

Abstract
Sepsis is a complex, multifactorial, rapidly progressive disease characterized by an overwhelming activation of the immune system and the countervailing antiinflammatory response. In the current study in murine peritoneal macrophages, chlorogenic acid suppressed endotoxin-induced high mobility group box 1 (HMGB1) release in a concentration-dependent manner. Administration of chlorogenic acid also attenuated systemic HMGB1 accumulation in vivo and prevented mortality induced by endotoxemia and polymicrobial sepsis. The mechanisms of action of chlorogenic acid included attenuation of the increase in toll-like receptor (TLR)-4 expression and suppression of sepsis-induced signaling pathways, such as c-Jun NH2-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB, which are critical for cytokine release. The protection conferred by chlorogenic acid was achieved through modulation of cytokine and chemokine release, suppression of immune cell apoptosis and augmentation of bacterial elimination. Chlorogenic acid warrants further evaluation as a potential therapeutic agent for the treatment of sepsis and other potentially fatal systemic inflammatory disorders.
INTRODUCTION
The dramatic rise in the incidence of sepsis, a systemic inflammatory response secondary to infection that leads to multiorgan failure and death, has been fuelled by an increase in the number of invasive surgical techniques performed during the past several decades. Because of deficits in our understanding of the pathophysiology of sepsis, it remains the most challenging problem in intensive care and a major cause of morbidity and mortality. The primary pathophysiological event in the septic response is the overactivation of the innate immune system and a countervailing immunosuppressed state (1). Recently, the importance of high mobility group box 1 (HMGB1) in sepsis was suggested by the observation that increased serum concentrations of HMGB1 were associated with decreased survival in septic patients (2). With Xigris® (activated protein C) having shown limited survival benefit in a restricted population (3), there is a substantial need for an effective therapy that will decrease morbidity and mortality associated with sepsis.
Chlorogenic acid is a phenolic compound found in many plants, and its antioxidant activity is more accessible than many other flavonoids (4). Phenolic compounds are ubiquitous in plants, and they are also present in beverages such as wine, tea and coffee (5). Previous studies have reported that chlorogenic acid regulates cytokine and chemokine release (6). Recent studies have reported that chlorogenic acid is protective against acute lung injury induced by lipopoly saccharide (LPS) (7), and chlorogenic acid was shown to protect animals challenged by intraperitoneal injection of LPS via the suppression of hepatic toll-like receptor (TLR)-4 mRNA expression (8). In addition to its antioxidant effects, chlorogenic acid suppresses cellular apoptosis and blocks the activation of nuclear factor (NF)-κB, activator protein (AP)-1 and mitogen-activated protein kinase (MAPK) in vitro(9,10).
In this study, we investigated the effects of chlorogenic acid treatment in experimental models of sepsis, and we demonstrated a decrease in sepsis- induced multiorgan failure and death with chlorogenic acid. Further study was undertaken to elucidate the mechanisms of action of chlorogenic acid. We characterized chlorogenic acid as a potential therapeutic agent for treating sepsis by evaluating its effect on cytokine and chemokine profiles, apoptotic loss of immune cells and signaling pathways. This work describes a novel strategy to counteract sepsis, which is currently the most challenging problem in intensive care.
MATERIALS AND METHODS
Cell Culture and LPS Stimulation
Three days after intraperitoneal injection of 2 mL thioglycollate broth (4.05 g/100 mL) (Difco Laboratories, Detroit, MI, USA) (2), peritoneal exudate cells were obtained by peritoneal lavage with 5 mL medium. Macrophages were pooled from three mice (male C57BL/6, Charles River Laboratories, Yokohama, Japan) and re-suspended in RPMI-1640 containing 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 IU/mL) and streptomycin (100 mg/mL) (RPMI-FBS). Adherent macrophages were gently washed 2 h before stimulation with bacterial endotoxin (LPS, Escherichia coli serotype O111:B4; Sigma-Aldrich, St. Louis, MO, USA). Peritoneal macrophage cytokine and chemokine release was determined after 16 h of LPS (100 ng/mL) stimulation in the presence or absence of various concentrations of chlorogenic acid (10, 20 and 40 μmol/L; Sigma-Aldrich).
Animals and Experimental Models of Sepsis
The in vivo components of this study were approved by the Sungkyunkwan University Animal Care Committee, and all experiments were conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (11). Polymicrobial sepsis was induced in male ICR mice (7–8 wks; Charles River Laboratories, Yokohama, Japan) by cecal ligation and puncture (CLP) (12). Briefly, animals were anesthetized with an intramuscular ketamine injection (55 mg/kg; Yuhan Corporation, Seoul, Korea) and xylazine injection (7 mg/kg; Boehringer Ingelheim, St. Joseph, MO, USA). Anesthetized mice were then opened with a midline abdominal incision, and the cecum was carefully exposed with care to avoid the surrounding blood vessels. The cecum was ligated distal to the ileocecal valve to avoid intestinal obstruction, and the cecal stump was punctured twice with a 20-gauge needle. A small amount of stool was extracted to ensure patency of the puncture sites. The cecum was placed back into its normal intra-abdominal position, and the wound was closed with two layers of running sutures. All animals received resuscitative normal saline (20 mL/kg body weight subcutaneously), an important step in demonstrating a hyper-dynamic circulatory state. Control mice received a sham operation consisting of a laparotomy with intestinal manipulation. However, in control mice, the cecum was neither ligated nor punctured. Endotoxemia was induced in ICR mice (7–8 wks) by intraperitoneal injection of LPS (10 mg/kg, Escherichia coli serotype O111:B4; Sigma-Aldrich). Immediately after the induction of sepsis, chlorogenic acid or ethyl pyruvate was administered intravenously, and animals were monitored for survival for up to 10 d. In parallel experiments, mice were euthanized to collect blood or tissue samples at indicated time points. Ethyl pyruvate was prepared in solution with sodium (130 mmol/L), potassium (4 mmol/L), calcium (2.7 mmol/L) and chloride (139 mmol/L) (pH 7.0) (20 mg/kg, 0.5 h intravenous infusion). Ethyl pyruvate, an aliphatic ester derived from pyruvic acid, is an inhibitor of HMGB1. Previous reports have shown that ethyl pyruvate significantly inhibits endotoxin-induced release of proinflammatory cytokines and that it protects mice against experimental sepsis, even when given as late as 12–24 h after the onset of the disease (13).
Measurement of Cytokine and Chemokine Levels
The levels of tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-1β, IL-2, IL-6, IL-10, IL-12, monocyte chemoattractant protein (MCP)-1 (BD Biosciences, San Jose, CA, USA), macrophage inflammatory protein (MIP)-2 (R&D Systems, Minneapolis, MN, USA) and HMGB1 (Shino-Test, Tokyo, Japan) were determined by using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions. For measurement of CLP-induced levels of cytokines and chemokines, serum samples were collected 1, 3, 6, 12, 18, 24 and 48 h after CLP and treatment with vehicle or chlorogenic acid (20 mg/kg).
Assessment of Serum Enzyme Activity and Histological Analysis
Serum lactate dehydrogenase (LDH), blood urea nitrogen (BUN), creatinine, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined by using a Hitachi 7600 automatic analyzer (Hitachi, Tokyo, Japan). Heart, kidney, liver and lung tissue samples were removed for histological analysis 24 h after CLP. These tissue samples were fixed in 10% neutral-buffered formalin and stained with hematoxylineosin for a blinded histological assessment. Histological changes were evaluated in nonconsecutive, randomly chosen fields at 200× magnification (Olympus BX51/Olympus DP71; Olympus, Tokyo, Japan).
Quantification of Intercellular Adhesion Molecule-1
To measure CLP-induced alterations in the serum levels of intercellular adhesion molecule (ICAM)-1, blood samples were collected from the abdominal inferior vena cava 6, 12, 18 and 24 h after CLP and treatment with vehicle or chlorogenic acid (20 mg/kg). To determine the change in the level of ICAM-1 in heart, kidney, liver and lung, the tissue samples were harvested 18 h after CLP and treatment with vehicle or chlorogenic acid (20 mg/kg). The levels of ICAM-1 present in serum or whole tissue homogen ates were determined quantitatively by an ELISA kit (R&D Systems) according to the manufacturer’s instructions.
TLR4 mRNA and Protein Expression
Total RNA isolation was carried out according to a previously described method (14). First, cDNA was synthesized through reverse transcription by using oligo dT-adaptor primer and avian myeloblastosis virus (AMV) reverse transcriptase. Polymerase chain reaction (PCR) amplification was performed with a 10-μL diluted cDNA sample. PCR was performed with a GeneAmp® PCR System 2700 (Applied Biosystems; Life Technologies, Carlsbad, CA, USA) with an initial denaturation step at 94°C for 5 min and a final extension at 72°C for 7 min. The following PCR amplification cycling conditions (denaturation, annealing and extension) were used: 30 cycles of 94°C for 30 s, 56°C for 30 s and 72°C for 30 s for TLR4; 26 cycles of 94°C for 30 s, 54°C for 30 s and 72°C for 60 s for β-actin. The TLR4 upstream primer was 5′-AGTGG GTCAA GGAAC AGAAG CAG-3′ and the downstream primer was 5′-CTTTA CCAGC TCATT TCTCA CCC-3′. To determine TLR4 protein expression, liver tissue samples were collected 6 h after CLP and homogenized in 1 mL lysis buffer (150 mmol/L NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate and 50 mmol/L Tris, pH 8.0). The homogenates were incubated on ice (60 min) and sonicated for 30 s. Proteins (50 μg) from each sample were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The separated proteins were transferred onto nitrocellulose membranes for 2 h and then incubated with primary antibody to TLR4 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. After washing three times with Tris-buffered saline (TBS)-0.05% Tween-20, we incubated the membranes with a peroxidase-conjugated goat anti-rabbit immuno globulin G secondary antibody (Jackson ImmunoResearch, Baltimore, MD, USA) for 1 h at ambient temperature. Signals were quantified using scanning densitometry and computer-assisted image analysis.
Mitogen-Activated Protein Kinase Signaling
Liver tissue samples collected 6 h after CLP were homogenized in lysis buffer (150 mmol/L NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mmol/L Tris, 1 mmol/L phenylmethylsulfonyl fluoride [PMSF], 40 mmol/L NaF and 4 mmol/L Na3VO4). Samples were then sonicated for 30 s and separated by gel electrophoresis. The separated proteins were transferred to nitrocellulose membranes by a Bio-Rad semidry transfer system for 2 h. After blocking for 1 h with TBS containing 0.1% Tween-20 and 5% skim milk, membranes were probed overnight at 4°C with primary antibodies to extracellular signal–regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), p38 MAPK, phosphorylated ERK, phosphorylated JNK and phosphorylated p38 MAPK (Cell Signaling Technology, Beverly, MA, USA) or β-actin. Peroxidase-conjugated AffiniPure goat anti-rabbit IgG and peroxidase-conjugated AffiniPure goat anti-mouse IgG (Jackson ImmunoResearch) were used as secondary antibodies. After washing, the blots were subjected to an enhanced chemiluminescence reaction and exposed to X-rays films. The intensity of the immunoreactive bands was determined by a densitometric analysis program.
NF-κB and IκB-α Activity
Nuclear and cytosol extracts were prepared from liver samples collected 6 h after CLP by NE-PER® Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer’s protocol (Pierce Biotechnology, Rockford, IL, USA). After the separated proteins were transferred to nitrocellulose membranes as described above, they were blocked and probed overnight at 4°C, with primary antibodies to NF-κB and an inhibitor of NF-κB (NF-κB inhibitor α [IκB-α]) (1:1,000; Santa Cruz Biotechnology). The intensity of the immunoreactive bands was determined as described above.
Detection of Apoptosis
Thymi and spleens obtained from experimental mice were gently ground by using a glass homogenizer to dissociate the cells and washed twice in phosphate-buffered saline (PBS). Blood samples acquired from experimental animals were lysed with FACS™ Lysing Solution (BD Biosciences) for lymphocyte subset analysis. Fluorescent-labeled monoclonal antibodies, rat anti-mouse cluster of differentiation (CD) 8 (FITC) antibody and rat anti-mouse CD4 (R-PE) antibody (Invitrogen; Life Technologies), were used to quantify lymphocyte subpopulations present in the thymus, spleen and blood. The proportion of cells undergoing apoptosis was determined by using a commercially available fluorescein-labeled annexin V kit (ApoTarget™; Annexin-V FITC Apoptosis Kit, BioSource International, Camarillo, CA, USA). Flow cytometric analysis was performed by using a Bryte-HS flow cytometer (Bio-Rad Laboratories, Hercules, CA, USA). Blood samples were collected, and differential leukocyte counts were obtained with an electronic cell counter (LH750; Beckman Coulter, Miami, FL, USA) 24 h after CLP with or without chlorogenic acid treatment.
In Vivo Bacterial Clearance, Phagocytosis and Bactericidal Activity
Mice were anesthetized and peritoneal fluid, blood, spleen and lung samples were acquired 24 h after CLP and treatment with vehicle or chlorogenic acid (20 mg/kg). After serial dilutions with PBS, peritoneal fluid, blood and tissue homogenates were cultured overnight on blood-agar base plates (Trypticase Soy Agar Deeps; BD Biosciences) maintained at 37°C, and colony-forming units (CFUs) were counted. Phagocytosis was determined by incubating peritoneal macrophages with 5 × 106 particles/mL zymosan and 600 mg/mL nitro blue tetrazolium (NBT) (15). The supernatant was removed after 1 h of incubation. The optical density of the reduction product of NBT was determined at 540 nm by using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). Nitric oxide (NO) production and hydrogen peroxide (H2O2) levels were determined at 24 h after chlorogenic acid treatment. Culture supernatants were collected and the accumulation of NO2− in culture supernatants was measured by using an assay system described previously (16). The level of NO2− is indicative of NO production. The level of H2O2 in culture supernatants was measured by using a commercially available hydrogen peroxide assay kit (Amplex® Red Hydrogen Peroxide Assay Kit; Molecular Probes; Life Technologies) according to the manufacturer’s instructions.
Statistical Analysis
Survival data were analyzed by the Kaplan-Meier log-rank test. All other data were analyzed by analysis of variance, and the Bonferroni test was used for post hoc comparisons. Differences between groups were considered statistically significant at P < 0.05. Results are presented as mean ± standard error of the mean (SEM).
RESULTS
Chlorogenic Acid Inhibits LPS-Induced HMGB1 Release and Modulates the Release of Cytokines and Chemokines in Murine Peritoneal Macrophages
Chlorogenic acid suppressed endotoxin-induced HMGB1 release in murine peritoneal macrophages in a concentration-dependent manner (Figure 1A). To further evaluate its role in modulating cytokine and chemokine release, the effect of chlorogenic acid on basal and LPS-induced TNF-α, IL-1β, IL-6, IL-12, MIP-2 and MCP-1 release was determined. Chlorogenic acid suppressed the release of MIP-2, TNF-α and IL-1β in LPS-stimulated macrophages (Figures 1B, D, E), but it did not affect the release of IL-6. However, chlorogenic acid increased the release of MCP-1 and IL-12, suggesting a possible role in activation of the Th1 response (Figures 1C, G).
Figure 1.

The effect of chlorogenic acid on in vitro cytokine and chemokine release. Cytokine and chemokine release from macrophages were determined after 16 h of LPS stimulation in the presence or absence of various concentrations of chlorogenic acid (CA). The effects of CA on basal and LPS-induced release of HMGB1 (A), MIP-2 (B), MCP-1 (C), TNF-α (D), IL-1β (E), IL-6 (F) and IL-12 (G) were determined. Data are presented as mean ± SEM; n = 8. Statistically significant differences from the control group (**P < 0.01). Statistically significant differences from the LPS-treated control group (+P < 0.05 and ++P < 0.01).
Chlorogenic Acid Protects Mice Against Lethal Endotoxemia
On the basis of the capacity of chlorogenic acid to modulate endotoxin-induced cytokine and chemokine release, the efficacy of chlorogenic acid for the treatment of experimentally induced lethal endotoxemia in mice was investigated. Endotoxemia was induced by intraperitoneal injection of LPS (10 mg/kg) and a significant improvement in murine survival was observed with the administration of chlorogenic acid at 10 mg/kg (P = 0.0078) and 20 mg/kg (P = 0.0001) (Figure 2A). When animals were treated with ethyl pyruvate (20 mg/kg), an effective inhibitor of HMGB1 (13), mortality was also significantly reduced (P < 0.0001).
Figure 2.

Chlorogenic acid protected against sepsis-induced mortality. Chlorogenic acid was administered at various doses after the induction of sepsis via an intraperitoneal injection of LPS or CLP. Mice were monitored for survival for up to 10 d. (A) LPS-injected mice were given chlorogenic acid or ethyl pyruvate. (B) CLP mice were given various doses of chlorogenic acid or ethyl pyruvate. The results of treatment groups were compared with the vehicle-treated group. n = 10–30 per group.
Chlorogenic Acid Protects Against Cecal Ligation and Puncture-Induced Mortality
The therapeutic effect of chlorogenic acid was confirmed in a well-established animal model of sepsis induced by CLP. Among the various animal models of sepsis, CLP more closely mimics clinical sepsis and is currently the most widely used model (17). CLP was performed as described above, and animals were treated with chlorogenic acid (1–40 mg/kg) or ethyl pyruvate (20 mg/kg) after CLP. Consistent with the results from the model of endotoxemia, chlorogenic acid provided significant protection against CLP-induced death at a dose of 10 mg/kg (P = 0.0051) and a higher level of mortality reduction at 20 mg/kg (P = 0.0003) (Figure 2B).
Effects of Chlorogenic Acid on CLP-Induced Organ Injury
To determine the effect of chlorogenic acid on sepsis-induced multiorgan dysfunction, we assessed serum enzyme levels and performed organ-specific histological analysis. The serum level of LDH was significantly increased by CLP, reaching a maximum 24 h after the induction of sepsis (2,178.8 ± 286.3 IU/L) (Figure 3A). The administration of chlorogenic acid (20 mg/kg) significantly attenuated this increase 12 and 24 h after CLP (1,385.4 ± 191.6 IU/L), and both doses of chlorogenic acid (10 and 20 mg/kg) markedly attenuated the increase in LDH 24 h after CLP (624.4 ± 110.7 and 726.3 ± 173.0 IU/L, respectively). CLP caused increases in the levels of BUN, creatinine, ALT and AST, with all serum enzyme levels reaching their peak 12–24 h after CLP (312.6 ± 14.2 mg/dL, 0.7 ± 0.1 mg/dL, 187.0 ± 11.0 U/L and 375.7 ± 40.2 U/L, respectively). Treatment with chlorogenic acid conferred protection against CLP-induced organ injury on the basis of both serum enzyme levels and histological analysis. Heart, kidney, liver and lung samples were removed for histological analysis 24 h after CLP. Histological analysis showed normal cell structure in the heart, kidney, liver and lung of sham-operated animals (Figures 3B–E). Significant histopathological changes (such as inflammatory cell infiltration, congestion, necrosis and degeneration) were observed in CLP-treated mice, and these pathological changes were ameliorated by the administration of chlorogenic acid (20 mg/kg). Chlorogenic acid treatment alone did not induce any histological alterations.
Figure 3.

Chlorogenic acid protected against CLP-induced organ injury. Assessment of serum enzyme activities and histological analysis were performed to evaluate the protective effects of chlorogenic acid on multiorgan dysfunction induced by sepsis. (A) Serum levels of LDH, BUN, creatinine, ALT and AST were determined at various time points after CLP. Histological analysis of heart (B), kidney (C), liver (D) and lung tissue (E) collected 24 h after CLP. Significant histopathological changes (such as inflammatory cell infiltration, congestion, necrosis and degeneration) were observed in the CLP group (200×, scale bar 50 μm), and these pathological changes were ameliorated by the administration of chlorogenic acid (20 mg/kg). The serum levels of soluble ICAM-1 were measured 6, 12, 18 and 24 h after CLP (F), and the changes in the level of ICAM-1 in heart, kidney, liver and lung tissues were determined 18 h after CLP (G). Treatment with chlorogenic acid (20 mg/kg) suppressed the increase in ICAM-1 levels induced by CLP. Data are presented as mean ± SEM; n = 8–10. Statistically significant differences compared with the sham group (*P < 0.05 and **P < 0.01). Statistically significant differences compared with the vehicle-treated CLP group (+P < 0.05 and ++P < 0.01).
Effects of Chlorogenic Acid on Intercellular Adhesion Molecule-1
The serum levels of soluble ICAM-1 increased significantly after the induction of sepsis by CLP. The level of serum ICAM-1 in vehicle-treated mice 6 h after CLP was twofold higher than the ICAM-1 level 6 h after sham surgery in control mice (Figure 3F). The ICAM-1 level in the CLP group continued to rise, and by 18 h after CLP, the level of ICAM-1 was 4.2-fold higher than that in the sham control mice. Treatment with chlorogenic acid (20 mg/kg) attenuated the increase in ICAM-1 induced by CLP, and 18 h after chlorogenic acid treatment, the level of ICAM-1 in chlorogenic acid–treated CLP mice was 46.5% of that observed in the vehicle-treated CLP group. Furthermore, in vehicle-treated CLP mice 18 h after CLP, the levels of ICAM-1 in the homogenates of heart, kidney, liver and lung tissues were increased by 1.5-, 2.6-, 2.7- and 1.4-fold, respectively, compared with sham control mice (Figure 3G). Chlorogenic acid treatment attenuated this increase, and in chlorogenic acid–treated CLP mice, the levels of ICAM-1 in heart, kidney, liver and lung were 86.6%, 71.7%, 80.7% and 73.8% of that observed in vehicle-treated CLP mice, respectively.
Effects of Chlorogenic Acid on CLP-Induced Release of Cytokines and Chemokines
We evaluated the systemic accumulation of HMGB1 after the induction of sepsis by CLP. Consistent with previous reports (18), HMGB1 levels peaked 24 h after CLP (134.2 ± 12.5 ng/mL) (Figure 4A). Administration of chlorogenic acid significantly attenuated circulating HMGB1 levels in septic mice. TNF-α and IL-1β levels peaked 3 h after CLP (967.5 ± 67.8 and 160.0 ± 9.1 pg/mL, respectively), whereas IL-6 levels peaked 6 h after CLP (9,821.2 ± 583.5 pg/mL). Treatment with chlorogenic acid significantly lowered TNF-α, IL-1β and IL-6 levels, and it decreased MIP-2 levels both 3 and 6 h after CLP (Figures 4B, D, E, F). Meanwhile, Th1 cytokines (IFN-γ, IL-2 and IL-12) were increased when mice were treated with chlorogenic acid after CLP, and the level of MCP-1 was augmented 3 h after CLP by chlorogenic acid (Figures 4C, G, H, I). Chlorogenic acid did not affect the Th2 cytokine IL-10 (Figure 4J).
Figure 4.

The effects of chlorogenic acid on CLP-induced cytokine and chemokine release. The levels of HMGB1 (A), MIP-2 (B), MCP-1 (C), TNF-α (D), (E) IL-1β (E), IL-6 (F), IFN-γ (G), IL-2 (H), IL-12 (I) and IL-10 (J) were determined after CLP, with or without treatment with chlorogenic acid (20 mg/kg). Data are presented as mean ± SEM; n = 8–10. Statistically significant differences compared with the sham group (*P < 0.05 and **P < 0.01). Statistically significant differences compared with the vehicle-treated CLP group (+P < 0.05 and ++P < 0.01).
Effect of Chlorogenic Acid on TLR4 Expression
To gain insights into the protective mechanisms of chlorogenic acid, we investigated its effect on TLR4. Recent in vitro evidence has suggested that TLR4 acts as a receptor for HMGB1 (19), and previous reports have demonstrated that blunting TLR4 gene and protein expression correlates with improved long-term survival in sepsis (20). In this study, TLR4 mRNA expression was quantified in heart, kidney, liver and lung tissue at various time points after CLP (Figures 5A–D). In the heart, kidney and liver at all measured time points, CLP induced a significant increase in TLR4 mRNA expression compared with the sham control mice. In the heart, chlorogenic acid administration markedly decreased TLR4 mRNA expression both 6 and 12 h after CLP. In the kidney and liver, the difference between TLR4 mRNA expression in the CLP group and chlorogenic acid–treated CLP group was significant from 3 to 24 h after CLP. In the lung, TLR4 mRNA expression was increased significantly 3 h after CLP and peaked 6 h after CLP. More than 6 h after CLP in the lung, TLR4 mRNA expression gradually decreased. Chlorogenic acid treatment did not affect TLR4 mRNA expression in the lung. The protein expression of TLR4 was increased 6 h after CLP in the liver, and this increase was attenuated by chlorogenic acid treatment.
Figure 5.

The effects of chlorogenic acid on TLR4 and signaling pathways. The influences of chlorogenic acid on TLR4 and MAPK activation were evaluated to understand the mechanisms underlying chlorogenic acid–mediated survival. TLR4 mRNA expression was quantified in heart (A), kidney (B), liver (C) and lung tissues (D) at various time points after CLP. (E) The protein expression of TLR4 was increased 6 h after CLP in the liver, and this increase was attenuated by chlorogenic acid treatment (20 mg/kg). The phosphorylation of JNK (F), p38 (G) and ERK (H) were assessed 6 h after CLP-induced sepsis. (I) NF-κB activation was determined in nuclear fraction of liver samples collected 6 h after CLP. (J) The decrease in the level of cytosolic IκB-α expression after CLP was significantly attenuated by the administration of chlorogenic acid (20 mg/kg). Data are presented as mean ± SEM; n = 6–8. Statistically significant differences compared with the sham group (*P < 0.05 and **P < 0.01). Statistically significant differences compared with the vehicle-treated CLP group (+P < 0.05 and ++P < 0.01).
Chlorogenic Acid Modulates Inflammatory Signaling Pathways
To further elucidate the mechanisms of action of chlorogenic acid, we determined its influence on MAPK activation. Phosphorylation of JNK, p38 and ERK was assessed 6 h after the induction of sepsis by CLP. We confirmed that CLP increased the phosphorylation of JNK, p38 and ERK in the liver compared with sham control mice (Figures 5F–H). The increased phosphorylation of JNK and p38 were attenuated by chlorogenic acid treatment. However, chlorogenic acid did not affect the phosphorylation of ERK, which is consistent with previous in vitro results (9). NF-κB activation was also increased after CLP, and this increase was blocked by chlorogenic acid treatment. The decrease in cytosolic IκB-α expression after CLP was significantly reversed by the administration of chlorogenic acid (Figures 5I, J).
Flow Cytometry and Apoptosis
The percentage of apoptotic lymphocytes was quantified by annexin V staining. An increase in lymphocyte apoptosis was observed 24 h after CLP, and chlorogenic acid effectively suppressed lymphocyte apoptosis (Figure 6). Table 1 shows percentages of apoptotic lymphocyte subsets. In septic mice, we observed a statistically significant decrease in the percentage of CD4+CD8− T cells.
Figure 6.
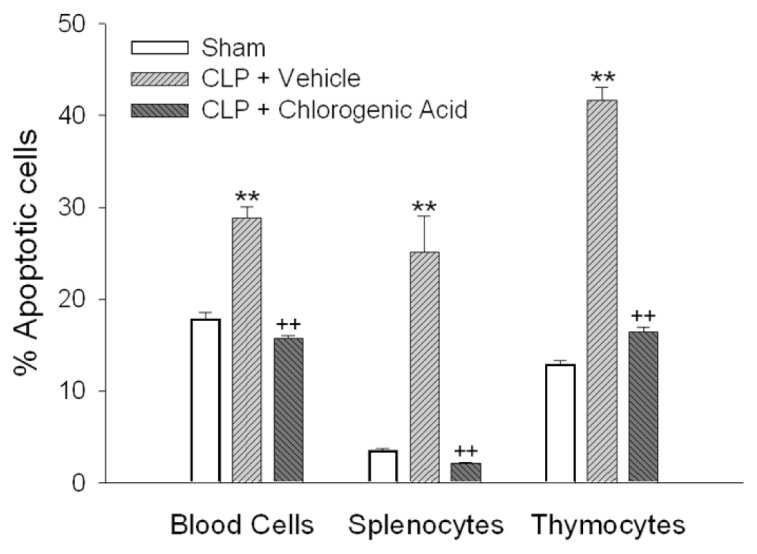
Lymphocyte apoptosis determined by annexin V staining. Lymphocytes were identified by characteristic forward and side scatter properties. The percentage of lymphocytes undergoing apoptosis was quantified by annexin V in the following experimental groups: sham, vehicle-treated CLP and chlorogenic acid (20 mg/kg)-treated CLP groups 24 h after CLP-induced sepsis. Data are presented as mean ± SEM; n = 5–7. Statistically significant differences compared with the sham group (**P < 0.01). Statistically significant differences compared with the vehicle-treated CLP group (++P < 0.01).
Table 1.
Percentage of various lymphocyte subpopulations.
| CD4+CD8− | CD4+CD8+ | CD4−CD8− | CD4−CD8+ | |
|---|---|---|---|---|
| Blood cells | ||||
| Sham | 33.6 ± 2.8 | 12.6 ± 1.8 | 50.5 ± 3.9 | 3.3 ± 0.7 |
| CLP | 11.4 ± 0.5b | 17.4 ± 2.7 | 67.2 ± 3.7a | 4.0 ± 1.2 |
| CLP + chlorogenic acid | 21.4 ± 2.1c | 10.9 ± 2.6 | 64.6 ± 5.2 | 3.1 ± 0.8 |
| Splenocytes | ||||
| Sham | 12.3 ± 1.1 | 11.3 ± 0.1 | 69.7 ± 1.0 | 6.7 ± 0.5 |
| CLP | 5.6 ± 0.9b | 10.4 ± 1.6 | 78.3 ± 1.6b | 5.8 ± 0.6 |
| CLP + chlorogenic acid | 11.5 ± 0.5c | 11.5 ± 0.6 | 69.7 ± 1.0c | 7.3 ± 0.6 |
| Thymocytes | ||||
| Sham | 12.3 ± 0.1 | 43.0 ± 1.0 | 34.1 ± 1.0 | 10.6 ± 0.1 |
| CLP | 3.3 ± 0.1b | 65.5 ± 0.8b | 22.8 ± 0.6b | 8.3 ± 0.2 |
| CLP + chlorogenic acid | 9.5 ± 0.4c | 42.6 ± 1.2c | 39.9 ± 1.6c | 8.1 ± 0.5 |
The effect of chlorogenic acid (20 mg/kg) on lymphocyte subpopulations was determined via flow cytometry and CD markers. The results are presented as mean ± SEM. n = 5–7 per group.
Statistically significant differences from the sham group (P < 0.05).
Statistically significant differences from the sham group (P < 0.01).
Statistically significant differences from the vehicle-treated CLP group (P < 0.01).
Chlorogenic Acid Enhances Bacterial Clearance In Vivo
We examined whether the administration of chlorogenic acid enhanced the clearance of CLP-induced bacteria. CFUs in peritoneal fluid, blood, lung and spleen were counted 24 h after CLP. In peritoneal fluid, blood and lung, chlorogenic acid administration significantly decreased the number of CFUs, but in the spleen, the decrease in CFUs induced by chlorogenic acid failed to reach statistical significance (Figures 7A–D). To investigate the effect of chlorogenic acid treatment on bactericidal activity, the levels of NO and hydrogen peroxide production by macrophages were measured. Chlorogenic acid at concentrations of 10, 20, and 40 μmol/L activated NO (1.4-, 1.6- and 2.0-fold, respectively) and hydrogen peroxide (1.2-, 1.5- and 1.6-fold, respectively) production. When measured with zymosan and NBT, chlorogenic acid at 10, 20 and 40 μmol/L increased the phagocytic activity of macrophages (1.3-, 1.3-and 1.5-fold, respectively).
Figure 7.

Chlorogenic acid enhanced in vivo bacterial clearance. CFUs were counted in peritoneal fluid (A), blood (B), lung (C) and spleen (D) 24 h after CLP to examine whether the administration of chlorogenic acid (20 mg/kg) enhanced the clearance of CLP-induced bacteria. After serial dilutions with PBS, peritoneal fluid, blood and tissue homogenates were cultured overnight on blood-agar base plates maintained at 37°C. Data are presented as mean ± SEM; n = 8. Statistically significant differences compared with the sham group (**P < 0.01). Statistically significant differences compared with the vehicle-treated CLP group (+P < 0.05 and ++P < 0.01).
DISCUSSION
Despite extensive research, the pathophysiology of sepsis is still poorly understood. Historically, sepsis was considered to be caused by hyperactivation of the inflammatory system in response to microbial infection or trauma. However, because of the failure of antiinflammatory therapies to improve prognosis in septic patients and the fact that most septic patients succumb after the hyperinflammatory phase, we began to place equal emphasis on the state of “immune paralysis” during sepsis. Immune paralysis occurs when important signaling pathways collapse and the immune system fails, leading to prolonged immunosuppression in the later stages of sepsis (21). Substances that act on various signaling cascades are likely to be more beneficial in the treatment of sepsis compared with drugs that target a single mediator. Therefore, in this study, we characterized chlorogenic acid as a potential therapy in sepsis.
The magnitude of the inflammatory response influences the development of tissue damage and multiorgan dysfunction in sepsis, and advances have been made in understanding the role of inflammatory mediators in the pathogenesis of sepsis. TNF and IL-1β, powerful pathological cytokines, cause cell degeneration, apoptosis and necrosis, leading to multiorgan dysfunction. Chlorogenic acid treatment significantly lowered TNF-α and IL-1β levels in vivo after the induction of sepsis as well as in vitro. IL-6 is an important pleiotropic acute reactant cytokine involved in inflammatory responses, with elevated plasma levels documented in numerous conditions such as shock, trauma and liver allograft rejection (22). However, IL-6 was found to have potent antiinflammatory properties, particularly in preventing injuries related to endotoxemia (23). Furthermore, IL-6 is produced by various types of lymphoid and nonlymphoid cells (24). Chlorogenic acid significantly reduced IL-6 level in septic mice, but not in a peritoneal macrophage treated by LPS. It is now evident that specifically targeting proinflammatory cytokines, such as TNF-α, IL-1β and IL-6, is not an effective treatment for sepsis because these cytokines are released early in the development of the inflammatory response, even before a patient is diagnosed with sepsis. Recently, HMGB1 was identified as a late mediator of lethal systemic inflammation in sepsis (25). HMGB1 is released from endotoxin-stimulated macrophages in the late phase of sepsis, approximately 10 h after TNF and IL-1β have peaked. A similar delay in HMGB1 accumulation is observed in the serum of animals exposed to endotoxin (2). Consistent with these findings, anti-HMGB1 therapies have shown the ability to protect against sepsis-induced mortality (26). The delayed kinetics and protective role of anti-HMGB1 therapies in vivo indicate that HMGB1 is an important therapeutic target for the treatment of sepsis (13). In this study, chlorogenic acid suppressed the accumulation of serum HMGB1 in septic mice and inhibited HMGB1 release from macrophages. Our results indicate that chlorogenic acid inhibits HMGB1 release from macrophages, attenuates circulating levels and improves survival in septic mice. The sources of circulating HMGB1 are secretion from monocytes and macrophages in response to LPS, TNF-α or IL-1β and release from damaged or necrotic cells (27).
Our study showed that the administration of chlorogenic acid in septic mice effectively reduced mortality. CLP is generally accepted as the most clinically relevant animal model for sepsis and has been a mainstay of basic sepsis research. In the CLP model, chlorogenic acid given at 20 mg/kg was a potent and efficacious therapy. The effect of chlorogenic acid was equivalent to the efficacy conferred by ethyl pyruvate, an HMGB1 inhibitor, which we also used in the current study. Chlorogenic acid ameliorated multiorgan dysfunction, enhanced bacterial elimination and inhibited the actions of LPS. Multiorgan dysfunction is the critical cause of death in patients with sepsis. Levels of LDH, BUN, creatinine and AST peaked 12 h after the induction of sepsis, whereas ALT peaked 24 h after CLP. The time-dependent profile of CLP-induced enzymes shows the differences in the vulnerability of various organs affected by sepsis. Histological analysis also confirmed that CLP was associated with substantially increased inflammatory cell infiltration and congestion. Although leukocyte migration to the site of infection is crucial for the local control of bacterial growth and the prevention of bacterial dissemination, during the septic response, excessive cellular infiltrates cause direct tissue damage by releasing lysosomal enzymes and superoxide-derived free radicals (28). The tissue damage induced by oxygen free radicals may lead to organ dysfunction. The previously reported potent antioxidant activity of chlorogenic acid may have provided protection against sepsis-induced multiorgan dysfunction (4). In addition to the ICAM-1 expression of the surface of cells, increased levels of circulating soluble form of ICAM-1 were found in septic patients and correlated with severity of sepsis, as well as subsequent organ failure and eventual outcome (29). The results of this study indicated that increased cellular adhesion, an important contributor to septic pathophysiology, was reversed by chlorogenic acid administration, and this decrease in cellular adhesion protected vital organs from sepsis-induced damage and dysfunction. Defects in monocyte function play an important role in sepsis-induced mortality. Enhanced in vivo bacterial elimination after CLP and increased phagocytosis suggested that chlorogenic acid stimulated macrophages, which may have contributed to improved innate immunity and increased survival in murine sepsis (30).
Previous studies have suggested that NF-κB and MAPK are involved in important signaling pathways involved in the proinflammatory functions of HMGB1 (31). The classic NF-κB pathway is essential for innate immunity, and the activation and nuclear translocation of NF-κB is associated with increased transcription of genes encoding adhesion molecules, cytokines, chemokines and growth factors. NF-κB, which resides in the cytoplasm, translocates to the nucleus when activated and induces gene transcription. In the presence of appropriate stimuli, NF-κB can be activated within minutes by the degradation of IκB-α. Stimuli that rapidly activate NF-κB include TNF-α, IL-1β, T-cell activation signals and stress inducers (32). The identification of TLRs as specific pattern recognition molecules and the finding that stimulation of TLRs results in the activation of NF-κB has improved our understanding of how several different factors activate NF-κB (33). Signaling via the TLR4 receptor is responsible for gram-negative bacteria-induced activation of IκB kinase (IKK), kinase complex phosphorylation, degradation of IκB, nuclear translocation of NF-κB and enhanced expression of NF-κB–dependent proinflammatory cytokine genes (34). In the systemic HMGB1-induced inflammation model, pattern recognition receptors, especially TLR4 and the receptor for advanced glycation end products (RAGE), were implicated in release of cytokines, activation of coagulation and neutrophil recruitment (35). Recent studies have also reported that TLR4 is required for HMGB1-dependent activation of macrophage TNF release (36). Studies have shown that HMGB1-induced enhancement of NF-κB–dependent transcription involves the IKK complex in a similar manner to LPS. The MAPK family represents a group of proteins that is involved in signal transduction from a variety of cellular stimuli. The JNK subgroup of MAPKs, also known as stress-activated protein kinases, are activated in response to environmental stresses, and JNK activation was reported in the liver after hemorrhagic shock (37). The engagement of TLR4 results in phosphorylation and activation of p38, and activated p38 modulates NF-κB transcriptional activity. In this manner, activated p38 plays a critical role in the HMGB1-induced enhancement of NF-κB activation. Therefore, we investigated the potential inhibitory effects of chlorogenic acid on CLP-induced activation of MAPKs and NF-κB. Our results showed that chlorogenic acid significantly inhibited NF-κB, JNK and p38 MAPK activation, but no ERK inhibitory effects were observed. Treatment with chlorogenic acid was also able to suppress the increase in TLR4 mRNA expression in the liver, heart and kidney of CLP mice.
In human sepsis, two immunologically distinct stages are observed: an early proinflammatory phase and a late, compensatory antiinflammatory phase. CLP-induced sepsis increases lymphocyte apoptosis, which causes immunosuppression during the late phase of human sepsis (1). Immunological cytokines are functionally categorized into two groups: those with Th1 properties or those with Th2 properties. Activated CD4+ T cells are programmed to secrete cytokines with inflammatory (Th1-type cytokines; IFN-γ, IL-2 and IL-12) or antiinflammatory (Th2-type cytokines; IL-4 and IL-10) properties (38). In addition to attenuating the abrupt release of proinflammatory cytokines such as TNF-α, IL-1β and IL-6, treatment with chlorogenic acid augmented serum Th1 cytokines during the late phase of sepsis without affecting Th2 cytokine release. A similar pattern of cytokine regulation was observed in LPS-stimulated macrophages incubated with chlorogenic acid. The differential effects of chlorogenic acid on different types of cytokines may be caused by distinct sets of target cells that chlorogenic acid affects. Accumulating data suggest that increases in Th1 cytokines are beneficial in the late-stage of sepsis when immuno-suppression predominates and can cause death (39). Decreased Th1 function was reported in patients with peritonitis, and defective T-cell proliferation and cytokine secretion correlates with mortality (40). Although the chlorogenic acid– induced changes in cytokine levels may not play a key role in the protective effect of chlorogenic acid because of the transient nature of these effects, we cannot exclude the possibility that the combined effect of the changes in cytokines may partially contribute to improved survival.
Previous research has shown that HMGB1 induces the secretion of chemokines MCP-1 and MIP-2 (41). MCP-1 belongs to the CC chemokine family and is under the control of NF-κB, Sp1 and early growth response-1 (42,43). MCP-1 recruits immune cells, such as monocytes and T lymphocytes, to sites of infection, and it is responsible for various inflammatory reactions (44). MIP-2 is a member of the CXC chemokine family of inflammatory and immunoregulatory cytokines. Increased MIP-2 expression was associated with neutrophil influx in various inflammatory conditions (45). In this study, the serum level of MCP-1 increased significantly 3 h after CLP, and the MCP-1 level steadily increased until 12 h after CLP. Treatment with chlorogenic acid augmented this increase 3 h after CLP. The level of MIP-2 increased after CLP, and chlorogenic acid suppressed this increase both 3 and 6 h after CLP. In LPS-stimulated macrophages incubated with chlorogenic acid, a similar pattern was observed. These changes in chemokines support the chlorogenic acid–induced changes in cytokine levels.
The programmed cell death of immune cells, responsible for the substantially impaired immune response observed in sepsis, is a novel and potentially important therapeutic target in the treatment of sepsis. In CLP, previous reports demonstrated profound thymyocyte apoptosis due to robust complement activation (46). Also, treatment with antibodies to C5a or its receptor (C5aR) was found to improve survival (47). Results obtained by flow cytometric analysis indicated that chlorogenic acid prevented immune cell apoptosis. Apoptosis after CLP is related to the appearance of C5a and its interaction with C5a receptors C5aR and C5L2 (48). However, further investigation is necessary to elucidate the exact mechanisms of the effect of chlorogenic acid on lymphocyte apoptosis. Further evidence to support our observations included the fact that administration of chlorogenic acid to mice undergoing CLP-induced sepsis markedly enhanced bacterial clearance and activated the phagocytic activity of macrophages in vitro.
CONCLUSION
The current study shows that chlorogenic acid may have therapeutic potential for the treatment of sepsis. Chlorogenic acid is an effective modulator of inflammatory mediators and an inhibitor of lymphocyte apoptosis. No adverse effects of chlorogenic acid were observed in this experimental system. Indeed, chlorogenic acid is relatively nontoxic, and the protective effects were demonstrated in therapeutically achievable and safe doses. However, extending the use of chlorogenic acid to human patients will depend on dosage. This work identifies chlorogenic acid as a new therapeutic agent for the treatment of sepsis, as a multistep therapeutic agent for use in combination with other immunomodulatory agents or as a supplement to other therapies. The promising results presented here may warrant prospective clinical trials.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 3.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–7. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niggeweg R, Michael AJ, Martin C. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nat Biotechnol. 2004;22:746–54. doi: 10.1038/nbt966. [DOI] [PubMed] [Google Scholar]
- 5.Stoclet JC, et al. Vascular protection by dietary polyphenols. Eur J Pharmacol. 2004;500:299–313. doi: 10.1016/j.ejphar.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 6.Olmos A, et al. Effects of plant alkyl-phenols on cytokine production, tyrosine nitration and inflammatory damage in the efferent phase of contact hypersensitivity. Br J Pharmacol. 2007;152:366–73. doi: 10.1038/sj.bjp.0707402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, et al. Chlorogenic acid protects mice against lipopolysaccharide-induced acute lung injury. Injury. 2010;41:746–52. doi: 10.1016/j.injury.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, et al. Protective effects of chlorogenic acid on acute hepatotoxicity induced by lipopoly saccharide in mice. Inflamm Res. 2010;59:871–7. doi: 10.1007/s00011-010-0199-z. [DOI] [PubMed] [Google Scholar]
- 9.Feng R, et al. Inhibition of activator protein-1, NF-kappaB, and MAPKs and induction of phase 2 detoxifying enzyme activity by chlorogenic acid. J Biol Chem. 2005;280:27888–95. doi: 10.1074/jbc.M503347200. [DOI] [PubMed] [Google Scholar]
- 10.Cho ES, et al. Attenuation of oxidative neuronal cell death by coffee phenolic phyto-chemicals. Mutat Res. 2009;661:18–24. doi: 10.1016/j.mrfmmm.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 11.Institute of Laboratory Animal Resources; Commission on Life Sciences; National Research Council. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academy Press; 1996. [cited 2012 Mar 15]. Available from: http://www.nap.edu/openbook.php?record_id=5140. [Google Scholar]
- 12.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulloa L, et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99:12351–6. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 15.Freire-Garabal M, et al. Effects of fluoxetine on the activity of phagocytosis in stressed mice. Life Sci. 2002;72:173–83. doi: 10.1016/s0024-3205(02)02207-5. [DOI] [PubMed] [Google Scholar]
- 16.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages: comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–12. [PubMed] [Google Scholar]
- 17.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–65. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 18.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JS, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 20.Williams DL, et al. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–18. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- 21.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebersole JL, Cappelli D. Acute-phase reactants in infections and inflammatory diseases. Periodontol 2000. 2000;23:19–49. doi: 10.1034/j.1600-0757.2000.2230103.x. [DOI] [PubMed] [Google Scholar]
- 23.Heremans H, Dillen C, Put W, Van Damme J, Billiau A. Protective effect of anti-interleukin (IL)-6 antibody against endotoxin, associated with paradoxically increased IL-6 levels. Eur J Immunol. 1992;22:2395–401. doi: 10.1002/eji.1830220932. [DOI] [PubMed] [Google Scholar]
- 24.Kishimoto T. The biology of interleukin-6. Blood. 1989;74:1–10. [PubMed] [Google Scholar]
- 25.Czura CJ, Tracey KJ. Targeting high mobility group box 1 as a late-acting mediator of inflammation. Crit Care Med. 2003;31:S46–50. doi: 10.1097/00003246-200301001-00007. [DOI] [PubMed] [Google Scholar]
- 26.Li W, et al. A cardiovascular drug rescues mice from lethal sepsis by selectively attenuating a late-acting proinflammatory mediator, high mobility group box 1. J Immunol. 2007;178:3856–64. doi: 10.4049/jimmunol.178.6.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 28.Salvemini D, Cuzzocrea S. Therapeutic potential of superoxide dismutase mimetics as therapeutic agents in critical care medicine. Crit Care Med. 2003;31:S29–38. doi: 10.1097/00003246-200301001-00005. [DOI] [PubMed] [Google Scholar]
- 29.Sessler CN, et al. Circulating ICAM-1 is increased in septic shock. Am J Respir Crit Care Med. 1995;151:1420–7. doi: 10.1164/ajrccm.151.5.7735595. [DOI] [PubMed] [Google Scholar]
- 30.Muller Kobold AC, et al. Leukocyte activation in sepsis; correlations with disease state and mortality. Intensive Care Med. 2000;26:883–92. doi: 10.1007/s001340051277. [DOI] [PubMed] [Google Scholar]
- 31.Yu M, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 32.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 33.Kopp EB, Medzhitov R. The Toll-receptor family and control of innate immunity. Curr Opin Immunol. 1999;11:13–8. doi: 10.1016/s0952-7915(99)80003-x. [DOI] [PubMed] [Google Scholar]
- 34.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 35.van Zoelen MA, et al. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–4. doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H, et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–7. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCloskey CA, Kameneva MV, Uryash A, Gallo DJ, Billiar TR. Tissue hypoxia activates JNK in the liver during hemorrhagic shock. Shock. 2004;22:380–6. doi: 10.1097/01.shk.0000140660.78744.bf. [DOI] [PubMed] [Google Scholar]
- 38.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 39.Weighardt H, et al. Impaired monocyte IL-12 production before surgery as a predictive factor for the lethal outcome of postoperative sepsis. Ann Surg. 2002;235:560–7. doi: 10.1097/00000658-200204000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heidecke CD, et al. Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am J Surg. 1999;178:288–92. doi: 10.1016/s0002-9610(99)00183-x. [DOI] [PubMed] [Google Scholar]
- 41.Fiuza C, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 42.Yan SF, et al. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–61. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 43.Ping D, et al. Sp1 binding is critical for promoter assembly and activation of the MCP-1 gene by tumor necrosis factor. J Biol Chem. 2000;275:1708–14. doi: 10.1074/jbc.275.3.1708. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimura T, Leonard EJ. Human monocyte chemoattractant protein-1 (MCP-1) Adv Exp Med Biol. 1991;305:47–56. doi: 10.1007/978-1-4684-6009-4_6. [DOI] [PubMed] [Google Scholar]
- 45.Standiford TJ, Kunkel SL, Greenberger MJ, Laichalk LL, Strieter RM. Expression and regulation of chemokines in bacterial pneumonia. J Leukoc Biol. 1996;59:24–8. doi: 10.1002/jlb.59.1.24. [DOI] [PubMed] [Google Scholar]
- 46.Huber-Lang MS, et al. Complement-induced impairment of innate immunity during sepsis. J Immunol. 2002;169:3223–31. doi: 10.4049/jimmunol.169.6.3223. [DOI] [PubMed] [Google Scholar]
- 47.Czermak BJ, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–92. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 48.Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol. 2008;76:1383–8. doi: 10.1016/j.bcp.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]