

Abstract
Background
Papillon-Lefèvre Syndrome (PLS) is a type IV genodermatosis caused by mutations in cathepsin C (CTSC), with a worldwide prevalence of 1–4 cases per million in the general population. In México, the prevalence of this syndrome is unknown, and there are few case reports. The diagnosis of twenty patients in the state of Sinaloa highlights the need to characterize this syndrome in Mexicans.
Methods
To understand the basis of PLS in Mexicans, the gene expression, enzymatic activity and mutational analysis of CTSC were assayed in nine PLS patients and their relatives. Frequencies of CTSC gene polymorphisms and HLA alleles were determined in these patients, their relatives, and the population.
Results
Patients showed normal CTSC gene expression, but a deep reduction (up to 85%) in enzymatic activity in comparison to unrelated healthy individuals. A novel loss-of-function mutation, c.203 T >; G (p.Leu68Arg), was found in all patients, and some carried the polymorphism c.458C >; T (p.Thr153Ile). Allelic frequencies in patients, relatives and controls were 88.89%, 38.24% and 0.25% for G (c.203 T >; G); and 11.11%, 8.82% and 9.00% for T (c.458C >; T). HLA-DRB1*11 was found significantly more frequent (P = 0.0071) in patients than controls (33.33% vs. 7.32%), with an estimated relative risk of 6.33.
Conclusions
The novel loss-of function mutation of CTSC gene (c.203 T >; G) found in patients correlated with their diminished enzymatic activity, and HLA-DRB1*11 was found to be associated with PLS. The study of more PLS patients may give more insights into the etiology of the disease as well as its prevalence in México.
Keywords: Papillon-Lefèvre Syndrome, Cathepsin C, Mutations, Enzymatic activity, HLA, Mexicans
Background
Papillon-Lefèvre Syndrome (PLS) is an autosomal recessive disorder that is caused by mutations in cathepsin C (CTSC) (OMIM #245000). PLS is characterized by palmoplantar hyperkeratosis and aggressive periodontitis, has a worldwide prevalence of 1–4 cases per million in the general population, and is often related with consanguinity [1-3]. Dermatological disorders initiate with erythema and after about six months, they progress to hyperkeratosis of soles, palms, knees and elbows [4]. This exacerbates in winter, leading to painful fissures. Aggressive periodontitis begins with the primary dentition, leading to periodontium destruction, and the premature loss of deciduous teeth by age of 6 years; afterwards gums heal, until the development of the permanent dentition, when periodontitis reappears and teeth are prematurely lost by the age of 16-years [5]. Besides these basic features, PLS patients can present with liver abscesses [6-9], increased incidence of pyogenic infections [10], or mental retardation [11]. Medication for hyperkeratosis and periodontal care improve quality of life [12,13].
The CTSC gene (11q14.2) spans 4.7 kb and has seven exons [14,15]. CTSC gene mutations have been reported as responsible for PLS [14], as well as similar conditions such as Haims-Munk Syndrome, and juvenile periodontitis [16,17]. CTSC is expressed at high levels in polymorphonuclear leukocytes, alveolar macrophages, skin, kidney, and placenta, and at moderate or low levels in a variety of other organs. Gene structure shows several possible tissue-specific regulatory elements.
CTSC (EC 3.4.14.1) or dipeptidyl peptidase I (DPPI) is a lysosomal cysteine protease that removes dipeptides from the free N-termini of protein and peptides. The CTSC gene encodes a polypeptide chain that folds into an exclusion domain, a propeptide, and two papain-like domains [18,19]. The active CTSC is a tetramer (200 kD) [20,21], while cathepsin B, H and L are monomers [21-25]. CTSC plays a major role in the cleavage of some hormones (including glucagon, gastrin and angiotensin II), and the activation of granule serine proteases from cytotoxic T lymphocytes, natural killer cells (granzymes A and B), mast cells (tryptase and chymase), and neutrophils (cathepsin G and elastase) [19,20,26-28].
CTSC deficiency has been associated with PLS-associated increased susceptibility to bacterial infection in gums and other sites; however, the heterogeneous severity of the periodontitis and the susceptibility to infections observed in PLS suggests the existence of compensatory pathways in most tissues [29]. The PLS phenotype also suggests a role for CTSC in epithelial differentiation or desquamation. Several works have shown a relationship between CTSC and the development of gingival fibroblasts, and the peeling of skin keratin in affected zones [30]. Aberrant epithelial differentiation may affect the junctional epithelium that binds the gingiva to the tooth surface, possibly weakening this mechanical barrier and allowing deeper colonization by periodontal pathogens [30].
The Major Histocompatibility Complex (MHC) loci (6p21.31) span nearly 3.6 Mb, and contains the most polymorphic human genes, with strong linkage disequilibrium. MHC products, known as Human Leukocyte Antigens (HLA) class I (A, B and C) and class II (DP, DQ and DR), play a key role in antigen presentation to T-cells during elicitation of adaptive immune responses [31]. Some PLS patients show an increased susceptibility to recurrent suppurative infections, and several works have attempted to associate HLA alleles with PLS and severe periodontitis [32-34].
In México, the prevalence of PLS is unknown. Sada-Tamayo et al. reported three cases of this syndrome in the Mexican state of Tamaulipas in 2007 [35]. We have found twenty PLS patients in the state of Sinaloa, indicating a high prevalence of the disease in this region. In this work, we report the clinical characteristics, gene expression, enzymatic activity and mutational analysis of CTSC in nine PLS patients (from seven different families) and their relatives. Frequencies of CTSC gene polymorphisms and HLA alleles were determined in these patients, their relatives and the population.
Methods
Study subjects
We have diagnosed twenty patients with Papillon-Lefèvre Syndrome at the Hospital Pediátrico de Sinaloa (HPS) level 2, but only nine were available to participate in this study. These nine patients were born to seven non-related families from the following locations at the northwestern Mexican state of Sinaloa: La Reforma (two families), Las Aguamitas, Higueras de Abuya, Sinaloa de Leyva, Navolato and Culiacán. Parents (n = 12) and siblings (n = 5) of the patients, and unrelated healthy individuals (n = 8) were included in the study as comparisons. Voluntary blood donors (n = 200) from the Instituto Mexicano del Seguro Social (IMSS) were included for population analysis. All subjects were natives of Sinaloa for two previous generations. The Ethical and Research Committee of HPS approved this study and subjects signed an informed consent.
Cell isolation
Peripheral blood mononuclear cells (PBMCs) and Polymorphonuclear cells (PMNs) were isolated by centrifugation on Polymorphprep (Axis Shield, Oslo, Norway) following manufacturer recommendations. Contaminating erythrocytes in pellets were lysed by adding 5 mL lipopolysaccharide (LPS)-free cold water for 45 seconds and immediately adding 5 mL of LPS-free cold physiological saline solution. Purity of cell fractions was assessed by directly counting Giemsa-stained smears. Cell viability was assessed by trypan blue exclusion-stain (Invitrogen, Carlsbad, CA, USA), and was always >;90% in all experiments of this work.
Enzymatic activity
Leukocyte pellets were disrupted in 100 μL lysis buffer (10 mM Na3PO4, 0.1% Trixon X-100) by one pulse of sonication (100 W potency) for 5 seconds in ice, using an ultrasonic probe Sonic Dismembrator 100 (Fisher Scientific, Pittsburgh, PA, USA). Samples were centrifuged at 2,500 g during 5 min at 4°C, and the supernatant were recovered for enzymatic assays. Proteins were quantified using the Bradford assay.
Enzymatic activity was assayed by mixing 20 μL of cell extract with 30 μL reaction buffer (100 mM Na3PO4, 2 mM NaCl, 2 mM DTT), 30 μL dilution buffer (10 mM Na3PO4, 0.1% Trixon X-100), and 100 μL of chromogenic CTSC-substrate (1.5 mM gly-phe-para-nitroanilide). Absorbencies were measured at 405 nm, samples were incubated at 37°C for 1 h, and read again. Dipeptidyl peptidase activity of samples, expressed as para-nitroanilide released (μmol/mg of protein * hour), was determined interpolating the difference between first and second reads in a standard curve (0.025 to 175 μmol) of pure para-nitroanilide (pNA) (Sigma-Aldrich, St. Louis, MO, USA). Dipeptidyl peptidase activity from healthy individuals was set up as 100% and used for comparison to all other groups.
Bovine spleen cathepsin C (0.5 μg/μL solution with 5 μg/μL BSA) (Sigma-Aldrich) was used to determine enzyme kinetics by measuring pNA release from the substrate. A reaction rate of 2.58 ±0.47 μmol of pNA released per minute was estimated for pure bovine spleen CTSC, and was similar to samples rates of 1.66 ±0.22. Reaction conditions and inhibitor (iodoacetic acid, Sigma-Aldrich) concentrations were determined from kinetic assays and others reports [36,37].
Quantitative analysis of gene expression by qPCR
Purified leukocyte pellets were homogenized in 1 mL of TRIZOL reagent (Invitrogen). Total RNA was extracted in accordance to manufacturer’s instructions, treated with DNase I (Invitrogen), and its integrity and concentration were confirmed by electrophoresis and spectrophotometry, respectively. RNA (2 μg) was annealed to Oligo-dT, and synthesis of single-strand complementary DNA (cDNA) was performed under standard conditions.
Quantitative Polymerase Chain Reaction (qPCR) was performed following conditions previously described [38], using a MiniOpticon Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Primers to amplify 273 bp (from exons 7 and 8) of human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene (5’-TCATCCAT GACAACTTTGGTATCG-3’; 5’-TGGCAGGTTTTTCTAGACGGC-3’) and 125 bp (from exons 6 and 7) of CTSC gene (5’-AGGAGGTTGTGTCTTGTAGCC-3’; 5’-AGTGCCTGTGTAGGGGAAGC-3’) were designed with the Primer Express 2.0 software (Applied Biosystems, Foster City, CA, USA). Relative expression was calculated with the 2(−Delta Delta C(T)) method [39]. Expression level from healthy individual was set up as 1.0 for all comparisons.
PCR-SSCP and sequencing
Genomic DNA was isolated from whole blood containing EDTA, using the salt precipitation method [40]. The seven exons of the CTSC gene, and their splice junctions, were amplified by PCR, under standard conditions using a PXE 0.2 thermocycler (Thermo Electron). Sequence of primers for exons 2 to 6 were taken from a previous report [14], and were designed for exons 1 (5’-CGCCTCGTGGTGGACTCAC-3’; 5’-GCTCAAGGG CAGAAAGGACG-3’) and 7 (5’-GCAAAGAATAATG GAGCAAAGAAGA-3’; 5’-AATTCCCCTTTACAACT GATGCA-3’).
Mutation scanning of CTSC exons was done by single-strand conformation polymorphism (SSCP), as described elsewhere [41,42]. Briefly, PCR product samples were diluted, 1:1 in loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol), heated at 95°C for 5 min and immediately placed on ice. Aliquots were subjected to electrophoresis on polyacrylamide gel at 300 V for 3.2 h, at 10°C (±0.5). Gels were silver stained to visualize the reannealed single-stranded PCR products.
Amplified exons with mobility shifts were analyzed to identify nucleotide changes by automated DNA sequencing. Obtained sequences were compared with human CTSC sequence reference of GenBank (NG_007952.1) using CLC sequence viewer V6.4 software (CLC bio, Aarhus, Denmark).
Protein homology analysis
Amino acid sequences of CTSC from human (NP_001805), rhesus monkey (XP_001104734), cattle (AAI02116), dog (AAD02704), house mouse (AAH67063), Norway rat (NP_058793), zebrafish (AAH64286), African clawed frog (NP_001080511), black tiger shrimp (ABW74905), Pacific white shrimp (ACK57788), Marsupenaeus japonicum (BAC57943) and Schistosoma japonicum (ACC32040), were obtained from GenBank. Sequences were aligned and analyzed using CLC sequence viewer (CLC bio).
Amino acid substitutions were analyzed with the PolyPhen-2 (Polymorphism Phenotyping v2) software (http://genetics.bwh.harvard.edu/pph2) to predict their possible functional effect, in a scale from 0.00 (for benign) to 1.00 (for deleterious).
PCR-RFLP
Exons 2 and 3 were amplified by PCR followed by restriction fragment length polymorphism (RFLP). A region of 339 bp from exon 2 of CTSC gene, containing the c.203 T >; G mutation, was amplified with the same primers used for SSCP, and digested with Acu I (1 U for 2 h at 37°C), to identify wild-type T allele (202 and 139 bp fragments) and the mutated G (loss of enzyme recognition site). In the same way, a region of 257 bp from exon 3, spanning the polymorphic site c.458C >; T, was amplified using a modified reverse-primer (5’-TCCTGAGAATTCTTAAGGTCAGC-3’) to generate a recognition site for Pvu II. After digestion (1 U for 2 h at 37°C), normal C allele generates a 235 bp and a 22 bp fragments, while allele T remains uncleaved. PCR products and digestion fragments were visualized in silver-stained polyacrylamide or GelRed-stained (Biotium, Hayward, CA, USA) agarose gels.
HLA typing
Molecular typing of HLA class I (HLA-A, HLA-B) and HLA class II (HLA-DRB1, HLA-DQB1), at low resolution procedure modality, was done by PCR sequence-specific primers (PCR-SSP) technique, with the AB/DR/DQ SSP UniTray kit, (Dynal, Invitrogen) following manufacturer’s instructions, and using GoTaq Flexi DNA Polymerase (Promega, Madison, WI, USA). Products were separated by electrophoresis in 2% agarose. UniMatch Plus 5.0 software (Invitrogen) was used to type the specific alleles.
Statistical analysis
Experimental data was obtained from triplicates and presented as mean ± SD. Comparisons in gene expression and enzymatic activity were performed with ANOVA and Bonferroni multiple comparison test, and P values of less than 0.05 were considered statistically significant. A population sample of 96 individuals was calculated to estimate the frequency of CTSC gene polymorphisms in the population. Differences in allele frequencies were evaluated by χ2 test or Fisher’s exact test. Significant P values (≤ 0.05) were corrected taking into account the number of alleles observed [43]. Odds ratios (OR) with 95% confidence intervals (CI) were used as the measure of association between specific alleles with PLS [44]. Hardy-Weinberg’s equilibrium was calculated by χ2 test. PASW v18.0 (SPSS inc., Chicago, IL, USA) and Arlequin v3.5.1.2 (Swiss National Science Foundation) software packages were used for analysis.
Results
Clinical evaluation
The nine PLS-diagnosed patients described in this work showed keratosis palmoplantaris, with affected knees and elbows, and aggressive periodontitis with variable presentation. They were born (first or second gestation) to non-consanguineous parents. Ages ranged from 1.5 to 61 years (median = 9), 55.56% were female and 44.44% male. Panoramic and periapical radiographs confirmed the alveolar bone loss in periodontal disease, and were used to follow-up with patients after dental treatments. Some patients showed increased susceptibility to pyoderma, such as furuncles and cold abscesses. Standard microbiologic tests showed the presence of Aggregatibacter actinomycetemcomitans and Staphylococcus aureus in lesions. Infections were treated with clindamycin with positive results. Periodontitis and hyperkeratosis severities did not correlate. Hyperkeratosis was treated with topical keratolytics containing salicylic acid and urea, resulting in notable improvement of condition. Routine blood and biochemical laboratory tests were within normal limits.
Case 1P1: 7 year-old female, showed overt palmoplantar hyperkeratosis, with fissures. She had only one deciduous tooth remaining, but her gums still showed signs of swelling and inflammation. She suffered recurrent folliculitis and furuncles. Case 1P2: 11 year-old female, with mild hyperkeratosis. She had severe periodontitis, and suffered recurrent bacterial skin infections. Case 1P3: 61 year-old male (uncle of the father of cases 1P1 and 1P2), with mild hyperkeratosis. He had lost all his permanent teeth, and gums appeared healthy. Case 2P: 14 year-old male (Figure 1A), showed overt hyperkeratosis and severe periodontitis. X-ray films of both hands discarded arachnodactyly, discarding Haims-Munk Syndrome. Case 3P: 6 year-old female, with mild hyperkeratosis. She developed periodontitis just after initiation of primary dentition. Case 4P: 12 year-old female, showed mild hyperkeratosis and severe periodontitis, and lost most of the permanent teeth. Case 5P: 12 year-old female (Figure 1B), with mild hyperkeratosis. She had periodontitis and loss of many permanent teeth, malformed and decaying teeth were observed. Case 6P: 3 year-old male (Figure 1C), showed overt hyperkeratosis. Primary dentition has not occurred yet and gums appeared healthy. Case 7P: 18-month-old male, first gestation, showed mild hyperkeratosis. Primary dentition has not occurred yet and gums appeared healthy.
Figure 1.
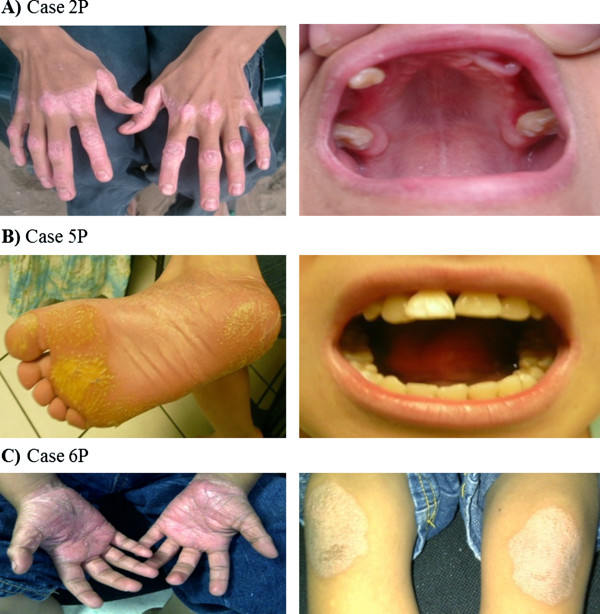
Representative clinical features of patients with Papillon-Lefèvre Syndrome. A) Case 2P: Overt hyperkeratosis in hands (dorsal view). With gingivitis, missing teeth and periodontal pocket. B) Case 5P: Mild hyperkeratosis of sole and mobile teeth. C) Case 6P: Psoriasiform lesions over palms and knees.
CTSC activity and gene expression
Enzymatic activity and gene expression (Table 1) were measured in PLS patients (n = 7), their relatives (n = 17) and unrelated healthy individuals (n = 8). Patients showed 14.89% (±4.96) of the enzymatic activity observed by controls (100%), while parents and siblings showed 47.60% (±19.74) and 38.42% (±24.90), respectively. This represents an enzymatic deficiency of nearly 85% in patients, and nearly 50% in parents and siblings.
Table 1.
Percentage of cathepsin C activity, gene expression, and genotypes in patients with Papillon-Lefèvre Syndrome and their relatives
| Family | Subject | % | SD | P | CE | SD | P | c.203 T >; G | c.458C >; T |
|---|---|---|---|---|---|---|---|---|---|
|
1 |
1P1 |
13.45 |
2.97 |
0.0001 |
0.25 |
0.05 |
0.0070 |
G/G |
C/C |
| |
1P2 |
- |
- |
- |
- |
- |
- |
G/G |
C/C |
| |
1P3 |
- |
- |
- |
- |
- |
- |
G/G |
C/C |
| |
1 F |
46.99 |
1.40 |
0.0041 |
1.62 |
0.05 |
0.0110 |
T/G |
C/C |
| |
1 M |
67.93 |
2.76 |
0.0489 |
0.31 |
0.11 |
0.0170 |
T/G |
C/C |
|
2 |
2P |
11.03 |
4.41 |
0.0061 |
1.81 |
0.09 |
0.0750 |
T/G |
C/T |
| |
2 F |
59.22 |
5.76 |
0.1580 |
2.99 |
1.07 |
0.1190 |
T/G |
C/C |
| |
2 M |
42.91 |
5.01 |
0.0561 |
0.42 |
0.09 |
0.1190 |
T/T |
C/T |
| |
2B1 |
39.26 |
3.25 |
0.0438 |
0.37 |
0.06 |
0.1000 |
T/T |
C/T |
| |
2B2 |
- |
- |
- |
1.33 |
0.01 |
0.1440 |
T/T |
C/C |
|
3 |
3P |
24.16 |
1.33 |
0.0000 |
1.39 |
0.66 |
0.4280 |
G/G |
C/C |
| |
3 M |
55.76 |
0.90 |
0.0000 |
0.44 |
0.07 |
0.0070 |
T/G |
C/C |
|
4 |
4P |
14.97 |
0.10 |
0.0013 |
0.19 |
0.01 |
0.0080 |
T/G |
C/T |
| |
4 M |
27.75 |
0.10 |
0.0018 |
1.40 |
0.08 |
0.0470 |
T/T |
C/T |
| |
4S |
72.64 |
0.16 |
0.0106 |
0.57 |
0.11 |
0.0560 |
T/G |
C/C |
| |
4B |
26.93 |
0.17 |
0.0017 |
0.29 |
0.11 |
0.0210 |
T/G |
C/C |
|
5 |
5P |
15.32 |
2.12 |
0.0000 |
0.15 |
0.00 |
0.0570 |
G/G |
C/C |
| |
5 F |
77.74 |
12.07 |
0.0608 |
2.55 |
1.81 |
0.2900 |
T/G |
C/C |
| |
5 M |
56.50 |
1.26 |
0.0016 |
0.46 |
0.05 |
0.1260 |
T/G |
C/C |
|
6 |
6P |
10.42 |
0.86 |
0.0000 |
0.34 |
0.12 |
0.1030 |
G/G |
C/C |
| |
6 F |
16.92 |
2.01 |
0.0000 |
1.37 |
0.57 |
0.0177 |
T/G |
C/C |
| |
6 M |
24.23 |
7.85 |
0.0000 |
1.32 |
0.39 |
0.0123 |
T/G |
C/C |
| |
6S |
14.84 |
3.61 |
0.0000 |
0.34 |
0.21 |
0.0002 |
T/G |
C/C |
|
7 |
7P |
- |
- |
- |
2.15 |
0.10 |
0.0410 |
G/G |
C/C |
| |
7 F |
- |
- |
- |
0.57 |
0.18 |
0.2220 |
T/G |
C/C |
| 7 M | - | - | - | 1.06 | 0.79 | 0.8020 | T/G | C/C |
%, percentage of enzymatic activity in relation to healthy individuals (set as 100%). CE, cathepsin C expression in relation to healthy individuals (set up as 1.0). P, p value from comparison to healthy individuals. (−), sample not available. Subjects: P, patient; F, father; M, mother; B, brother: S, sister. SD, standard deviation.
Quantification of CTSC expression showed no significant differences (P >; 0.05) among all groups. Relative expression in comparison to controls (set up as 1.0) was 0.90 (±0.86), 1.21 (±0.86) and 0.58 ±0.43 for patients, parents and siblings, respectively.
Mutational analyzes of CTSC
The coding sequences and splice junctions of CTSC gene were analyzed by SSCP in nine PLS patients and their relatives. There were changes in reannealed products in exons 2 and 3 from patients and relatives (Figure 2A). As expected, there were no changes in any exon in eight unrelated healthy individuals. On sequencing (nt 177 to 228 of exon 2, and nt 433 to 484 of exon 3), nucleotide changes were found at positions 203 (T >; G) of exon 2 and 458 (C >; T) of exon 3 (Figure 2B). All sequence variants were submitted to GenBank nucleotide database (JQ763324-77) and to dbSNPs (rs199474831).
Figure 2.
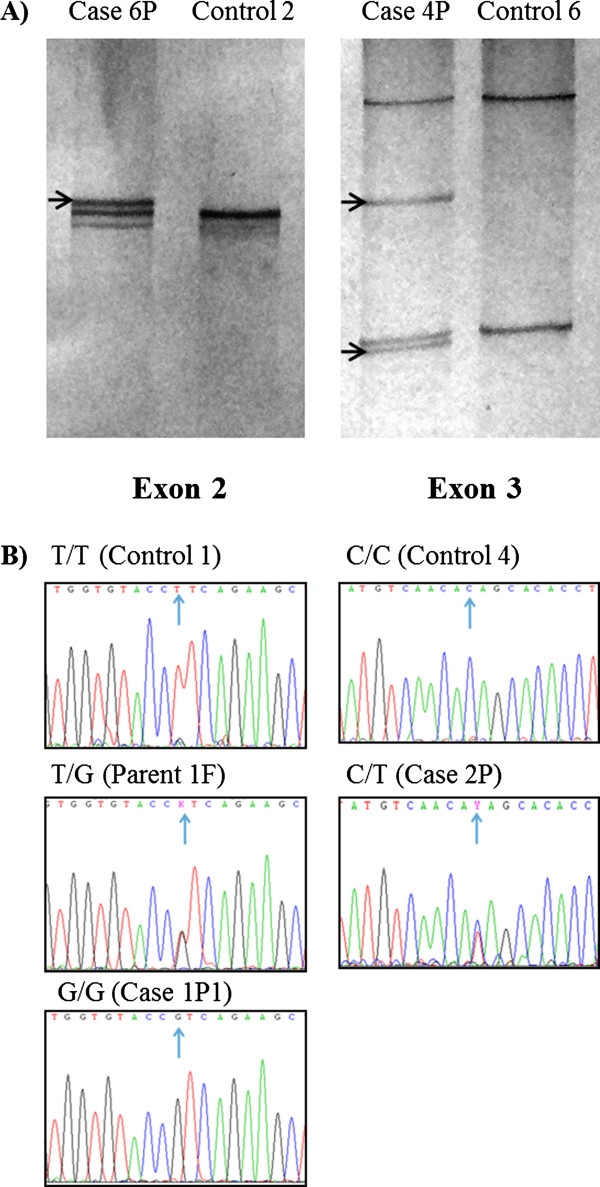
Mutational analysis of Cathepsin C gene. A) Representative results of CTSC gene exon scanning by single-strand conformation polymorphism (SSCP). Reannealed products, in silver-stained polyacrylamide gels, of exons 2 (left) and 3 (right) in cases 6P and 4P showed differences (arrows) against controls 2 and 6. B) Representative electropherograms of exon 2 showing genotypes (arrows) homozygote normal T/T (control 1), heterozygote T/G (parent 1 F) and homozygote mutated G/G (case 1P1), for mutation c.203 T >; G. Exon 3, showing genotypes (arrows) homozygote normal C/C (control 4) and heterozygote C/T (case 2P) for polymorphism c.458C >; T.
Cases 2P and 4P were found to be compound heterozygous for mutations c.203 T >; G and c.458C >; T, while all other patients were found to be homozygous (G/G) for mutation c.203 T >; G. All parents and four siblings were carriers of either c.203 T >; G or c.458C >; T, but not affected (Table 1).
Protein homology analysis
DNA sequence analysis revealed that c.203 T >; G mutation is located in the exclusion domain of DPPI, resulting in a change of the amino acid Leucine for Arginine at residue 68 (p.Leu68Arg). The presence of Arginine at this site may interfere with the insertion of the dipeptide substrate, subsequently blocking the enzymatic activity. In fact, homology analysis showed that Leucine 68 is a highly conserved residue, and analysis of this substitution with the PolyPhen-2 software predicted a damaging mutation, with a score of 1.00.
The substitution c.458C >; T results in a change of the amino acid Threonine for Isoleucine at residue 153 (p.Thr153Ile); this residue is not highly conserved in evolution and is located in the pro-region of heavy chain, which is removed during DPPI activation. PolyPhen-2 software predicted a probably benign mutation with a score of 0.00.
Population frequencies of CTSC gene polymorphisms
PCR-SSCP analyzes of CTSC exons 2 and 3 were done in individuals from the population (n = 200). Positive samples were confirmed for mutations c.203 T >; G and c.458C >; T by PCR-RFLPs (Figure 3).
Figure 3.
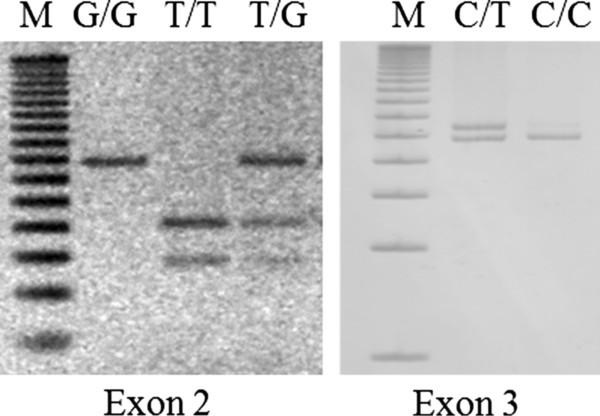
Gene polymorphisms analysis. PCR-RFLPs products of genotypes homozygous normal T/T, heterozygous T/G and homozygous mutated G/G for mutation c.203 T >; G of exon 2 (left). Genotypes homozygous normal C/C and heterozygous C/T, for c.458C >; T polymorphism of exon 3 (right). M, 50 bp DNA ladder (Invitrogen).
Allelic frequencies in PLS patients (n = 9), their relatives (n = 17) and controls were 88.89%, 38.24% and 0.25% for G (c.203 T >; G), respectively; and 11.11%, 8.82% and 9.00% for T (c.458C >; T). Genotypes frequencies of c.203 T >; G were 77.78%, 0.0% and 0.0% for G/G; 22.22%, 76.47% and 0.5% for T/G; and 0.0%, 23.53% and 99.5% for T/T. For c.458C >; T genotypes, frequencies were 22.22%, 17.65% and 18.0% for C/T; and 77.78%, 82.35, and 82.0% for C/C (Table 2). The distributions of c.203 T >; G and c.458C >; T polymorphisms in the groups were in Hardy-Weinberg equilibrium (all P >; 0.05).
Table 2.
Allelic (af) and genotypic (gf) frequencies of cathepsin C polymorphisms in patients with Papillon-Lefèvre Syndrome, their relatives and controls
| Polymorphism | Patients | Relatives | Controls | |||
|---|---|---|---|---|---|---|
|
c.203 T >; G |
|
|
|
|
|
|
| Alleles |
n = 18 |
af |
n = 34 |
af |
n = 400 |
af |
| G |
16 |
0.8889 |
13 |
0.3824 |
1 |
0.0025 |
| T |
2 |
0.1111 |
21 |
0.6176 |
399 |
0.9975 |
| Genotypes |
n = 9 |
gf |
n = 17 |
gf |
n = 200 |
gf |
| G/G |
7 |
0.7778 |
0 |
0.0000 |
0 |
0.0000 |
| T/G |
2 |
0.2222 |
13 |
0.7647 |
1 |
0.0050 |
| T/T |
0 |
0.0000 |
4 |
0.2353 |
199 |
0.9950 |
|
c.458C >; T |
|
|
|
|
|
|
| Alleles |
n = 18 |
af |
n = 34 |
af |
n = 400 |
af |
| T |
2 |
0.1111 |
3 |
0.0882 |
36 |
0.0900 |
| C |
16 |
0.8889 |
31 |
0.9118 |
364 |
0.9100 |
| Genotypes |
n = 9 |
gf |
n = 17 |
gf |
n = 200 |
gf |
| T/T |
0 |
0.0000 |
0 |
0.0000 |
0 |
0.0000 |
| C/T |
2 |
0.2222 |
3 |
0.1765 |
36 |
0.1800 |
| C/C | 7 | 0.7778 | 14 | 0.8235 | 164 | 0.8200 |
For c.203 T >; G there were significant differences in allelic (P = 0.0000; OR = 3192.00; 95% CI = 274.92-37061.06) and genotypic (P = 0.0000) frequencies between patients and population controls and in alleles (P = 0.0000; OR = 247.00; 95% CI = 30.83-1978.58) and genotypes (P = 0.0000) between patients’ relatives and controls. For c.458C >; T alleles and genotype distributions there were no significant differences between all groups (all P >; 0.05).
The initial calculated sample size for population analysis was 96, however, an individual carrying a heterozygous T/G genotype for c.203 T >; G was found, and therefore a final sample of 200 individuals was used.
Immunogenetics
HLA alleles were typed in PLS patients (n = 9), their relatives (n = 15) and population controls (n = 41) (Table 3). The more common HLA alleles found in patients and controls were 33.33% vs. 14.63% (P = 0.0873; OR = 2.92; 95% CI = 0.92-9.26) for HLA-A*24, and 16.67% vs. 29.27% (P = 0.2755) for A*02. HLA-B*35 was found in 33.33% vs. 20.73% (P = 0.2500), and B*44 was found in 11.11% vs. 10.98% (P = 0.9867). Frequencies of HLA-DRB1*11 were 33.33% vs. 7.32% (P = 0.0071; OR = 6.33; 95% CI = 1.75-22.89), and 22.22% vs. 24.39% (P = 0.8454) for DRB1*04. Frequencies for HLA-DR53, DR52 and DR51 were 44.44% vs. 41.46% (P = 0.8165), 33.33% vs. 21.95% (P = 0.3059), and 22.22% vs. 24.39% (P = 0.8454), respectively. For HLA-DQB1*0301, frequencies were 33.33% vs. 24.39% (P = 0.2715), and 27.08% vs. 17.07% (P = 0.1745) for DQB1*0302. Significant deviations from Hardy–Weinberg equilibrium were not detected in the distribution of HLA alleles (all P >; 0.05).
Table 3.
Allele frequencies (af) of the more common HLA class I and II molecules found in patients with Papillon-Lefèvre Syndrome, their relatives and controls
|
Alleles |
Patients |
|
Relatives |
|
Controls |
|
|---|---|---|---|---|---|---|
| (N = 18) | (N = 30) | (N = 82) | ||||
|
HLA-A |
|
af |
|
af |
|
af |
| A*24 |
6 |
0.3333 |
8 |
0.2667 |
12 |
0.1463 |
| A*02 |
3 |
0.1667 |
4 |
0.1333 |
24 |
0.2927 |
| A*33 |
3 |
0.1667 |
2 |
0.0667 |
1 |
0.0122 |
| A*31 |
2 |
0.1111 |
5 |
0.1667 |
8 |
0.0976 |
| A*03 |
2 |
0.1111 |
2 |
0.0667 |
2 |
0.0244 |
|
HLA-B |
|
|
|
|
|
|
| B*35 |
6 |
0.3333 |
9 |
0.3000 |
17 |
0.2073 |
| B*65 |
3 |
0.1667 |
2 |
0.0667 |
2 |
0.0244 |
| B*44 |
2 |
0.1111 |
2 |
0.0667 |
9 |
0.1098 |
| B*51 |
1 |
0.0556 |
2 |
0.0667 |
4 |
0.0488 |
| B*60 |
1 |
0.0556 |
2 |
0.0667 |
3 |
0.0366 |
|
HLA-DRB1 |
|
|
|
|
|
|
| DRB1*11 |
6 |
0.3333 |
6 |
0.2000 |
6 |
0.0732 |
| DRB1*04 |
4 |
0.2222 |
9 |
0.3000 |
20 |
0.2439 |
| DRB1*01 |
3 |
0.1667 |
6 |
0.2000 |
13 |
0.1585 |
| DRB1*15 |
3 |
0.1667 |
4 |
0.1333 |
3 |
0.0366 |
| DRB1*07 |
1 |
0.0556 |
2 |
0.0667 |
8 |
0.0976 |
|
HLA-DQB1 |
|
|
|
|
|
|
| DQB1*0301 |
7 |
0.3889 |
9 |
0.3000 |
20 |
0.2439 |
| DQB1*0302 |
4 |
0.2222 |
9 |
0.3000 |
14 |
0.1707 |
| DQB1*0501 |
4 |
0.2222 |
4 |
0.1333 |
9 |
0.1098 |
| DQB1*0601 |
2 |
0.1111 |
2 |
0.0667 |
8 |
0.0976 |
| DQB1*0201 | 1 | 0.0556 | 5 | 0.1667 | 14 | 0.1707 |
Discussion
Here we described the clinical profile of nine patients (belong to seven different families) with Papillon-Lefèvre Syndrome (PLS), from the northwestern Mexican state of Sinaloa. They showed keratosis palmoplantaris and aggressive periodontitis with variable expression, which are the classical features of this syndrome [45]. In contrast to the nearly 30% of consanguinity of PLS cases reported in different countries [2,3], our PLS patients were born to non-consanguineous parents, and their families were not related.
PLS patients showed a DPPI activity reduction up to 85%, while parents and siblings showed a partial reduction of nearly 50%. This enzymatic deficiency was not due to diminished CTSC gene expression levels, as no significant differences were found between all groups. In agreement with our findings, more than 80% of all PLS cases reported show CTSC deficiency due to gene mutations [14,34]. Zhang et al. found less than 10% of the normal enzymatic activity in PLS patients and 50% in parents [46]. To our knowledge, there are no reports of CTSC messenger quantification using qPCR technique for this purpose in the literature.
Cases 2P and 4P were found to be compound heterozygous for c.203 T >; G and c.458C >; T, while all other cases were found to be homozygous for c.203 T >; G. Of note, all parents, as well as four siblings, carried either c.203 T >; G or c.458C >; T. These findings could explain the reduction in DPPI activity observed in these subjects, and seems to confirm the autosomal recessive mode of inheritance of PLS.
According to the Human Gene Mutation Database (HGMD), 77 variants of the CTSC gene have been reported. The c.203 T >; G substitution we report here, is a novel loss-of function mutation, and associates with the diminished enzymatic activity observed in our PLS patients. In accordance, frequency of allele G (c.203 T >; G) was higher in patients and relatives in comparison to population controls (all P ≤ 0.05).
The substitution c.458C >; T has been reported as both a common polymorphism in some populations, and as causal mutation in PLS patients [47-49]. c.458C >; T has been reported as causal mutation in one Australian PLS patient (compound heterozygous for c.458C >; T and c.199_222del) [49]. However, a genotypic study for the c.458C >; T polymorphism in the Spanish population reported frequencies of 81% for homozygote normal and 19% for heterozygote [47]. In accordance, allele T (c.458C >; T) was found relatively frequent in our population (9%), with no significant differences between the studied groups, and PolyPhen-2 software predicted a probably benign mutation. Therefore, the results of cases 2P and 4P remain inconclusive as the pathogenic variant other than c.203 T >; G remains unidentified in the coding region.
Some patients in this study showed increased susceptibility to bacterial skin infections. This condition seems to be due to an impairment of the immune system, which is involved in the pathoetiology of PLS. Therefore, we genotyped HLA class I and II alleles of PLS patients, their relatives and controls to evaluate any particular HLA association with this disease. We found fourteen HLA-A, twenty-nine HLA-B, twelve HLA-DRB1 and six HLA-DQB1 alleles (online Additional file 1), with HLA-A*02, B*35, DRB1*04, and DQB1*0301 being the most frequent in the population. A common HLA-A/B/DR/DP haplotype was not found in the families, confirming the lack of relationship among them.
With exception of HLA-DRB1*11, our results are in agreement with other reports showing no particular association of HLA alleles with PLS [32,34]. We found that HLA-DRB1*11 allele was significantly more frequent (P = 0.0071) in our PLS patients than in controls (33.33% vs. 7.32%, respectively), resulting in an estimated relative risk index of 6.33. Interestingly, this allele has been also associated with scleroderma in Caucasian patients [50]. HLA-A*24(9), HLA-B*35, DRB1*04 and DR51 showed higher frequency in our PLS patients than in controls, but the differences were not statistically significant. HLA-A*24(9) DRB1*04 and DR51 alleles have been associated with different kinds of periodontitis in other countries [33,51-53].
Mexicans, Mestizos in general, have a high degree of genetic heterogeneity due to centuries of mixing with Native Americans, Africans and Europeans. In the South and Central parts of Mexico, the predominant HLA type is more Amerindian than European or African, but in Sinaloa, half of the most common HLA haplotypes are believed to be of European origin. Allelic frequencies of the population found in this work were in accordance with a previous report [54]. An important difference is that our more extensive molecular study had included, for the first time, the side-by-side comparison of PLS families with unrelated, healthy controls.
Although the exact pathogenesis of PLS is still unknown, biochemical and genetic analysis allows for genetic counseling and opportune treatment, especially for those patients with familial history of this disease. Furthermore, some authors believe that if treatment is started at an early age, patients would possibly have normal adult dentition [55]. One of the studied cases (7P) is only 18-months-old, and will be under treatment and observation to avoid serious complications.
While in México the general prevalence of this syndrome is still unknown, and the number of reported cases is very low, the finding so far of 20 patients in the state of Sinaloa highlights the need for more studies of this nature. Due to the concentrated number of PLS patients found in our population, none of them previously described, is would be possible to argue that there is a founder effect for CTSC gene mutation. An haplotype analysis of DNA markers in CTSC gene region can help to determine if c.203 T >; G mutation has this founder effect.
Conclusion
In conclusion, our work describes the immunogenetics, and the biochemical and molecular characterization of CTSC in nine PLS patients and their families. We describe the identification of a novel loss-of function mutation in CTSC gene (c.203 T >; G) causing PLS in Mexican patients; and the association of the HLA-DRB1*11 allele with this syndrome. The study of more PLS patients may give more insights into the etiology of the disease as well as its prevalence in México.
Consent
Written consent was obtained from pateints’ parents for publication of this work.
Competing interests
Authors declare that they have no competing interests.
Authors’ contributions
LOFC, JEDA and JDMS diagnosed and treated the patients, and gave genetic counseling to the parents. JGRQ and JGRN conducted laboratory and biochemical test and interpreted results. EAM and MAM collected the data relative to this report. RPR and JEDA wrote the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Complete HLA list. Allele frequencies (af) of HLA class I and II molecules, in studied subjects from Sinaloa, México.
Contributor Information
José G Romero-Quintana, Email: geovannirom@yahoo.com.mx.
Luis O Frías-Castro, Email: lofc57@gmail.com.
Eliakym Arámbula-Meraz, Email: eliakymarambula@hotmail.com.
Maribel Aguilar-Medina, Email: maribela2@excite.com.
Jesús E Dueñas-Arias, Email: jedanet@yahoo.com.
Jesús D Melchor-Soto, Email: jedanet@yahoo.com.
José G Romero-Navarro, Email: gromero@uas.uasnet.mx.
Rosalío Ramos-Payán, Email: ramospayan@yahoo.com.mx.
Acknowledgements
This work was supported by CONACYT (106152) and PROFAPI-UAS grants. Authors thank Dr. Sanchez-Schmitz G. (The Division of Infectious Diseases, Children's Hospital Boston, and Harvard Medical School, Harvard University) and Dr. Julio Granados (División de Inmunogenética, Instituto Nacional de Ciencias Médicas y Nutrición. México DF), for their critical readings.
References
- Papillon MM, Lefèvre P. Deux cas de kératodermie palmaire et plantaire symetrique familiale (maladie de Méléda) chez le frère et la soeur. Coexistence dans les deux cas d’altérations dentaires graves. Bull Soc Fr Dermatol Syph. 1924;31:82–87. [Google Scholar]
- Gorlin RJ, Sedano H, Anderson VE. The syndrome of palmar-plantar hyperkeratosis and premature periodontal destruction of teeth. J Pediatr. 1964;65:895–908. doi: 10.1016/S0022-3476(64)80014-7. [DOI] [PubMed] [Google Scholar]
- Cury VF, Costa JE, Gomez RS, Boson WL, Loures CG, Marco LD. A novel mutation of the cathepsin C gene in Papillon-Lefevre syndrome. J Periodontol. 2002;73:307–312. doi: 10.1902/jop.2002.73.3.307. [DOI] [PubMed] [Google Scholar]
- Gorlin R. Of palms, soles, and gums. J Med Genet. 2000;37(2):81–82. doi: 10.1136/jmg.37.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MMJ, Levin LS. Syndromes of the head and neck. 3. New York: Oxford University Press; 1990. pp. 853–854. [Google Scholar]
- Posteraro A. Papillon-Lefèvre syndrome. J Ala Dent Assoc. 1992;76(3):16–19. [PubMed] [Google Scholar]
- Khandpur S, Reddy BS. Papillon-Lefèvre syndrome with pyogenic hepatic abscess: a rare association. Pediatr Dermatol. 2001;18(1):45–47. doi: 10.1046/j.1525-1470.2001.018001045.x. [DOI] [PubMed] [Google Scholar]
- Almuneef M, Al Khenaizan S, Al Ajaji S, Al-Anazi A. Pyogenic liver abscess and Papillon-Lefèvre syndrome: not a rare association. Pediatrics. 2003;111(1):85–88. doi: 10.1542/peds.111.1.e85. [DOI] [PubMed] [Google Scholar]
- Tosti A, Manuzzi P, Bardazzi F, Costa A. Is etretinate dangerous in Papillon-Lefèvre syndrome? Dermatologica. 1988;176(3):148–150. doi: 10.1159/000248692. [DOI] [PubMed] [Google Scholar]
- Oğuzkurt P, Tanyel FC, Büyükpamukçu N, Hiçsönmez A. Increased risk of pyogenic liver abscess in children with Papillon-Lefevre syndrome. J Pediatr Surg. 1996;31(7):955–956. doi: 10.1016/S0022-3468(96)90420-0. [DOI] [PubMed] [Google Scholar]
- Borroni G, Pagani A, Carcaterra A, Pericoli R, Gabba P, Marconi M. Immunological alterations in a case of Papillon-Lefèvre syndrome with recurrent cutaneous infections. Dermatologica. 1985;170:27–30. doi: 10.1159/000249491. [DOI] [PubMed] [Google Scholar]
- Lundgren T, Renvert S. Periodontal treatment of patients with Papillon-Lefèvre syndrome: a 3-year follow-up. J Clin Periodontol. 2004;31:933–938. doi: 10.1111/j.1600-051X.2004.00591.x. [DOI] [PubMed] [Google Scholar]
- Lundgren T, Crossner C-G, Ocaron Ran Twetman S, Ullbro C. Systemic retinoid medication and periodontal health in patients with Papillon-Lefèvre syndrome. J Clin Periodontol. 1996;23:176–179. doi: 10.1111/j.1600-051X.1996.tb02073.x. [DOI] [PubMed] [Google Scholar]
- Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG. et al. Loss-of-function mutations in the cathepsin C gene result in priodontal disease and palmoplantar keratosis. Nat Gen. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Bowden DW, Michalec MD, Callison SA, Walker SJ, Zhang Y, Firatli E. Mutations of the cathepsin C gene are responsible for Papillon-Lefèvre syndrome. J Med Genet. 1999;36(12):881–887. [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Michalec MD, Zhang Y, Firatli E, Van Dyke TE, Stabholz A, Zlotogorski A, Shapira L, Soskolne WA. Haim-Munk syndrome and Papillon-Lefèvre syndrome are allelic mutations in cathepsin C. J Med Genet. 2000;37(2):88–94. doi: 10.1136/jmg.37.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Michalec MD, Zhang Y, Marazita ML, Cooper M, Yassin OM, Nusier M, Walker S. Localisation of a gene for prepubertal periodontitis to chromosome 11q14 and identification of a cathepsin C gene mutation. J Med Genet. 2000;37(2):95–101. doi: 10.1136/jmg.37.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao NV, Rao GV, Hoidal JR. Human dipeptidyl-peptidase I. Gene characterization, localization, and expression. J Biol Chem. 1997;272(15):10260–10265. doi: 10.1074/jbc.272.15.10260. [DOI] [PubMed] [Google Scholar]
- Cigić B, Dahl SW, Pain RH. The residual pro-part of cathepsin C fulfills the criteria required for an intramolecular chaperone in folding and stabilizing the human proenzyme. Biochemistry. 2000;39:12382–12390. doi: 10.1021/bi0008837. [DOI] [PubMed] [Google Scholar]
- Dahl SW, Halkier T, Lauritzen C, Dolenc I, Pedersen J, Turk V, Turk B. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry. 2001;40:1671–1678. doi: 10.1021/bi001693z. [DOI] [PubMed] [Google Scholar]
- Paris A, Strukelj B, Pungercar J, Renko M, Dolenc I, Turk V. Molecular cloning and sequence analysis of human preprocathepsin C. FEBS Lett. 1995;369:326–330. doi: 10.1016/0014-5793(95)00777-7. [DOI] [PubMed] [Google Scholar]
- Nakano A, Nomura K, Nakano H, Ono Y, LaForgia S, Pulkkinen L, Hashimoto I, Uitto J. Papillon-Lefèvre syndrome: mutations and polymorphisms in the cathepsin C gene. J Invest Dermatol. 2001;116(2):339–343. doi: 10.1046/j.1523-1747.2001.01244.x. [DOI] [PubMed] [Google Scholar]
- Turk D, Janjić V, Stern I, Podobnik M, Lamba D, Dahl SW, Lauritzen C, Pedersen J, Turk V, Turk B. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001;20(23):6570–6582. doi: 10.1093/emboj/20.23.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolenc I, Turk B, Pungercic G, Ritonja A, Turk V. Oligomeric structure and substrate induced inhibition of human cathepsin C. J Biol Chem. 1995;270(37):21626–21631. doi: 10.1074/jbc.270.37.21626. [DOI] [PubMed] [Google Scholar]
- Wolters PJ, Raymond WW, Blount JL, Caughey GH. Regulated expression, processing, and secretion of dog mast cell dipeptidyl peptidase I. J Biol Chem. 1998;273:15514–15520. doi: 10.1074/jbc.273.25.15514. [DOI] [PubMed] [Google Scholar]
- Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. 1999;96(15):8627–8632. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepel C, Brömme D, Herzog V, Brix K. Cathepsin K in thyroid epithelial cells: sequence, localization and possible function in extracellular proteolysis of thyroglobulin. J Cell Sci. 2000;113(24):4487–4498. doi: 10.1242/jcs.113.24.4487. [DOI] [PubMed] [Google Scholar]
- McDonald JK, Zeitman BB, Reilly TJ, Ellis S. New observations on the substrate specificity of cathepsin C (dipeptidyl aminopeptidase I). Including the degradation of beta-corticotropin and other peptide hormones. J Biol Chem. 1969;244:2693–2709. [PubMed] [Google Scholar]
- Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. 2004;173:7277–7281. doi: 10.4049/jimmunol.173.12.7277. Baltimore, Md: 1950. [DOI] [PubMed] [Google Scholar]
- Nuckolls GH, Slavkin HC. Paths of glorious proteases. Nat Genet. 1999;23:378–380. doi: 10.1038/70472. [DOI] [PubMed] [Google Scholar]
- Abbas AK, Lichtman AH, Pillai S. Complejo Principal de Histocompatibilidad. 6. St. Louis, Mo: El Sevier Saunders; 2009. pp. 96–112. (Inmunología Celular y Molecular). [Google Scholar]
- Farjadian S, Kiyanimanesh N, Abbaszadegan A, Lotfazar M. HLA class I gene polymorphism in Iranian patients with Papillon-Lefevre Syndrome. Iran J Immunol. 2007;4:241–245. doi: 10.22034/iji.2007.17204. [DOI] [PubMed] [Google Scholar]
- Amer A, Singh G, Darke C, Dolby AE. Association between HLA antigens and periodontal disease. Tissue Antigens. 1988;31:53–58. doi: 10.1111/j.1399-0039.1988.tb02063.x. [DOI] [PubMed] [Google Scholar]
- Farjadian S, Lotfazar M, Ghaderi A. Analysis of human leukocyte antigen class II gene polymorphism in Iranian patients with Papillon-Lefevre syndrome: a family study. Iran J Immunol. 2008;5:171–176. doi: 10.22034/iji.2008.17163. [DOI] [PubMed] [Google Scholar]
- Sada Tamayo JJ, Villagrán Uribe J. Experiencias en niños con el Síndrome Papillón-Lefevre. Rev Mex Pediatr. 2007;74:3. [Google Scholar]
- Maes MB, Lambeir AM, Gilany K, Senten K, Van der Veken P, Leiting B, Augustyns K, Scharpe S, De Meester I. Kinetic investigation of human dipeptidyl peptidase II (DPPII)-mediated hydrolysis of dipeptide derivatives and its identification as quiescent cell proline dipeptidase (QPP)/dipeptidyl peptidase 7 (DPP7) Biochem J. 2005;386:315–324. doi: 10.1042/BJ20041156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadot M, Colmant C, Wattiaux-De Coninck S, Wattiaux R. Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem J. 1984;219:965–970. doi: 10.1042/bj2190965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Medina M, Ramos-Payan R, Arambula-Meraz E, Sanchez-Torres L, Favila-Castillo L. Parasitaemia levels in Plasmodium chabaudi infected-mice modify IFN-gamma and IL-10 expression after a homologous or heterologous challenge. Parasite Immunol. 2010;32:267–274. doi: 10.1111/j.1365-3024.2009.01183.x. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. San Diego, Calif. [DOI] [PubMed] [Google Scholar]
- Gustincich S, Manfioletti G, Del Sal G, Schneider C, Carninci P. A fast method for high-quality genomic DNA extraction from whole human blood. Biotechniques. 1991;11(3):298–300–302. [PubMed] [Google Scholar]
- Hongyo T, Buzard GS, Calvert RJ, Weghorst CM. 'Cold SSCP': a simple, rapid and non-radioactive method for optimized single-strand conformation polymorphism analyses. Nucleic Acids Res. 1993;21(16):3637–3642. doi: 10.1093/nar/21.16.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, Cafarchia C, Beveridge I, Zhu X. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1:3121–3128. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Furusho JK, Uscanga LF, Vargas-Alarcon G, Rodriguez-Perez JM, Zuniga J, Granados J. Polymorphisms in the promoter region of tumor necrosis factor alpha (TNF-alpha) and the HLA-DRB1 locus in Mexican mestizo patients with ulcerative colitis. Immunol Lett. 2004;95:31–35. doi: 10.1016/j.imlet.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Woolf B. On estimating the relation between blood group and disease. Ann Hum Genet. 1955;19:251–253. doi: 10.1111/j.1469-1809.1955.tb01348.x. [DOI] [PubMed] [Google Scholar]
- Ullbro C, Crossner CG, Nederfors T, Alfadley A, Thestrup-Pedersen K. Dermatologic and oral findings in a cohort of 47 patients with Papillon-Lefevre syndrome. J Am Acad Dermatol. 2003;48:345–351. doi: 10.1067/mjd.2003.197. [DOI] [PubMed] [Google Scholar]
- Zhang YHP, Moretti AJ, Bouwsma OJ, Fisher EM, Dudlicek L, Pettenati MJ, Hart TC. Biochemical and mutational analyses of the cathepsin c gene (CTSC) in three North American families with Papillon Lefèvre syndrome. Hum Mutat. 2002;20(1):75–80. doi: 10.1002/humu.9040. [DOI] [PubMed] [Google Scholar]
- Allende LM G-PM, Moreno A, Corell A, Carasol M, Martínez-Canut P, Arnaiz-Villena A. Cathepsin C gene: First compound heterozygous patient with Papillon-Lefèvre syndrome and a novel symptomless mutation. Hum Mutat. 2001;17(2):152–153. doi: 10.1002/1098-1004(200102)17:2<152::AID-HUMU10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Lefèvre CB-BC, Jobard F, Bouadjar B, Stalder JF, Cure S, Hoffmann A, Prud'Homme JF, Fischer J. Novel point mutations, deletions, and polymorphisms in the cathepsin C gene in nine families from Europe and North Africa with Papillon-Lefèvre syndrome. J Invest Dermatol. 2001;117(6):1657–1661. doi: 10.1046/j.0022-202x.2001.01595.x. [DOI] [PubMed] [Google Scholar]
- Hart PS, Zhang Y, Firatli E, Uygur C, Lotfazar M, Michalec MD, Marks JJ, Lu X, Coates BJ, Seow WK. et al. Identification of cathepsin C mutations in ethnically diverse papillon-Lefèvre syndrome patients. J Med Genet. 2000;37(12):927–32. doi: 10.1136/jmg.37.12.927. 2000, 37(12):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladman DD, Kung TN, Siannis F, Pellett F, Farewell VT, Lee P. HLA markers for susceptibility and expression in scleroderma. Journal Rheumatol. 2005;32:1481–1487. [PubMed] [Google Scholar]
- Klouda PT, Porter SR, Scully C, Corbin SA, Bradley BA, Smith R, Davies RM. Association between HLA-A9 and rapidly progressive periodontitis. Tissue Antigens. 1986;28:146–149. doi: 10.1111/j.1399-0039.1986.tb00474.x. [DOI] [PubMed] [Google Scholar]
- Firatli E, Kantarci A, Cebeci I, Tanyeri H, Sonmez G, Carin M, Tuncer O. Association between HLA antigens and early onset periodontitis. J Clin Periodontol. 1996;23:563–566. doi: 10.1111/j.1600-051X.1996.tb01825.x. [DOI] [PubMed] [Google Scholar]
- Stein J, Reichert S, Gautsch A, Machulla HK. Are there HLA combinations typical supporting for or making resistant against aggressive and/or chronic periodontitis? J Periodontal Res. 2003;38:508–517. doi: 10.1034/j.1600-0765.2003.00683.x. [DOI] [PubMed] [Google Scholar]
- Barquera R, Zuniga J, Hernandez-Diaz R, Acuna-Alonzo V, Montoya-Gama K, Moscoso J, Torres-Garcia D, Garcia-Salas C, Silva B, Cruz-Robles D. et al. HLA class I and class II haplotypes in admixed families from several regions of Mexico. Mol Immunol. 2008;45:1171–1178. doi: 10.1016/j.molimm.2007.07.042. [DOI] [PubMed] [Google Scholar]
- Nazzaro V, Blanchet-Bardon C, Mimoz C, Revuz J, Puissant A. Papillon-Lefèvre syndrome. Ultrastructural study and successful treatment with acitretin. Arch Dermatol. 1988;124(4):533–539. doi: 10.1001/archderm.1988.01670040035018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete HLA list. Allele frequencies (af) of HLA class I and II molecules, in studied subjects from Sinaloa, México.