values of Ωa and Ωr and then made histograms of the Ωathresholds that would be required for each cluster to be reabsorbed into some other cluster (Figure 3). If there are errors with moderate statistical deviations from our model, we expect that these will show up as a tail of increasingly small p-values that will disappear smoothly as we lower the Ωa threshold. Thus, we looked for the first large gap in these histograms that would suggest all such model departures had been captured. Such a gap occurs at Ωa=10−15 for the Divergent data, Ωa=10−40 for the Artificial data, and Ωa=10−100for the Titanium data. We used these values in the analysis that follows, but also clustered all three data sets with Ωa=10−100and found that the results were unchanged (see appendix Appendix 2: Ωa robustness). This suggests that Ωa=10−100is a reasonable default value to use when clustering diversity at this scale, even though higher resolution may be achieved by the method outlined above. For non-test metagenome data that is more diverse and less oversampled, we have seen evidence that using much larger values of Ωa(such as .01) may be possible without compromised accuracy, but in such cases it is always advisable to make histograms of the type above to ensure that there is not an excess of clusters that would vanish if Ωa were lowered slightly.
Figure 3.
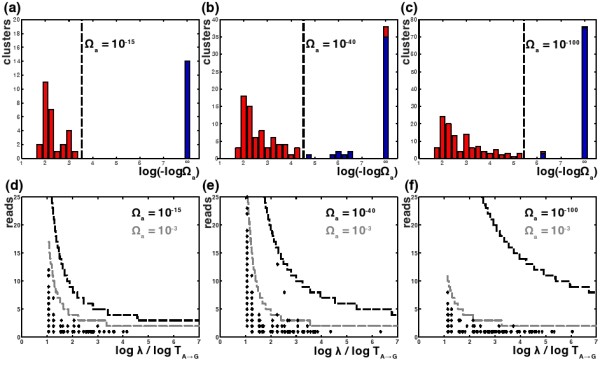
Ad hoc Ωa choices for the Divergent (a) and (d), Artificial (b) and (e), and Titanium (c) and (f) data sets. (a)-(c) are histograms of the Ωathreshold at which each cluster derived from a run of DADA with Ωa=Ωr=10−3rejoins some other nearby cluster. Genuine genotype counts are shown in blue and false positive counts are shown in red. The first gaps in these histograms were used to pick Ωathresholds for reclustering the data, and are indicated by vertical dashed lines. (d)-(f) show the Ωadiscrimination lines for the largest cluster in each data set (with 2294, 5479, and 1095 reads) for Ωa=10−3and the associated ad hoc Ωavalues.
We did not observe any significant departures in these data from our model that would affect the read p-values, and it was therefore possible to maintain the interpretation of Ωras a significance threshold. As a result, for these data, which contain ≤50 preclusters that were clustered separately by DADA, we set Ωr=10−3 so that the probability of having a false positive would be ≤5%for each data set.
False negatives and false positives
The purpose of DADA (and AmpliconNoise) is the inference of the genotypes present in the underlying sample from a set of noisy (error-containing) sequencing reads. There are two types of errors that such an algorithm can make: false positives in which a sample genotype is inferred that was not present in the sample, and false negatives in which the algorithm fails to infer a sample genotype that was present in the sequencing reads. The tradeoff between false positives and false negatives in the two algorithms can be controlled by the algorithmic parameters, depending on which type of error presents more of a problem to the user.
We present, in Table 1, a comparison of the false positives and false negatives for DADA and AmpliconNoise applied to the control data sets described above. Note, however, one important detail: these algorithms are designed to remediate substitution and indel errors, not all possible errors. In particular, we found that contaminants, chimeras, and pathological homopolymer errors contributed to these sequencing data sets. Using ad hoc methods, discussed in appendix Appendix 1: chimeras, contaminants, and missing or incorrect Sanger sequences, we accounted for these additional error sources, and did not penalize either algorithm for them.
Table 1.
False positives and false negatives
| DADA | AmpliconNoise | ||||
|---|---|---|---|---|---|
|
Sample |
False Pos |
False Neg |
False Pos |
False Neg |
|
| Divergent |
0 |
0 |
2 |
0 |
|
| Artificial |
1 |
2 |
8 |
7 |
|
| Titanium (s10) |
6 |
0 |
8 |
9 |
|
| Titanium (s25) | 23 | 4 | |||
DADA is more accurate in its inference of the sample genotypes than is AmpliconNoise on every data set. The difference is especially strong among false negatives, where DADA successfully identifies virtually all sample genotypes; DADA’s only two false negatives, both in the Artificial set, result from pathological alignment issues between sequences that differ only in the last two bases.
The differences in the nature of the false positives and negatives made by DADA and AmpliconNoise are shown in Figure 4. DADA produces one false positive in the Artificial data: a sequence with 268 reads that is a single substitution from a sample genotype with only 210 reads. Due to its vast abundance, this is unlikely to be an error, and we speculate it may represent a polymorphism that arose early in the growth of this clone. AmpliconNoise produces eight false positives in the Artificial data: all sequences with 1-4 reads that are (except for one) 1-4 substitutions away from clusters with a few hundreds of reads. Although these sequences were not atypical errors as judged by DADA due to their low abundances, AmpliconNoise calls them real as a result of setting a narrow error radius that is needed to prevent additional false negatives. The differences between the errors made by the two algorithms is less clear in the Titanium data set, but DADA outperforms AmpliconNoise in both FPs and FNs. We included AmpliconNoise’s results for the Titanium data set for both parameter settings included in Q11: the σ=.04 clustering (s25), which produces only four false negatives, leads to many false positives similar in nature to those of the Artificial clustering – low abundances and a small number of substitutions away from large clusters; the σ=.1 (s10) clustering produces many fewer false positives but misses nine sample genotypes.
Figure 4.

Nature of false positives and false negatives of DADA and AmpliconNoise on Artificial and Titanium data sets. False positives are characterized by the number of reads associated with the falsely inferred genotype r, the distance to the nearest real sequence d, and the number of reads associated with that nearest real sequence R. False negatives are characterized by the number of reads that matched the missing genotype r, the distance from that missing genotype to the nearest inferred genotype d, and the number of reads associated with that nearest inferred genotype R.
Speed
We evaluated the speed of DADA applied to the Artificial data, which was used to profile AmpliconNoise in Q11 (Table 2). ESPRIT was run on a single core of an AMD Phenom II 3.2GHz running Ubuntu and DADA was run on a MacBook Pro with an Intel Core 2 Duo 2.4 GHz.
Table 2.
CPU times for clustering artificial community
| Function | CPU time (seconds) |
|---|---|
| ESPRIT |
|
| kmerdist |
74.80 |
| needledist |
924.68 |
| Total |
1002.68 |
| DADA |
|
| N-W alignments |
97.81 |
| read p-values |
58.84 |
| Total |
296.64 |
| ESPRIT+DADA |
1.30×103 |
| AmpliconNoise | 7.57×104 |
The CPU times for a few significant subroutines within DADA and ESPRIT as well as their total CPU times.
DADA is currently written in MATLAB, but sequence alignments and the construction of indel families were bottlenecks that we reimplemented as MEX (Matlab executable) C programs. The majority of the time to run our denoising pipeline on the Artificial data set is spent on ESPRIT’s performance of pairwise alignments during the single-linkage pre-clustering step (needledist). A newer version of ESPRIT promises to be released soon that may dramatically lower this time [18]. If additional speedups are needed as data sets grow, it should be possible to replace the global alignments of ESPRIT by banded alignments that would be guaranteed to produce the same clusters if the width of the band is equal to the cluster radius, and would have have roughly linear (in sequence length) rather than quadratic, running time. Nonetheless, for these data DADA already gives a 60-fold speedup over AmpliconNoise, making jobs that required a 64-core cluster to run AmpliconNoise appropriate for a laptop running DADA.
As read lengths continue to grow, we expect the time complexity of DADA to be affected in two primary ways. First, because the complexity of the Needleman-Wunsch alignment algorithm used by both DADA and ESPRIT scales with the product of the lengths of the input sequences [19] there will be a quadratic slowdown with increasing read length unless heuristics are employed. On data sets comparable to those analyzed here, alignments consume the majority of algorithmic time and this scaling will dominate in the near future. Second, our current implementation for computing read p-values has both time and space complexity that grows very rapidly with read length (it is asymptotically ). This was not strongly limiting for these data, but in case it should become so as reads become longer, we have explored the use of a continuous approximation for the error probabilities that may alleviate this problem.
PCR substitution probabilities: symmetries and nearest-neighbor context-dependence
DADA not only infers sample genotypes, it also infers the substitution error probabilities caused by the amplification and sequencing processes. The substitution error probabilities inferred by DADA for all three data sets exhibit an approximate symmetry under complementation of the two bases involved. For example, the A→G probability is close to the T→C probability. This symmetry is expected if substitution errors predominantly arise during PCR amplification because substitution errors during PCR can be the result of either of two different mis-pairing events (from when the sequence is being copied to the opposite strand, or from when it is being copied back), and complementary substitution errors share causal mis-pairing events (see Figure 5 for a schematic). As it was not imposed, and the identities of the original genotypes were not known to the algorithm, the symmetry is a highly non-trivial check on DADA’s ability to learn error probabilities without training data. Additionally, the inferred substitution probabilities were similar across the data sets, and especially so between Divergent and Artificial, which were generated with the same PCR protocols.
Figure 5.

Two paths to the same error. Different mispaired bases (red) produce the same double stranded product once paired with complementary bases (green) so that each path leads to an ATG→AGG substitution error on one strand and a CAT→CCT on the other. The probability of these two errors is therefore expected to be very similar.
We also found that the nearest-neighbor nucleotide context affects the probability of substitution errors. We therefore introduced context-dependent substitution probabilities into DADA, allowing for dependence on the nucleotides immediately preceding and following the substituted nucleotide. Such probabilities are expected to exist in reverse complementary pairs for the same reason given for the context-independent case (again, see Figure 5); the lir→ljr probability is expected to be similar to the probability where denotes the reverse complement of lir. The degree of symmetry in the inferred probabilities, both context-independent and context-dependent, are shown in Figure 6 for all three data sets.
Figure 6.
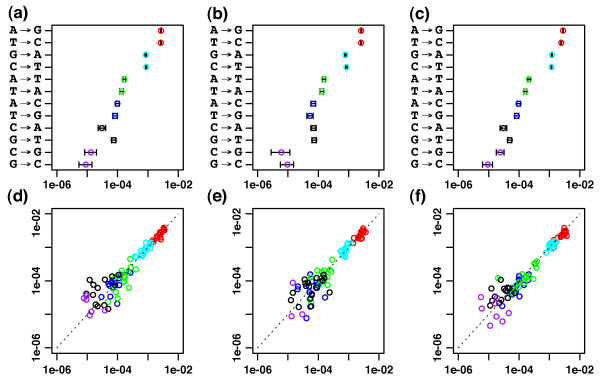
Error probability symmetries for Divergent (a) and (d), Artificial (b) and (e), and Titanium (c) and (f) data sets. (a)-(c): context-independent substitution error probabilities inferred by DADA with 95% confidence intervals based on binomial sampling error. Note the approximate symmetry between i→jand probabilities (which show up contiguously along the y-axis), where denotes the complement of nucleotide i. (d)-(f): All 96 reverse-complementary pairs of context-dependent error probabilities inferred by DADA for each data set. For each pair, the probability of the error away from an A or C is plotted on the x-axis and the error probability away from T or G is plotted on the y-axis. The pairing between these probabilities – seen by the tendency to lie along the diagonal – is stronger for the largest probabilities, which have the least sampling noise. The colors signify complementary pairs of errors red = (A→G,T→C) cyan=(C→T,G→A) green=(A→T,T→A) black=(C→A,G→T) blue=(A→C,T→G) purple=(C→G,G→C).
The magnitude of context-dependence for these data was moderate (most context-dependent probabilities differed by <50% from context-independent ones) as seen in the spread of points along the diagonal in Figure 6d,e,f . As a result, maintaining separate probabilities for different contexts did not affect the inferred sample genotypes. Nonetheless, that DADA was robust to significant variation in its parameters is a strong check on the stability of its sample inference.
We have worked with data for which context-dependence is large and has a strong effect on clustering. Therefore, we leave use of context-dependence as an optional feature of DADA, either as a consistency check, or when justified by the amount and nature of the data. But a caution is in order: with modest sized data sets, or if the sequences are too similar, use of the context-dependent rates could result in over-fitting and calling too many errors. However if this did occur, the expected complementarity symmetry of the inferred error probabilities would be unlikely to obtain unless the sequences were read in both directions.
Discussion
DADA explicitly incorporates read abundance when deciding whether sequences are genuine or errors; if there are many identical reads of a sequence, DADA will be more likely to infer an underlying sample genotype, even if individually those reads would be consistent with being an error from a nearby genotype. Furthermore, DADA implicitly assumes, via the error model, that reads near highly abundant sequences are far more likely to be errors. In contrast, previous methods have typically treated each read independently. AmpliconNoise partially incorporates abundance by weighting the prior probability that a read belongs to a cluster by the frequency of that cluster, but this is weaker than DADA, where dependence on cluster size shows up in a binomial coefficient (see Methods), especially for high-abundance errors. By using both sequence identity and abundance in this way, DADA is able to disentangle real diversity from errors at finer scales than previous methods, even when tuned to be very conservative.
However, full incorporation of abundance information makes DADA sensitive to early-stage PCR effects and the mis-estimation of error probabilities. The problem of early-stage effects is particularly pronounced in these data: when clustered with Ωa=Ωr=10−3, the Artificial data produces 68 false positives (we would have expected no false positives if the model assumptions were not violated). The majority of these sequences have 2-5 reads and 2-4 errors. Such problems would be typical of moderately oversampled PCR, the regime in which initial sample molecules are typically sampled multiple times, allowing a single error during early stages to show up in more than one read.
In lieu of an abundance statistic that appropriately compensates for this affect, we deal with this problem by lowering the sensitivity of the algorithm by tuning down Ωa. Further, because the probability given to each sequence scales as the error probabilities to the power of the number of reads (see Methods), if certain error parameters are larger than estimated in certain contexts, then the statistical significance of an error with many reads can be substantially overestimated. This problem gets progressively worse for deeper data sets, as all one-away errors begin to take on many reads. In anticipation of this problem, we have introduced nearest-neighbor context-dependence of error rates (see Methods). These had no impact on the final clustering for the test data presented, but in other data sets with larger context-dependent effects, we found a reduction in diversity estimates when context-dependence was included (data not shown).
DADA is a divisive hierarchical clustering algorithm: all sequences are assigned to a single cluster that is subdivided until the clustering fits an error model. Previous methods, including AmpliconNoise and simple OTU-clustering, have predominantly taken the opposite, agglomerative approach, which starts with too many clusters and merges them until some condition is met. This gives DADA a practical advantage, as the computational and space requirements (especially the number of alignments to perform and store) scale with the square of the initial number of clusters [20]. The typical problem of divisive methods – that the number of possible splittings is too large – is handled in DADA by seeding new clusters with sequences that are identified as not being errors and allowing other sequences, e.g. errors associated with the new clusters, to relocate if they become more probable by doing so.
Finally, DADA uses unsupervised learning to acquire error probabilities from the data that it is given. As PCR protocols vary in their choice of polymerase and number of rounds, these parameters vary by data set, perhaps greatly. This makes the universality of DADA’s approach especially attractive, and will be important as new sequencing methods come into use such as longer read-length and paired-end Illumina that commonly make substitution as well as indel errors [21]. While it now relies on training data to establish error parameters, AmpliconNoise could be embedded in the same procedure of estimating error probabilities after each successive round of clustering, but this would multiply the computation requirements by a factor of the number of rounds of re-estimation, compounding the problem of its slower speed.
Conclusions
OTUs serve as a rough analogue for microbes of the more clearly defined taxonomic groups of higher organisms. However, the repurposing of the OTU concept to the problem of inferring sample genotypes from error-prone metagenomic sequence data has serious and inherent shortcomings. The absence of an error model causes estimates of diversity, especially species richness, to depend strongly on experimental variables such as the size of the data set, the length of the region sequenced, and the details of the PCR/sequencing chemistry. These shortcomings are not amenable to simple fixes; it is not possible to separate real diversity from errors using an OTU approach when the diversity and the errors exist at similar scales (as measured by Hamming distance), as is the case in many metagenomic studies. PyroNoise and AmpliconNoise have demonstrated the usefulness of denoising sequence data with statistical, physically-based error models. These methods are based on the classical statistical technique of expectation-maximization. We have presented an alternate approach, DADA, which is more targeted to the particular task of producing conservative estimates of diversity from noisy sequence data. It is much faster and more capable of resolving fine-scale diversity while maintaining a lower false positive rate.
We did not achieve our goal of complete freedom from ad hoc parameters in this work. Even though Ωa, our input parameter, has a simple probabilistic meaning that is data set independent, there are corrections to our PCR model, and as a result Ωa takes on an ad hoc quality in this analysis. Nonetheless, Ωacan be coarsely tuned from the data itself in the way shown. Alternatively, for conservative diversity estimates, Ωa may be set to very small values (such as 10−100), and the resolution of the algorithm may be directly quantified. DADA not only guesses what is there, but knows what would have been missed if it were present, making Ωa ad hoc but not arbitrary.
Much work remains to be done, and it is not yet clear how the algorithms will fare with extremely rich fine-scale diversity as occurs for the antibody repertoire of B-cells and T-cells of the human immune system [22,23]. DADA must be equipped with statistics that correctly describe the abundance distribution of sequencing errors when a realistic model of PCR is used in which some reads are the result of shared lineages. More sophisticated methods for chimera detection that explicitly parameterize the chimera formation process analogously to the substitution and indel processes are also needed. Finally, these methods must be fully adapted and tested on sequencing platforms other than Roche’s 454.
Methods
General notation
From a sequencing data set , where rx is the number of individual reads of each distinct sequence sx, we would like to construct an estimate of the set of genotypes in the sample that gave rise to . With this aim, we construct a partition of the sequences {sx} where each is a collection of sequences hypothesized to have originated from a common Gα, and notate the number of reads assigned to Gαby . Because each sx can reside in only one Bα and it is assumed that Gα is the source of all sx in Bα, this framework does not allow for multiple Gα to contribute reads to the same sx. Allowing the latter is likely to affect only in special cases and adds complications.
Treatment of insertions and deletions: the construction of indel families
In addition to substitution errors, reads acquire insertions and deletions (indels) during amplification and sequencing. Both substitutions and indels could be used to parameterize an error model, but here we focus on substitutions and do not attempt to characterize the statistics of indels. Instead, we collapse together all the reads of sequences within each Bαthat differ from each other only by the location of indels in their alignments to Gα, forming subsets of each Bα that we call indel families. We call the indel families , where each sy refers either to a subset of some Bα or the sequence identical with Gα except for the substitution errors of its constituents, and ry is the number of reads in the family. The ry of each indel family will be used to test whether agrees with an error model, i.e. whether the substitution errors observed on the families in each Bα was not too improbable under an error model of substitution errors.
Alignments between sequences and each Gα in this paper took place with a scoring matrix of 5 + logT(to make them comparable with NCBI BLAST’s NUC.4.4 matrix [24]), where T, introduced below, is a matrix of substitution error probabilities. We used a gap penalty of −4 and a hompolymer gap penalty of −1. The gap penalty had to be less than half the smallest mismatch score or alignments would favor a pair of indels to that mismatch. The worst mismatch score tended to be about −6, and so −4 was chosen as a gap penalty to allow as many gaps as possible without making any mismatches prohibited within alignments.
The independence between substitution errors on different reads implies a binomial distribution for the number of reads of each family
If the occurrence of substitution errors on different reads are independent events, then each read of genotype Gα has an i.i.d. one-trial multinomial distribution with parameters Λ={λyα}, which we call the genotype error probabilities, to belong to each indel family sy. Λ also parameterizes the probability distribution for Ry, the number of reads of family y: if sy⊆Bα, then because Ry is the sum of ρα Bernoulli random variables each with success probability λyα, it follows the binomial distribution, Ry∼Bin(λyα,ρα). The assumption of independence between reads does not hold if early round PCR errors may be sampled multiple times in the final sequence data. Then, if we condition on having observed a particular error on some other read, the probability to observe it additional times is increased.
Λ may be constructed from simple nucleotide transition matrices
If the occurrence of substitution errors on different sites of the same read are independent events that do not depend upon the absolute position of the sites, then we can write each λyα in terms of a homogeneous Markov chain T, whose elements we call nucleotide error probabilities. The simplest useful model of this sort is the 4×4 transition matrix Tij=P(j|i) with, for example P(C|A) the probability for taking nucleotide A in the sample to C in the data (i,j will always index nucleotides), and this is the model used by AmpliconNoise. These probabilities generate the genotype error probabilities Λ via
| (1) |
where αn and yn denote the nth nucleotides of Gα and sy (also let λxα=λyα for all x|sx∈sy, used in Algorithm Algorithm 1).
If the nucleotide error probabilities at each site can depend upon the nearest-neighbor flanking nucleotides, we can keep track of a transition matrix T(L,R) for each possible (L,R) pair of flanking nucleotides such that , with P(ATC|AGC) the error probability for taking AGC to ATC. This generalization, which we call context-dependence, increases the number of free parameters from 12 to 192: there are 16 possible pairs of flanking nucleotides each with a 12 free parameter stochastic matrix. DADA may be run with or without context-dependence, but due to the risk of overfitting and because we saw no substantial effect on the outcome of clustering when it was used, context-dependence was not used to produce the results presented in this paper.
Assessing fit with an error model via tail probabilities
In order to assess whether and fit the error model Λ, we introduce two statistics, pyand qα: py is the probability of having seen at least ryreads of sy given that we saw at least one and qαis the probability of having seen at least one read with a genotype error probability at least as small as the smallest genotype error probability of an observed indel family in Bα, .
py: the abundance p-value
Call the number of reads of sygiven that we observed at least one:
Given the definition of pyabove,
We refer to this as the abundance p-value because it evaluates the probability of having observed more extreme abundances under the null hypothesis that each rywas generated by the error model. Because one abundance p-value is generated for each indel family, we use a Bonferroni correction and compare each pywith , where Ωa is a joint significance threshold that is provided to DADA by the user.
If we had not conditioned on having observed at least one read of each family, then the unobserved families would not have born any significance (they would all have py=1), but before looking at the data these families could have been significant. This would create a difficulty in choosing an appropriate multiple hypothesis correction; a naive Bonferroni correction of with Lα the length of Gα, which treats all possible families as tested hypotheses, would deprive the p-value of any statistical power. Conditioning on Ry>0 and evaluating only the observed sequences avoids this complication. However, any family with ry=1 obtains py=1 regardless of the smallness of λyα, which necessitates our second statistic, qα.
qα: the read p-value
For each cluster Bα, we compute the probability qα, which we call the read p-value, that there is at least one read with a genotype error probability at least as small as . Let lαbe a random variable representing the smallest genotype error probability when ρα reads of Gα are generated according to Λ. Then
where e iterates over all sequences, and λeαare the genotype error probabilities of these sequences. Evaluating the sum in this form would be computationally wasteful; instead we iterate over sets of sequences that share the same types of substitution errors. We index these sets by 4×4 off-diagonal matrices γ whose elements γi≠jspecify the number of i’s on a genotype that appear as j’s on the sequence. The genotype error probability for sequences of type γ away from genotype Gαis λ0α×λγ with the probability of having no errors (where nαi is the number of nucleotides of type i on Gα) and , which is independent of α. We also need the number of distinct sequences of type γfor each Gα, which we call the degeneracymγ(α) of γ on Gα. This is computed by taking a product over multinomial coefficients: . Rewritten as a sum over γ, qαbecomes
Vectors of λγ and mγ(α) can be computed starting with γrepresenting the more common errors and extended to more rare errors as needed to compute p-values for smaller . Finally, rather than maintaining vectors of mγ(α) for each α, we keep one for each of a small number of possible base compositions and interpolate between the qαthat would result from each of these in order to approximate the qα that would result from the exact base composition of Gα. Because one qα is generated for each Bα, the qα are then compared with , where Ωr is another joint significance threshold provided to DADA by the user, in order to determine whether any are too small to be the result of errors.
Maximum likelihood estimate (mle) of error probabilities
After forming a partition of that fits the error model generated by T, DADA updates T to its maximum likelihood estimate given this partition. The likelihood of T given and is
where αn and yn denote the nth aligned nucleotides of Gαand sx. For the case without context-dependence, the likelihood may be rewritten as
where Nij is the total number of js in that result from is in according to . The maximum likelihood equations for the off-diagonal elements of T, , are solved by maximum likelihood estimate
where Ni=∑jNij (the diagonal are set by normalization). Analogously, for the context-dependent case, the mle estimate is
where i≠j, L,R are any pair of left and right flanking nucleotides, NLijR is the total number of LjR codons in that result from LiR codons in according to , and NLiR = ∑jNLijR.
Algorithm for inferring the sample genotypes and error probabilities
The p-values py and qα are the basis for an algorithm that alternatively updates and the nucleotide error probabilities T, which may be specified by the user as either context-independent or dependent, denoting their values after t iterations by and Tt, in order to improve the likelihood of the data. This is similar to the hard-EM algorithm except that the partition at each step is the result of a model-based divisive hierarchical clustering approach and does not maximize the probability of given Tt. The algorithm requires two user inputs, Ωaand Ωr, which are the joint significance thresholds for the abundance and read p-values.
Algorithm 1
DADA Sequence clustering algorithm [15]
T0= , where is the trivial partition containing the entire .
t←1
repeat
repeat
if
start a new cluster within containing the most statistically inconsistent family.
repeat
update
(or if context-dependence is on)
each sxjoins Bαwhere
until is unchanged
update {py} and {qα}
until and
t←t + 1
untilT has converged
There are three levels of nesting, each beginning with a repeat statement in Algorithm Algorithm 1. From outer to inner, we give a qualitative description of their purpose:
1. Starting with T0, the maximum likelihood nucleotide error probabilities given the trivial partition of all sequences into a single cluster, the outermost loop iteratively updates and T until T converges. We have observed cases where T does not completely settle down but fluctuates within a small basin of attraction. To deal with such cases, DADA terminates if T ever returns to a previously held value or ||Tt−Tt−1|| <∊, where the tolerance ∊ = 1e − 9 is used as a default and may be altered by the user. If convergence has not been reached in ten rounds, DADA terminates with a warning message.
2. For each Tt, the next loop begins with the trivial partition, , and adds blocks to until the {py} and {qα} do not allow rejection of the error model at joint significance levels Ωaand Ωs. New are seeded by the sequences in families with the smallest p-values. If statistically significant families exist under both p-values, then those significant under the abundance p-value take priority for starting new clusters. This approach avoids the need to put an explicit penalty on the number of blocks of , instead aiming for the smallest under which the current error model cannot be rejected.
3. After adding a new block to , the innermost loop raises the probability of the data by reassigning each sequence to the block that would produce (under the error model) the largest expected number of reads of that sequence. The putative genotype of a cluster, Gα, is also updated if a cluster Bαhas a new consensus sequence. This continues until sequences cease changing clusters.
Appendix 1: chimeras, contaminants, and missing or incorrect Sanger sequences
There are disagreements between the Sanger sequences of the clonal isolates used to construct the data sets and the denoised sequences of DADA and AmpliconNoise that are due to sources other than PCR substitutions and pyrosequencing errors. These include contamination, chimeric sequences that result from the co-amplification of genomes with regions over which they exactly match, Sanger errors, the absence of any reads of two sample genotypes, and disagreements between the Sanger sequences and majority of the 454 reads about the lengths of several homopolymers (for example, not a single 454 read matched four of the Sanger sequences while many were identical except for the presence of a single deletion on a G homopolymer). In order to evaluate the relative performance of DADA and AmpliconNoise as denoising algorithms, it was necessary to identify which disagreements between Sanger and denoised sequences were due to these sources and which, falling outside these categories, were due to algorithmic shortcomings. We chose criteria for classifying errors of these types and applied them to the sequences denoised by DADA and AmpliconNoise (Table 3).
Table 3.
Additional sources of noise and false positives
| |
DADA |
AmpliconNoise |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Denoised | Clone | Chim | Contam | Other | Denoised | Clone | Chim | Contam | Other |
| Divergent |
43 |
23 |
18 |
2 |
0 |
51 |
23 |
23 |
3 |
2 |
| Artificial |
65 |
50 |
14 |
0 |
1 |
73 |
44 |
21 |
0 |
8 |
| Titanium (s10) |
274 |
80 |
185 |
3 |
6 |
163 |
71 |
82 |
2 |
8 |
| Titanium (s25) | 304 | 76 | 203 | 2 | 23 | |||||
For each data set and both algorithms: the total number of denoised sequences, the number that matched one of the Sanger sequenced clones, the number classified as chimeras, the number classified as contaminants, and all other false positives.
We began by correcting possible errors in the Sanger sequences. In the Divergent and Artificial data sets there were disagreements between very high abundance denoised sequences and their nearest neighbor Sanger sequences (12/23 Divergent Sanger sequences and 63/90 Artificial Sanger sequences). The denoised sequences were a consensus of thousands of pyrosequencing reads and did not differ from the Sanger sequences near homopolymers; rather, all disagreements were non-homopolymer related deletions near the starts of the reads. It was confirmed (Chris Quince, personal communication) that all bases of Sanger sequences aligning to sites within 13 nucleotides (nts) of the forward primer of the pyrosequencing reads had been removed in Q11, and so were likewise removed in all our analysis. In the Titanium data, the denoised sequences closest to eight of the Sanger sequences had over 100 reads but differed from them by one or two homopolymer deletions at several long homopolymers. DADA and AmpliconNoise (s10 and s25) agreed on the presence of the deletions in all of these sample genotypes, and there were more copies of the error-containing sequences than the Sanger sequences in the raw data (Table 4), suggesting either an error probability greater than 50% for the combined amplification/pyrosequencing process or problems with the Sanger sequences. Therefore, we did not consider these disagreements to be false positives or false negatives for either algorithm.
Table 4.
Titanium genotypes with more error-containing than Sanger sequence matching reads
| Reads of nearby pyrosequence | Reads of sanger sequence | Errors |
|---|---|---|
| 5 |
0 |
G8→G7 (301), |
| |
|
G5→G4 (354) |
| 70 |
0 |
G6→G5 (133) |
| 75 |
1 |
C7→C6 (313) |
| 21 |
18 |
G6→G5 (133) |
| 14 |
0 |
G6→G5 (133) |
| 80 |
0 |
G6→G5 (133) |
| 77 |
3 |
G6→G5 (133) |
| 80 | 1 | G6→G5 (133) |
The difference between the Sanger and 454 sequences and their locations are given. For example, G6→G5 (133) means that a homopolymer beginning at the 133rdbase with 6 Gs in a Sanger sequence showed up as 5 Gs in most of the pyrosequence reads. All disagreements consist of deletions in the 454 reads on long G/C homopolymers relative to the Sanger sequences, and the majority of the disagreements occurred on one locus, site 133.
Next we identified chimeras: sequences consisting of two sections with one section a close match to one sample genotype and the other a close match to a second sample genotype. These can be produced in substantial quantities by PCR [25]. Analogously to Q11, for each denoised sequence we computed the Hamming distance to the nearest Sanger sequence and to the nearest exact chimera by considering all possible breakpoints between all pairs of sequences of higher abundance (a chimera will have fewer reads than its parents unless it acquires substantial PCR bias). For a denoised sequence to be classified as a chimera, we required that it be at least 3 nts closer to the nearest exact chimera than the nearest sample genotype and within 5 nts of the optimal chimera (also analogous to the procedure used in Q11). We waived the 3 nt improvement criteria for denoised sequences that were identical to exact chimeras, which occurred for some particularly highly abundant chimeras between closely related sample genotypes. All data sets had a large number of chimeras amongst their denoised reads, with Titanium having more chimeras than sample genotypes (both algorithms), highlighting how essential accurate chimera identification is in tandem with the correction of PCR and sequencing errors.
Finally, we found several sequences too far from any sample genotypes or exact chimeras to be explained by being errors away from either. Some of these sequences were similar to previously observed sequences found on GenBank (Table 5). We classified as a contaminant any sequence within 2 nts of a GenBank sequence and at least 5 nts closer to a GenBank sequence than any sample genotype or chimera. We found a mixture of contaminants likely to come from the original sample (lake water bacteria), and contaminants that may have entered the sample during processing and sequencing (bacteria previously observed in human skin, a human mouth, and soil samples). These contaminants were not previously mentioned in Q11 but were straightforward to detect when looking at DADA’s denoised sequences, in part because having a smaller pool of algorithmic false positives makes identifying contaminants much easier.
Table 5.
Contaminants
| Accession | Reads/Frequency | DGB | Dsample | Dchim | Source | Type | DADA | AN |
|---|---|---|---|---|---|---|---|---|
| Divergent |
||||||||
|
FR697039 |
14/4×10−4 |
0 |
11 |
10 |
Lake Water |
Bacterium |
Y |
Y |
|
EU633742 |
1/3×10−5 |
1 |
9 |
9 |
Showerhead |
Methylobacter |
Y |
Y |
|
JF515955 |
1/3×10−5 |
1 |
8 |
8 |
Soil |
Nitrosomonadaceae |
N |
Y |
| Titanium |
||||||||
|
FJ004768 |
77/3×10−3 |
2 |
39 |
27 |
Soil |
Bacterium |
Y |
Y |
|
JF190756 |
1/4×10−5 |
1 |
40 |
27 |
Human Skin |
Bacterium |
Y |
Y |
| JQ462329 | 2/8×10−5 | 0 | 7 | 5 | Human Mouth | Bacterium | Y | N |
DGB, Dsampleand Dchimare the Hamming distances to the given GenBank entry, the nearest sample genotype, and the optimal chimera for each putative contaminant denoised sequence. No entries are given for the Artificial data set because no likely contaminants were found in the denoised sequences of either algorithm. The last two columns show which contaminants were present in the denoised sequences of each algorithm (“Y” if found and “N” if not found).
In classifying false negatives, we sought to evaluate the ability of the algorithms to detect the presence of genuine diversity in the pyrosequencing reads. However, not all clones used to construct the samples in Q11 had exact matches amongst the pyrosequencing reads: one Sanger sequence in the Artificial data set (#69 in Q11) was 29 nts away from the nearest 454 read and one Sanger sequence in the Titanium data set (#66 in Q11) was 61 nts away from the nearest 454 read. We assumed that these were missing from the 454 data, and they do not contribute to the false negatives of either algorithm. Further, a number of clones were identical to each other up to the point of truncation of the pyrosequencing reads. Finally, a number of the Titanium clones differed from each other only by the presence of Ns, bases that Sanger was unable to resolve. In such cases, we collapsed clones together and assumed the non-N containing Sanger sequence was correct. Table 6 gives the number of distinct (up to Ns) clones that are present in the data, and how many had been used to construct the sample.
Table 6.
Detectable genotypes
| Sample | Genotypes | Present and distinct |
|---|---|---|
| Divergent |
23 |
23 |
| Artificial |
90 |
50 |
| Titanium | 91 | 80 |
The number of genotypes used to construct the sample and the number that were present and distinct and so could detected by a denoising algorithm.
Several aspects of this post-processing pipeline – especially contaminant identification– utilize knowledge of the sample genotypes and do not constitute useful methods for non-test data. Our approach to chimera identification does not utilize sample genotype information, but requires more development to be applied to non-test data: it does not search for higher-order chimeras that are combinations of three or more parental sequences, the criteria for being labelled as a chimera do not scale with error rates and read lengths, and no attempt has been made to realistically model the chimera formation process. Our goal has been only the isolation of errors due to PCR and sequencing error.
Appendix 2: Ωarobustness
To assess whether DADA’s performance in this paper was the result of the fine-tuning of Ωa, we evaluated each data set under all three Ωavalues. The results are given in Table 7 and demonstrate that the same results would have been achieved by using Ωa = 10−100 for all three data sets.
Table 7.
False positives and false negatives for each data set with Ωa = {10−15,10−40,10−100} and Ωr = 10−3
| |
Divergent |
|
Artificial |
|
Titanium |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ωa | False Pos | False Neg | False Pos | False Neg | False Pos | False Neg | ||||
| 10−15 |
0 |
0 |
10 |
2 |
7 |
0 |
||||
| 10−40 |
0 |
0 |
1 |
2 |
6 |
0 |
||||
| 10−100 | 0 | 0 | 1 | 2 | 6 | 0 |
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MJR designed the algorithm, wrote the software, performed the analyses, and wrote the paper. BJC, DSF, and SPH provided assistance and advice with algorithm design, comparative analysis, and the paper. All authors read and approved the final manuscript.
Contributor Information
Michael J Rosen, Email: mjrosen@stanford.edu.
Benjamin J Callahan, Email: benjc@stanford.edu.
Daniel S Fisher, Email: dsfisher@stanford.edu.
Susan P Holmes, Email: susan@stat.stanford.edu.
Acknowledgements
Thanks to Chris Quince and Sue Huse for providing and helping with clarifications about test data. Thanks also to Yijun Sun for help with the ESPRIT clustering algorithm. Finally, thanks again to Chris Quince and to Kabir Peay for useful comments on the manuscript. This work was supported in part by the NSF under grant DMS-1120699. SH is supported by grant NIH-R01GM086884.
References
- Cheung MK, Au CH, Chu KH, Kwan HS, Wong CK. Composition and genetic diversity of picoeukaryotes in subtropic coastal waters as revealed by 454 pyrosequencing. ISME J. 2010;4:1053–1059. doi: 10.1038/ismej.2010.26. [DOI] [PubMed] [Google Scholar]
- Iwai S, Chai B, Sul WJ, Cole JR, Hashsham SA, Tiedje JM. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 2010;4:279–285. doi: 10.1038/ismej.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeria LCRS, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J, Rosado AS. Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J. 2010;4:989–1001. doi: 10.1038/ismej.2010.35. [DOI] [PubMed] [Google Scholar]
- Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes P, Simmons SL, Denef VJ, Banfield JF. The dynamic genetic repertoire of microbial communities. FEMS Microbiol Rev. 2009;33:109–132. doi: 10.1111/j.1574-6976.2008.00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, Affourtit J, Egholm M, Henrissat B, Knight R, Gordon JI. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. PNAS. 2010;107:7503–7507. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored rare biosphere. PNAS. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunan V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wu L, Deng Y, Zhi X, Jiang Y, Tu Q, Xie J, Nostrand JDV, He Z, Yang Y. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011;5:1303–1313. doi: 10.1038/ismej.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IA, Read LF, Sloan WT. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinf. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael Rosen. DADA website. 2012. http://sites.google.com/site/dadadenoiser.
- Sun Y, Cai Y, Yu F, Farrell MF, McKendree W, Farmerie W. ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009;37:e76. doi: 10.1093/nar/gkp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Sun Y. ESPRIT-Tree: hierarchical clustering analysis of millions of 16S rRNA pyrosequences in quasilinear computational time. Nucleic Acids Res. 2011;39:e95. doi: 10.1093/nar/gkr349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequences of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Fraley C, Raftery AE. How many clusters? which clustering method? answers via model-based cluster analysis. Comput J. 1998;41:578–588. doi: 10.1093/comjnl/41.8.578. [DOI] [Google Scholar]
- Yang X, Aluru S, Dorman KS. Repeat-aware modeling and correction of short read errors. BMC Bioinf. 2011;12:S52. doi: 10.1186/1471-2105-12-S1-S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, Simen BB, Hanczaruk B, Nguyen KD, Nadeau KC, Egholm M, Miklos DB, Zehnder JL, Fire AZ. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Translational Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sanders CM, Yang Q, Schroeder HW, Wang E, Babrzadeh F, Gharizadeh B, Myers RM, Hudson JR, Davis RW, Han J. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. PNAS. 2009;107:1518–1523. doi: 10.1073/pnas.0913939107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd Lowe. NUC.4.4 score matrix, NCBI. 1992. ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4.
- Lahr DJG, Katz LA. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. BioTechniques. 2009;47:857–866. doi: 10.2144/000113219. [DOI] [PubMed] [Google Scholar]