

Abstract
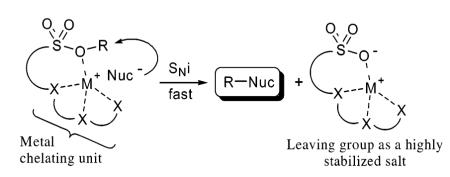
The synthesis and unique reactivity of a series of arylsulfonate-based nucleophile assisting leaving groups (NALG) containing oligomeric ether units (including crown ethers) attached to the arylsulfonyl ring in the ortho orientation are described. The reactions of a variety of these ether-containing alkyl sulfonates with metal halides proceeded at substantially greater rates than electronically similar sulfonates. These ether-containing leaving groups also displayed marked selectivity for lithium halides relative to the corresponding sodium and potassium salts in nucleophilic displacement reactions.
Introduction
Although reactions mediated by nucleophilic mechanisms have proved overwhelmingly useful in synthesis, prohibitively slow reaction kinetics are often observed with sterically hindered secondary substrates and weak or poorly soluble nucleophiles. A number of strategies are routinely employed to expedite nucleophilic reactions including the use of highly stabilized leaving groups1 to increase the electrophilicity of the substrate and ionophilic phase transfer agents to transport poorly soluble nucleophilic salts into the solution phase. A variety of useful phase transfer agents have been developed such as quaternary ammonium salts,2 macrocyclic ethers,3 and podands.4 We envisioned that electrophiles containing sulfonate leaving groups could be rendered more reactive by modifying them to contain a cation chelating moiety that would attract nucleophilic metal salts to the site of attack.5 In this chelated form, the nucleophilic portion of the salt is localized near the electrophilic center, thus decreasing the entropic barrier relative to intermolecular reactions. The chelated metal cation would also be expected to stabilize the developing negative charge on the oxygens of the sulfonate leaving group in the transition state.6 In addition, by attaching an ion-selective moiety to the leaving group, the electrophilic substrate should be more reactive toward nucleophiles containing a specific metal countercation potentially leading to the development of salt-specific leaving groups.7 In this report, we describe the unique reactivity of arylsulfonate-based nucleophile assisting leaving groups (NALG) that contain either acyclic or macrocyclic ether units attached to the aryl ring ortho to the sulfonate.
Results and Discussion
Poly(ethylene glycol)-containing arylsulfonyl chlorides 1 were prepared in good yields by treating sulfobenzoic acid anhydride with phosphorus pentachloride8 followed by the addition of the methyl poly(ethylene glycol) unit (Scheme 1). Arylsulfonyl chlorides 1 were then reacted with 3-phenylpropanol to give the corresponding sulfonate esters 2 in yields ranging from 72 to 96%. Sulfonate esters 2 (except for n = 0) were exceptionally stable to silica gel chromatography. Sulfonate esters 2 were reacted with lithium, sodium, and potassium bromide, giving 3-phenyl-1-bromopropane as the substitution product (Table 1). Methyl ester sulfonate 2a (n = 0) serves as a baseline to help determine the rate enhancing role of the various oligomeric ether side chains in compounds 2b–2e.
SCHEME 1.
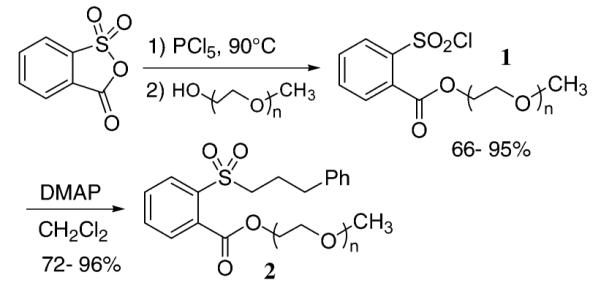
Synthesis of Polyethylene Glycol Containing Arylsulfonate Esters
TABLE 1.
Rates of Metal Bromide Additions to Arylsulfonates 2 in Acetone To Give 3-Phenyl-1-bromopropane
| reaction time in hours (% isolated yield) |
|||||
|---|---|---|---|---|---|
| n | compd no. | LiBr (rt) | LiBr (reflux) | NaBr (reflux) | KBr (reflux) |
| 0 | 2a | – | 0.75 (96) | 3 (94) | 15 (92) |
| 1 | 2b | 12 (96) | 0.3 (97) | 15 (93) | 24 (57) |
| 2 | 2c | 5 (96) | 0.1 (97) | 16 (92) | 24 (50) |
| 3 | 2d | 3.3 (93) | <0.1 (93) | 3.6 (94) | 12 (98) |
| 4 | 2e | 2.25 (96) | <0.1 (96) | 2.5 (95) | 6 (94) |
The reaction of LiBr with 2a gives nearly complete conversion to product at 4 times the rate of NaBr. The increasing number of ethylene glycol units correlates well with a decrease in reaction time required for LiBr to convert substrate 2 to product. In the case of 2e, which contains a tetraethylene glycol unit, the rate of reaction with LiBr is approximately 15 times faster than with 2a (n = 0).
Perhaps more striking is the increase in the reaction rate of LiBr relative to NaBr and KBr. The presence of ethylene glycol units increases the relative rate of LiBr versus NaBr additions to 50-fold for 2b (n = 1) and 160-fold for 2c (n = 2). The trend seems to level off with additional ethylene glycol units. Additionally, the ethylene glycol units appear to impose a retarding effect on the rate of NaBr and KBr except with substrate 2e.
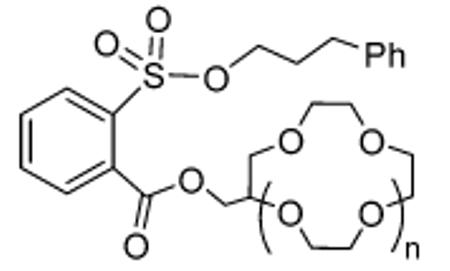
We also examined two macrocyclic ether containing sulfonates for potential rate enhancement and salt selectivity in the metal bromide addition reaction (Table 2). The reaction rates of LiBr with substrates 3a and 3b were comparable and nearly 50 times faster than the baseline methylester sulfonate 2a (Table 1). In terms of selectivity, the 12-crown-4-containing 3a reacts with LiBr in acetone to form product in 1.3 h at room temperature. However, with sodium or potassium bromide, the displacement reaction required 4 and 8 h under refluxing conditions, respectively. This corresponds to a selectivity of greater than 200-fold for lithium bromide over sodium and potassium bromide.9 To further investigate the rate enhancement afforded by the NALG approach, we prepared several diethylene glycol-containing sulfonates of secondary alcohols (4a–c).
TABLE 2.
Rates of Metal Bromide Additions in Acetone to Crown Ether-Containing Sulfonates To Give 3-Phenyl-1-bromopropane
| reaction time in hours (% yield) |
||||
|---|---|---|---|---|
| n | compd no. | LiBr (rt) | NaBr (reflux) | KBr (reflux) |
| 1 | 3a | 1.3 (98) | 4.4 (97) | 8.6 (92) |
| 2 | 3b | 0.75 (92) | 0.37 (93) | 2.1 (89) |
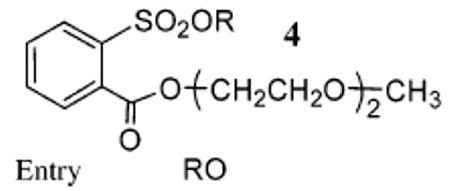
These derivatives were prepared in excellent yields (>96%) starting from 1 (n = 2). We also prepared the tosylates of these alcohols for comparison (Table 3).10 In each case, the rate enhancement afforded by each NALG substrate relative to the corresponding tosylate was very substantial. For example, substrate 4b reacted with LiBr in 1 h to give brominated product in 97% yield, whereas the same reaction with the analogous tosylate required 24 h to achieve a similar yield.
TABLE 3.
Comparison of Rates of Lithium Chloride and Bromide Addition to NALG Sulfonate Esters 4 versus Analogous Tosylates
| reaction time in hours (% yield)a |
||||
|---|---|---|---|---|
| Entry | RO | LiCl | LiBr | |
| 1 |
|
4a | 8(94) | 2(96) |
| 2 | TsOR | 22(54)b | 11(81) | |
| 5 |

|
4b | 6(96) | 1(97) |
| 6 | TsOR | 24(5)b | 24(95) | |
| 7 |
|
4c | - | 16(98) |
| 8 | TsOr | - | 70(0)b | |
Isolated yield.
Significant starting material recovered.
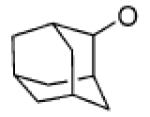
In the case of the 2-adamantyl NALG substrate 4c, we expected little or no reactivity toward metal halide salts since backside nucleophilic displacement is essentially precluded due to steric crowding.11 Nonetheless, we observed a 98% conversion of 4c in 16 h to the 2-bromoadamantane with LiBr in refluxing acetone. No side products involving rearrangement of the adamantine nucleus were observed.12 The corresponding 2-adamantyl tosylate gave no reaction even after 70 h in refluxing acetone. Even the highly electrophilic 2-adamantyl trifluoromethyl sulfonate (not shown) failed to give the bromide product when treated with LiBr in refluxing acetone for extended periods. The 2-adamantyl bromination reaction of 4c likely proceeds via a front-side SNi-type mechanism.13 Reactions involving the SNi mechanism are generally mediated by four-centered cyclic transition states and have been used to obtain alkyl chlorides from alkyl chlorosulfites and chloroformates14 and, more recently, from oxachlorocarbenes.15 However, to our knowledge, the bromination of 2-adamantyl NALG 4c is the first example of an SNi reaction involving a macrocyclic transition state.
To more clearly understand the cation binding geometry of the NALG, triethylene glycol NALG sodium salt 5 was crystallized and analyzed by X-ray crystallography (Figure 1).16 The crystal structure reveals that 5 forms a dimer in the solid phase. As anticipated, three of the side arm ether oxygens of 5 are coordinated to the sodium cation, whereas both the ester carbonyl and one of the sulfonyl oxygens coordinate the sodium of the adjacent monomer. Interestingly, the ester carbonyl is nearly orthogonal and therefore poorly conjugated to the phenyl ring. The nonconjugated carbonyl geometry would indicate that the chelation arms of the various NALGs discussed in this article may not be the most ideal design to the extent that the dimer formation of sodium sulfonate 5 reflects the solution-phase chelation geometry.
FIGURE 1.
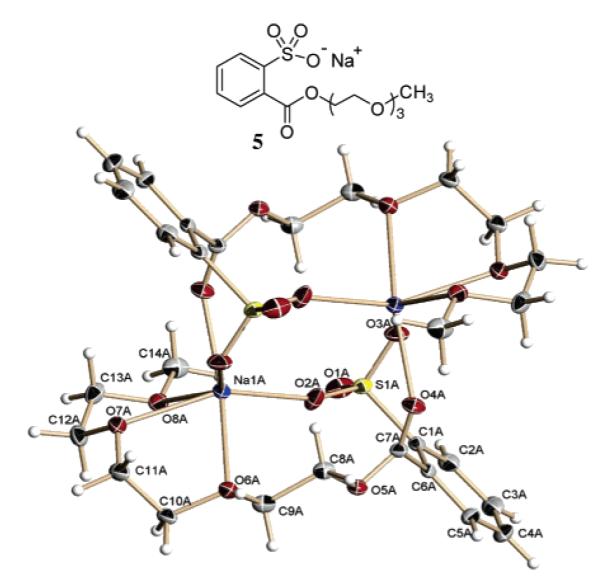
Crystal structure of the sodium salt of triethylene glycol-containing NALG 5.
Conclusion
Arylsulfonate leaving groups containing either acyclic or macrocyclic ether units in the ortho orientation significantly enhance SN2-type reactions involving metal halides. The observed rate enhancement is likely the result of the ability of the cation chelating unit both to attract the metal cation of the incoming nucleophile and to stabilize the newly forming leaving group through internal chelation. The reactivity differences in the bromination reactions involving lithium, sodium, and potassium bromide suggest that leaving groups can be designed to favor certain nucleophilic salts. The design of new NALGs possessing chelating moieties of greater binding affinity and cation specificity is currently underway. In addition, the application of NALG technology to a broader variety of nucleophilic reactions will also be pursued.
Experimental Section
Representative Procedure: Preparation of 2-Oxobutyl 2-(Chlorosulfonyl)benzoate (1b). A mixture of o-sulfobenzoic acid anhydride (1.23 g, 6.6 mmol) and phosphorus pentachloride (3.68 g, 13.2 mmol) was heated at 90 °C for 6 h. The oil was allowed to cool, dissolved in ether, and rinsed with ice water to remove unreacted phosphorus pentachloride. The solvent was evaporated in vacuo, leaving 2.1 g of oil. The crude oil (1.5 g, 6.2 mmol) was then dissolved in excess methoxyethanol (2.5 g, 32 mmol) and heated to 60 °C for 30 h. The reaction mixture was purified by flash chromatography by eluting with a hexane/acetone (85:15 v/v) to yield 1b as white solid: (1.6 g, 87%); mp 34–36 °C; IR (neat) 1737 (C=O), 1370, 1198 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.15 (dd, 1H, J = 1.02 and 7.68 Hz), 7.80–7.68 (m, 3H), 4.54–4.52 (m, 2H), 3.75–3.72 (m, 2H), 3.39 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 166.1, 141.7, 136.4, 132.7, 130.5, 129.3, 70.1, 65.9, 69.2; HRMS (EI) [M + Na]+: calcd for C10H11ClO5SNa 300.9908, found 300.9919.
2,5-Dioxoheptyl 2-(Chlorosulfonyl)benzoate (1c): (8.8 g, 95%); IR (neat) 1731 (C=O), 1353, 1196 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.14 (dd, 1H, J = 0.9 and 7.74 Hz), 7.80–7.69 (m, 2H), 4.57–4.54 (m, 2H), 3.86–3.84 (m, 2H), 3.67–3.65 (m, 2H), 3.55–3.53 (m, 2H), 3.36 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 166.0, 141.6, 135.4, 132.4, 131.6, 130.4, 129.2, 72.0, 70.6, 68.7, 65.9, 59.2; HRMS (EI) [M + H]+: calcd for C12H16ClO6S 323.0356, found 323.0350.
2,5,8-Trioxodecyl 2-(Chlorosulfonyl)benzoate (1d): (6.3 g, 88%); IR (neat) 1732 (C=O), 1356, 1196 (SO2) cm−12; 1HNMR (400 MHz, CDCl3) δ 8.16 (dd, 1H, J = 1.09 and 8.31 Hz), 7.82–7.70 (m, 3H), 4.57–4.54 (m, 2H), 3.87–3.84 (m, 2H), 3.70–3.62 (m, 6H), 3.54–3.52 (m, 2H), 3.36 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 166.1, 141.7, 135.4, 132.5, 131.7, 130.5, 129.2, 72.1, 70.8, 70.8, 68.8, 65.4, 59.3 HRMS (EI) [M + H]+: calcd for C14H20ClO7S 367.0613, found 367.0622.
2,5,8,11-Tetraoxotridecyl 2-(Chlorosulfonyl)benzoate (1e): (1.2 g, 66%); IR (neat) 1731 (C=O), 1373, 1196 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.18 (dd, 1H, J = 1.0 and 7.8 Hz), 7.83–7.7 (m, 3H), 4.57–4.55 (m, 2H), 3.87–3.85 (m, 2H), 3.72–3.62 (m, 10H), 3.57–3.53 (m, 2H), 3.38 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 166.1 141.7 136.4, 132.6, 131.7, 130.5, 129.2, 72.1, 70.8, 70.8, 70.7, 66.8, 66.5, 59.3; HRMS (EI) [M + H]+: calcd for C16H24ClO8S 411.0100, found 411.0113.
Representative Procedure: Preparation of [2-Methyl-(1,4,7,10-tetraoxocyclododecyl)] 2-(Chlorosulfonyl)benzoate (1f). A mixture of 2-chlorosulfonyl benzoyl chloride (1.42 g, 6 mmol) and 2-hydroxymethyl 12-crown-4 were taken in minimum amount of dichloromethane (2 mL), and the reaction mixture was heated to reflux for 35 h. The crude reaction mixture was purified by flash chromatography by eluting with a hexane/acetone (35:65 v/v) to yield 1f as an oil (0.525 g, 42%) with 0.36 g of 2-hydroxymethyl 12-crown-4 and 0.89 g of 2-chlorosulfonyl benzoyl chloride. IR (neat) 1723 (CdO), 1381, 1197 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.17 (dd, 1H, J = 1.38 and 7.35 Hz), 7.83–7,71 (m, 3H), 4.48 (dd, 1H, J = 4.82 and 11.61 Hz), 4.41 (dd, 1H, J = 5.72 and 11.56 Hz), 4.05–4.00 (m, 1H), 3.88–3.61 (m, 14H); 13CNMR (100.75 MHz, CDCl3) δ 167.0, 141.6, 136.5, 132.4, 131.7, 130.3, 129.2, 76.8, 71.0, 70.8, 70.7, 70.6, 70.5, 70.3, 70.3, 70.2, 69.9, 66.3; HRMS (EI) [M + Na]+: calcd for C16H21ClO8SNa 431.0538, found 431.0539.
[2-Methyl-(1,4,7,10,13-pentaoxocyclopentadecyl)] 2-(Chlorosulfonyl)benzoate (1g): (0.51 g, 37%); IR (neat) 1739 (C=O), 1366, 1192 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.16 (dd, 1H, J = 0.76 and 8.16 Hz), 7.83–7.70 (m, 3H), 4.58 (dd, 1H, J = 4.17 and 11.62 Hz, 1H), 4.41 (dd, 1H, J = 5.88 and 11.64 Hz), 4.02–3.96 (m, 1H), 3.87–3.56 (m, 18H); 13CNMR (100.75 MHz, CDCl3) δ 166.0, 141.6, 135.5, 132.5, 131.7, 130.4, 129.2, 77.2, 71.1, 70.9, 70.8, 70.7, 70.6, 70.5, 70.5, 70.3, 70.2, 70.1, 70.1, 69.9, 66.5, 39.4; HRMS (EI) [M + Na]+: calcd for C18H25ClO8SNa 475.0805, found 475.0852.
Preparation of 3-Phenylpropyl 2-(Methylcarboxy)-1-benzosulfonate (2a). To a dichloromethane solution (10 mL) of methyl 2-(chlorosulfonyl) benzoate (0.35 g, 1.5 mmol) was added DMAP (0.21 g, 1.8 mmol) under argon. The reaction mixture was cooled to 0 °C before the addition of phenyl propanol (0.0.5 g, 3.75 mmol). The resulting mixture was warmed to room temperature and stirred for 1 h. The solvent was evaporated in vacuo. The sulfonate ester 3a was purified by flash chromatography. Elution with a hexane/acetone (85: 15 v/v) mixture yielded a gummy material in 41% (0.4 g) yield. IR (neat): 1737 (CO), 1355, 1170 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.00 (dd, J = 1.0 and 7.7 Hz), 7.72–7.59 (m, 3H), 7.26–7.15 (m, 3H), 7.10–1.08 (m, 2H), 4.16 (t, J = 6.2 Hz, 2H), 2.69 (t, J = 7.34 Hz, 2H), 2.05–1.98 (m, 2H), 3.96 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 167.5, 140.5, 134.32, 133.7, 133.5, 131.0, 130.0, 129.6, 128.7, 128.6, 126.3, 70.7, 53.5, 31.6, 30.8; HRMS (EI) [M + H]+: calcd for C17H19ClO5S 335.0975, found 335.0940.
Representative Procedure: Preparation of 3-Phenyl-propyl 2-(2-Oxobutylcarboxy)-1-benzosulfonate (2b). To a dichloromethane solution (10 mL) of 1b (0.536 g, 1.92 mmol) was added DMAP (0.25 g, 2.08 mmol) under argon. The reaction mixture was cooled to 0 °C before the addition of phenylpropanol (0.47 g, 3.48 mmol). The resulting mixture was warmed to room temperature and stirred for 1 h. The solvent was evaporated in vacuo. The crude sulfonate ester was then purified by flash chromatography by eluting with hexane/ acetone (70:30 v/v) to give 2a as a gummy material (0.45 g, 72%); IR (neat) 1739 (C=O), 1369, 1196 (SO2) cm−1 ; 1HNMR (400 MHz, CDCl3) δ 7.98 (dt, 1H, J = 0.7 and 7.8 Hz), 7.69–7.67 (m, 2H), 7.64–7.58 (m, 1H), 7.24–7.13 (m, 3H), 7.08 (d, 2H, J = 6.84 Hz), 4.52–4.49 (m, 2H), 4.16 (t, 2H, J = 6.2 Hz), 3.73–3.70 (m, 2H), 3.37 (s, 3H), 2.7 (t, 2H, J = 7.3 Hz), 2.04–1.97 (m, 2H); HRMS (EI) [M + H]+: calcd for C19H23O6SNa 401.1035, found 401.1040.
3-Phenylpropyl 2-(2,5-Dioxoheptylcarboxy)-1-benzo-sulfonate (2c): (0.6 g, 95%); IR (neat) 1733 (C=O), 1350, 1166 (SO2) cm−1 ; 1HNMR (400 MHz, CDCl3) δ 7.98 (d, J = 7.3 Hz, 1H), 7.70–7.68 (m, 2H), 7.63–7.58 (m, 1H), 7.24–7.07 (m, 5H), 4.54–4.52 (m, 2H), 4.16 (t, J = 6.2 Hz, 2H), 3.84–3.82 (m, 2H), 3.66–3.63 (m, 2H), 3.53–3.51 (m, 2H), 3.55 (s, 3H), 2.69 (t, J = 7.3 Hz), 1.97–2.04 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 167.0, 140.6, 134.1, 133.7, 133.5, 131.0, 129.9, 129.8, 128.6, 128.6, 126.3, 72.0, 70.8, 70.6, 68.9, 65.5, 59.2, 31.5, 30.7; HRMS (EI) [M + H]+: calcd for C21H27O7S 423.1477, found 423.1469.
3-Phenylpropyl 2-(2,5,8-Trioxodecylcarboxy)-1-benzo-sulfonate (2d): (0.7 g, 87%); IR (neat) 1736 (C=O), 1369, 1196 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.01 (dt, 1H, J = 0.83 and 7.83 Hz), 7.69 (dt, 2H, J = 0.96 and 4.87 Hz), 7.65–7.60 (m, 1H), 7.26–7.21 (m, 2H), 7.19–7.15 (m, 1H), 7.10–7.08 (m, 2H), 4.54–4.52 (m, 2H, 4.16 (t, 2H, J = 6.24 Hz), 3.85–3.82 (m, 2H), 3.69–3.61 (m, 6H), 3.54–3.51 (m, 2H), 3.36 (s, 3H), 2.71 (t, 2H, J = 7.31 Hz), 2.05–1.98 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 167.0, 140.6, 134.1, 133.8, 133.5, 131.0, 130.0, 129.8, 128.7, 126.4, 72.1, 70.9, 70.7, 68.9, 65.6, 59.2, 31.6, 30.7; HRMS (EI) [M + H]+: calcd for C23H30O8SNa 489.1559, found 489.1561.
3-Phenylpropyl 2-(2,5,8,11-Tetraoxotridecylcarboxy)-1-benzosulfonate (2e): (0.75 g, 83%); IR (neat) 1728 (C=O), 1371, 1197 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.00 (dt, 1H, J = 0.82 and 7.84 Hz), 7.70–7.69 (m, 2H), 7.64–7.59 (m, 1H), 7.25–7.08 (m, 5H0, 4.53–4.51 (m, 2H), 3.67–3.61 (m, 10H), 3.54–3.52 (m, 2H), 3.36 (s, 3H), 2.70 (t, 2H, J = 7.3 Hz), 2.04–1.97 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 167.0, 140.6, 134.2, 133.8, 133.8, 133.7, 133.5, 131.0, 130.1, 130.0, 129.8, 129.8, 128.7, 128.6, 128.6, 126.4, 72.1, 71.2, 71.0, 70.8, 70.7, 70.6, 70.6, 68.9, 68.8, 65.6, 59.3; HRMS (EI) [M + Na]+: calcd for C25H34O9SNa 533.1816, found 533.1836.
3-Phenylpropyl [2-Methyl-(1,4,7,10-tetraoxocyclodode-cylcarboxy)]-1-benzosulfonate (3a): (0.51 g, 82%); IR (neat) 1728 (C=O), 1373, 1198 (SO2) cm−1 ; 1HNMR (400 MHz, CDCl3) δ 7.99 (d, 1H, J = 7.73 Hz), 7.72–7.36 (m, 3H), 7.25–7.21 (m, 2H), 7.19–7.15 (m, 1H), 7.08 (dd, 1H, J = 5.12 an 11.52 Hz), 4.17 (t, 2H, J = 6.23 Hz), 4.03–3.98 (m, 1H), 3.88–3.82 (m, 2H), 3.59–3.78 (m, 14H), 2.70 (t, 2H, J = 7.34 Hz), 2.05–1.98 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 166.9, 140.6, 134.2, 133.8, 133.4, 131.1, 129.9, 129.7, 128.7, 128.7, 126.4, 77.1, 71.3, 71.0, 70.9, 70.8, 70.5, 70.2, 66.0, 31.6, 30.7; HRMS (EI) [M + Na]+: calcd for C25H32O9SNa 531.1659, found 531.1673.
3-Phenylpropyl [2-Methyl-(1,4,7,10,13-pentaoxocyclo-pentadecylcarboxy)]-1-benzosulfonate (3b): (0.2 g, 39%); IR (neat) 1725 (C=O), 1357, 1196 (SO2) cm−1 ; 1HNMR (400 MHz, CDCl3) δ 7.96 (d, 1H, J = 7.87 Hz), 7.69–7.57 (m, 3H), 7.22–7.05 (m, 5H), 4.50 (dd, 1H, J = 4.31 and 11.43 Hz), 4.35 (dd, 1H, J = 5.79 and 11.59 Hz), 4.13 (t, J = 6.19 Hz, 2H), 3.96–3.91 (m, 1H), 3.83–3.54 (m, 18H), 2.66 (t, 2H, J = 7.45 Hz), 2.01–1.94 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 166.9, 140.6, 133.7, 133.5, 131.0, 129.9, 129.8, 128.7, 128.6, 126.3, 79.8, 71.6, 71.2, 71.2, 71.0, 70.9, 70.9, 70.7, 70.6, 70.6, 70.4, 70.1, 66.2, 63.1, 31.6, 30.7; HRMS (EI) [M + Na]+: calcd for C27H36O10SNa 575.1921, found 575.1927.
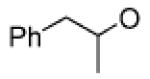
1-Phenyl-2-propyl 2-(2,5-dioxoheptylcarboxy)-1-benzosulfonate (4a): (0.62 g, 93%); IR (neat) 1734 (C=O), 1355, 1176 (SO2) cm−1 ; 1H NMR (400 MHz, CDCl3 δ 7.78 (d, J = 7.9 Hz, 1H), 7.60–7.59 (m, 2H), 7.50–7.45 (m, 1H), 7.19–7.07 (m, 5H), 4.94 (qdd, J = 6.3, 6.3, 6.7 Hz, 1H), 4.53–4.51 (m, 2H), 3.84–3.81 (m, 2H), 3.67–3.64 (m, 2H), 3.55–3.52 (m, 2H), 3.61 (s, 3H), 3.00 (dd, J = 6.3, 13.7 Hz, 1H), 2.82 (dd, J = 6.8, 13.7, 1H), 1.32 (d, J = 6.2 Hz, 3H); 13CNMR (100.75 MHz, CDCl3) δ 167.1, 136.3, 135.1, 133.3, 131.1, 130.9, 129.7, 129.5, 129.4, 128.6, 126.9, 82.2, 72.0, 70.65, 68.9, 65.5, 59.2, 43.0, 20.7; HRMS (EI) [M + H]+: calcd for C21H27O7S 423.1477, found 423.1485.
4-Decyl 2-(2,5-Dioxoheptylcarboxy)-1-benzosulfonate (4b): (0.61 g, 96%); IR (neat) 1732 (C=O), 1365, 1156 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.00 (d, J = 7.5 Hz, 1H), 7.67–7.58 (m, 3H), 4.72 (tt, J = 6.1, 6.0 Hz, 1H), 4.53–4.50 (m, 2H), 3.84–3.81 (m, 2H), 3.67–3.65 (m, 2H), 3.55–3.53 (m, 2H), 3.36 (s, 3H), 1.65–1.5 (m, 5H), 1.36–1.09 (m, 9H), 0.84 (t, J = 6.6 Hz, 3H), 0.82 (t, J = 7.4, 3H); 13CNMR (100.75 MHz, CDCl3) δ 167.1, 135.7, 133.3, 130.7, 129.6, 129.4, 86.0, 72.0, 70.6, 68.9, 65.4, 59.2, 36.4, 34.3, 31.7, 29.1, 22.1, 18.1, 14.2, 14.0; HRMS (EI) [M + H]+: calcd for C22H37O7S 445.2260, found (M + H)+ 445.2263.
Synthesis of 2-Adamantyl 2-(2,5-Dioxoheptylcarboxy)-1-benzosulfonate (4c). To a pyridine solution (10 mL) of 1 (0.9 g, 2.8 mmol) was added 2-adamantanol (0.85 g, 5.5 mmol) at 0 °C. The reaction mixture was slowly warmed to room temperature and allowed to stir for 16 h. The pyridine was removed in vacuo, resulting in a gummy material. The crude material was purified by flash chromatography by elution with a hexane/acetone (70:30 v/v) to give 6 (1.18 g, 96%) as a gummy material. IR (neat) 1734 (C=O), 1360, 1185 (SO2) cm−1; 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.4 Hz, 1H), 7.67–7.58 (m, 3H), 4.83 (m, 1H), 4.54–4.51 (m, 2H, 3.67–3.65 (m, 2H), 3.55–3.53 (m, 2H), 3.36 (s, 3H), 2.10–2.03 (m, 4H), 1.83–0.180 (m, 4H), 1.69–1.62 (m, 4H), 1.53–1.50 (m, 2H); 13C NMR (100.75 MHz, CDCl3) δ 167.0 (CO), 135.34, 133.1, 130.8, 129.7, 129.4, 87.8, 72.0, 70.6, 68.9, 65.4, 59.2, 37.3, 36.5, 32.9, 31.3, 27.0, 24.7; HRMS (EI) [(M + 2H) – C10H15]+: calcd for C12H17O7S 305.0695, found 305.0699.
Representative Procedure: Reaction of NALG Sulfonate 2c with NaBr. Aryl sulfonate ester 2c (0.42 g, 1.00 mmol) was dissolved in acetone, and NaBr (0.4 g, 4.0 mmol) was added to it. The reaction mixture was stirred at room temperature/reflux conditions. The reactions were monitored carefully by TLC until the disappearance of starting material. The solvent was evaporated in vacuo. The reaction mixtures were washed with water to remove excess inorganic salts, extracted in EtOAc, and dried over sodium sulfate. The organic layer was evaporated, giving pure desired products. Yields and reaction times are given in Tables 1, 2, and 3.
Supplementary Material
Acknowledgment
We thank the National Institute of Mental Health (66963-01) and the Florida Centers of Excellence in Marine Biotechnology (contribution P200412) for financial support. An acknowledgment is also made to the National Science Foundation Division of Undergraduate Education (0311369) for the 400 MHz NMR used in this study.
Footnotes
Supporting Information Available: Copies of 1H NMR spectra for compounds 1–4 and X-ray crystallographic data (CIF) for compound 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Zehavi U. J. Org. Chem. 1975;40:3870. doi: 10.1021/jo00914a011. [DOI] [PubMed] [Google Scholar]; (b) Wu M-C, Anderson L, Slife CW, Jensen LJ. J. Org. Chem. 1974;39:3014. [Google Scholar]
- (2).Wang M-L, Wu H-S. J. Org. Chem. 1990;55:2344. [Google Scholar]
- (3) (a).Pedersen CJ. J. Am. Chem. Soc. 1967;89:7017. [Google Scholar]; (b) Pedersen CJ, Frensdorff HK. Angew. Chem., Int. Ed. Engl. 1972;11:16. doi: 10.1002/anie.197200161. [DOI] [PubMed] [Google Scholar]; (c) Stott PE, Bradshaw JS, Parish WW. J. Am. Chem. Soc. 1980;102:4810. [Google Scholar]
- (4) (a).Kimura Y, Regan SL. J. Org. Chem. 1982;47:2493. [Google Scholar]; (b) Kimura Y, Regan SL. J. Org. Chem. 1983;48:195. [Google Scholar]; (c) Solov’ev VP, Baulin VE, Strakhova NN, Kazachenko VP, Belsky VK, Varnek AA, Volkova TA, Wipff G. J. Chem. Soc., Perkin Trans. 1998;2:1489. [Google Scholar]
- (5) (a).Cacciapaglia R, Di Stefano S, Mandolini J. Org. Chem. 2002;67:521. doi: 10.1021/jo016149q. [DOI] [PubMed] [Google Scholar]; (b) Cacciapaglia R, Mandolini L. Pure Appl. Chem. 1993;65:533. [Google Scholar]; (c) Alfimov MV, Gromov SP, Fedorov Yu. V., Fedorova OA, Vedernikov AI, Churakov AV, Kuz’mina LG, Howard JAK, Bossmann S, Braun A, Woerner M, Sears DF, Jr., Saltiel J. J. Am. Chem. Soc. 1999;121:4992. [Google Scholar]
- (6).Gobbi A, Landini D, Maia A, Secci D. J. Org. Chem. 1995;60:5954. [Google Scholar]
- (7).Lippard SJ. “Grand Challenges” in Chemistry. Chem. Eng. News. 2000 Aug 7;78:64. [Google Scholar]
- (8).Loev B, Kormendy M. J. Org. Chem. 1962;27:1703. doi: 10.1021/jo01349a015. [DOI] [PubMed] [Google Scholar]
- (9).The selectivity is probably higher since, at room temperature, the reaction of 3a with NaBr gives less than a 25% product conversion after 5 days.
- (10).For a more appropriate comparison, we prepared the methyl ester sulphonates of these secondary alcohols but found that their reaction with lithium halide tended to occur at the methyl position.
- (11).Kozikowski AP, Lee J. Tetrahedron Lett. 1988;29:3053. and references therein. [Google Scholar]
- (12).Sinnott ML, Storesund HJ, Whiting MC. Chem. Commun. 1969:1000. [Google Scholar]
- (13).March J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 4th ed Wiley-Interscience; New York: 1992. pp. 308–326. [Google Scholar]
- (14) (a).Lee CC, Clayton JW, Lee DG, Finlayson AJ. Tetrahedron. 1962;18:1395. [Google Scholar]; (b) Lewis ES, Herndon WC, Duffy DC. J. Am. Chem. Soc. 1961;83:1959. [Google Scholar]
- (15).Moss RA, Fu X, Tian J, Sauers R, Wipf P. Org. Lett. 2005;7:1371. doi: 10.1021/ol050185k. [DOI] [PubMed] [Google Scholar]
- (16).The sodium salt was chosen after considerable attempts with the lithium analogue demonstrated that this salt was too hygroscopic for efficient crystal growth.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.