

To the editor
Wiskott–Aldrich syndrome (WAS) is a rare X-linked disorder caused by mutations in the WAS gene, and it is characterized by the triad of thrombocytopenia, eczema, and susceptibility to infection1. WAS exhibits extensive clinical variability, and the genotypes of WAS patients are also highly variable1. Mutations impairing but not abolishing WAS protein expression can cause X-linked thrombocytopenia (XLT), an attenuated form of WAS with minimal or no immunodeficiency1. Autoimmune complications are exceedingly common in WAS/XLT and affect 40–70% of patients according to retrospective cohort studies1. Glomerulonephritis is a frequent complication of WAS/XLT. It is found in 3.5–19% of cases and can progress to chronic renal failure requiring renal transplantation2. Immunoglobulin A (IgA) nephropathy (IgAN) has been diagnosed in a majority of WAS/XLT patients2, 3.
Previous reports revealed that aberrantly O-glycosylated IgA which exhibits galactose deficiency in the O-linked glycans in the hinge region of the heavy chain may play a pivotal role in the pathogenesis of these IgAN4. We have previously reported that Was-knockout mice develop proliferative glomerulonephritis reminiscent of human IgAN5. We measured serum levels of aberrantly glycosylated IgA in Was- knockout mice using lectin-binding assays with elderberry (Sambucus nigra) bark and ricinus communis agglutinin I, which recognize terminal sialic acid and galactose residues6. These results indicated aberrant IgA production may be critically involved in the pathogenesis of glomerulonephritis in these mice5. Furthermore, aberrant O-glycosylation of serum IgA with characteristics similar to that observed in IgAN was detected in a WAS-carrier patient who presented with Henoch-Schönlein purpura (HSP)6. In this report, we evaluated the role of aberrant IgA production in the development of autoimmunity and glomerulonephritis in WAS/XLT patients.
Serum samples were obtained from 26 patients with WAS or XLT and 20 age matched control. The clinical characteristics of the patients with WAS/XLT are shown in Table e1. Eleven patients presented with autoimmune complications including IgAN, vasculitis, arthritis, colitis, autoimmune hemolytic anemia, and autoimmune thrombocytopenia. To examine the terminal galactosylation of IgA molecules, we used lectin-binding assays with Helix aspersa (HAA), which recognizes terminal N-acetylgalactosamine (GalNAc) residues7 (for more information, see the Methods section in this article's Online Repository.)
Table e1.
| patient | age(yr) | sex | diagnosis | autoimmune diseases |
|---|---|---|---|---|
| 1 | 1 | Male | XLT | no |
| 2 | 2 | Male | XLT | no |
| 3 | 24 | Male | XLT | no |
| 4 | 10 | Male | XLT | no |
| 5 | 22 | Male | XLT | no |
| 6 | 16 | Male | XLT | no |
| 7 | 30 | Male | XLT | no |
| 8 | 2 | Male | XLT | no |
| 9 | 8 | Male | XLT | no |
| 10 | 1 | Male | XLT | no |
| 11 | 15 | Male | XLT | no |
| 12 | 25 | Male | WAS | no |
| 13 | 3 | Male | WAS | no |
| 14 | 4 | Male | WAS | no |
| 15 | 1 | Male | WAS | no |
| 16 | 18 | Male | XLT | IgA nephropathy |
| 17 | 23 | Male | WAS | Autoimmune hemolytic anemia |
| 18 | 6 | Male | WAS | Autoimmune thrombocytopenia |
| 19 | 22 | Male | WAS | systemic vasculitis |
| 20 | 28 | Male | WAS | IgA nephropathy |
| 21 | 13 | Male | WAS | Colitis |
| 22 | 23 | Male | WAS | IgA nephropathy |
| 23 | 1 | Male | WAS | Vasculitis |
| 24 | 41 | Male | WAS | Arthritis |
| 25 | 28 | Male | WAS | IgA nephropathy |
| 26 | 35 | Male | WAS | IgA nephropathy |
The serum levels of galactose-deficient IgA in WAS/XLT patients were significantly higher than those in the control group (p<0.001) (Figure 1A). Interestingly, the levels of galactose-deficient IgA were not significantly higher in WAS patients than in XLT patients. However, galactose-deficient IgA levels were significantly higher in WAS/XLT patients with autoimmune complications than in patients without autoimmune complications (p<0.05) (Figure 1A). In addition, galactose-deficient IgA levels were significantly higher in WAS/XLT patients older than 10 years compared to age matched control. (p<0.0001) (Figure 1B). In WAS/XLT patients, we found that galactose-deficient IgA increased in an age-dependent manner (Figure 1C).
Figure 1. Increased serum galactose-deficient IgA levels and CIC containing IgA and IgG in WAS/XLT.
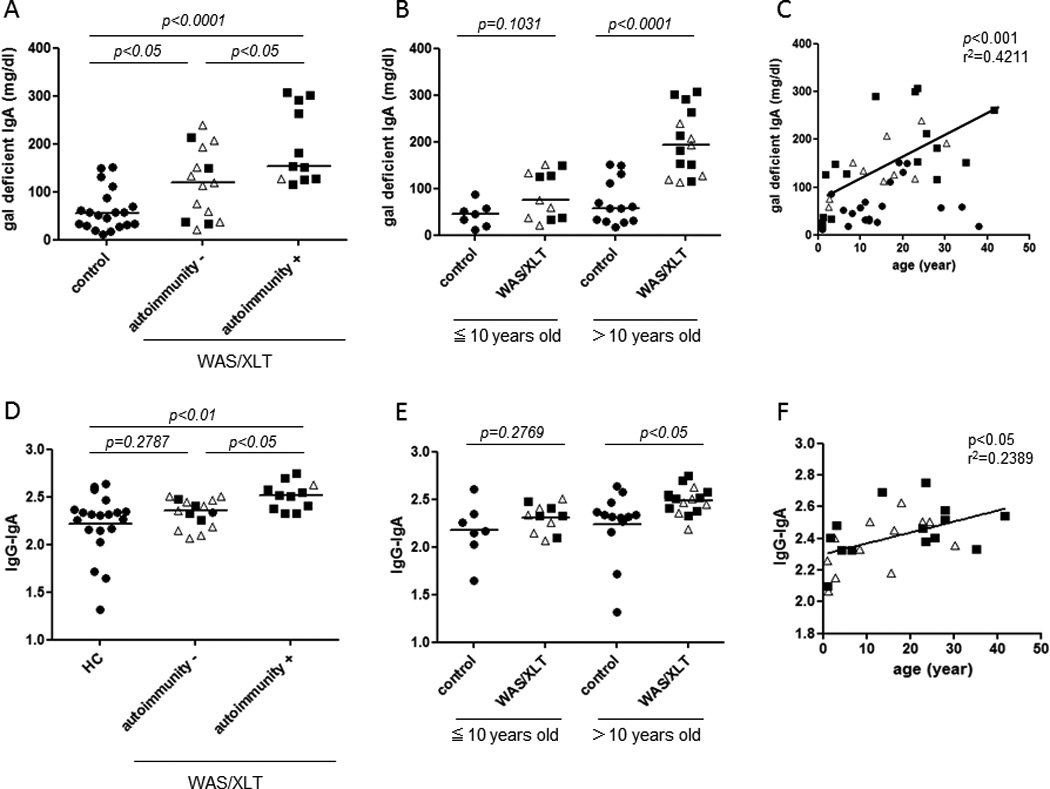
The bars represent the median values. Open triangles, XLT; filled squares, WAS.
A. Serum levels of galactose-deficient IgA
B,C. Correlation between serum galactose-deficient IgA levels and age.
D. Serum levels of IgG-IgA CIC
E,F. The correlation between serum levels of IgG-IgA CIC and age.
Next, levels of CIC containing IgA and IgG (IgG-IgA CIC) in serum were determined by sandwich ELISA. These levels were found to be significantly higher in WAS/XLT patients with autoimmune disorders than in the control group (p<0.01) (Figure 1D). Notably, IgG-IgA CIC levels were significantly higher in WAS/XLT patients than in patients without such complications (p<0.05) (Figure 1D). In addition, IgG-IgA CIC levels were significantly higher in WAS/XLT patients older than 10 years of age compared to age-matched controls (p<0.05) (Figure 1E). Similarly to galactose-deficient IgA levels, the elevation of IgG-IgA CICs was age-dependent (Figure 1F). Interestingly, IgG-IgA CIC levels were positively correlated with the serum levels of galactose-deficient IgAs (Supplementary Figure).
Patient 16 underwent BMT3. After BMT, he achieved full donor chimerism and his clinical symptoms were improved. Laboratory findings 24 months after BMT were as follows: platelets increased from 8.7×109/L to 23.2×109/L; serum IgA levels decreased from 264 mg/dl to 131 mg/dl; urinalysis results changed from 3+ proteinuria and sediment containing over 100 red blood cells/high-power field with casts, to normal findings. The IgA-dominant immune deposits in the renal mesangium and glomerular injury significantly improved after BMT (Figure 2A) and the serum levels of galactose-deficient IgA also markedly decreased (Figure 2B).
Figure 2. Improved XLT-associated IgAN and decreased the production of aberrant IgA after BMT.

A, B: Immunofluorescence (IF) studies of kidney biopsy specimens from patient 16.
C: Changes of the levels of galactose-deficient IgA after BMT
In this study, we demonstrated that serum galactose-deficient IgA levels and the levels of IgG-IgA CIC increased in WAS/XLT patients in an age dependent manner. Interestingly, these levels significantly increased in WAS/XLT patients with autoimmune manifestations. These findings suggest that increased levels of galactose-deficient IgA with age might be one of the factors contributing to the development of autoimmunity in WAS/XLT. Furthermore, we observed that IgA-dominant immune deposits in the renal mesangium and glomerular injury in an XLT patient (patient 16) were significantly reduced after BMT. Galactose-deficient IgA levels were also markedly decreased after BMT. These findings indicate that aberrant IgA production in WAS/XLT may result from the immune dysregulation associated with the syndrome and lead to the development of IgAN as one of the several autoimmune complications of WAS that can be corrected with immune reconstitution by BMT.
Some of our WAS/XLT patients without autoimmune complications showed increased serum levels of galactose-deficient IgA and IgG-IgA CIC. It is possible that increase in IgA and IgG-IgA CIC serum levels precede the clinical presentation of IgAN. Therefore, careful monitoring is considered for these patients.
Altered oligosaccharide biosynthesis in lymphocytes from WAS patients has been extensively studied8; however, the specific mechanisms responsible for the aberrant glycosylation of IgA remain unclear. Previous research suggested the possibility of age-dependent increases in the B cell populations that produce aberrant IgA5. Interestingly, our results showed aberrant glycosylation of IgA and production of IgG-IgA CIC in WAS/XLT patients are also increased in an age-dependent fashion. Another possibility is that IgA molecules may be influenced by other factors induced by WASp deficiency, including Th2-dominant changes in cytokine balance5, 9. Further studies, however, are necessary to validate this hypothesis and clarify the mechanisms responsible for the increased aberrant IgA production in WAS patients.
Overall, our observations suggest that aberrant IgA production plays a role in the pathogenesis of the renal disease observed in WAS patients, perhaps through the accumulation of IgA-containing CIC and consequent glomerular injury.
In conclusion, our findings indicate that aberrant IgA production due to mutations in the WAS gene may be critically involved in the development of autoimmune-mediated glomerulonephritis. Further studies are required to clarify the specific mechanisms responsible for the aberrant glycosylation of IgA in WAS.
Supplementary Material
Acknowledgement
This work was supported by Grant-in-Aid for Young Scientists from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by intramural funds of the National Human Genome Research Institute, NIH, Bethesda, MD, USA. We thank Ms. Chikako Sakai and Mr. Hitoshi Moriuchi for their excellent technical assistance. We also wish to acknowledge Dr. Hiro Matsukura who performed renal biopsy in patient 16.
Abbreviations
- BMT
bone marrow transplantation
- BSA
bovine serum albumin
- CIC
circulating immune complexes
- ELISA
enzyme-linked immunosorbent assay
- HAA
Helix aspersa
- HRP
horse radish peroxidase
- HSP
Henoch-Shönlein purpura
- IgA
immunoglobulin A
- IgAN
IgA nephropathy
- OD
optical density
- PBS
phosphate-buffered saline
- WAS
Wiskott–Aldrich syndrome
- WASp
WAS protein
- XLT
X-linked thrombocytopenia
Footnotes
Disclosure
The authors have no financial conflicts of interest to disclose.
References
- 1.Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia RM. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113:6288–6295. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- 2.Liu CH, Wu KH, Lin TY, Wei CC, Lin CY, Chen XX, Lee WI. Wiskott-Aldrich syndrome with IgA nephropathy: a case report and literature review. Int Urol Nephrol. 2012 doi: 10.1007/s11255-012-0178-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3.Matsukura H, Kanegane H, Miya K, Ohtsubo K, Higuchi A, Tanizawa T, et al. IgA nephropathy associated with X-linked thrombocytopenia. Am J Kidney Dis. 2004;43:e7–e12. doi: 10.1053/j.ajkd.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 4.Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgAN consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu M, Nikolov NP, Ueno K, Ohta K, Siegel RM, Yachie A, et al. Development of IgA nephropathy-like glomerulonephritis associated with Wiskott�Aldrich syndrome protein deficiency. Clin Immunol. 2012;142:160–166. doi: 10.1016/j.clim.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasseur C, Allen AC, Demini�re C, Aparicio M, Feehally JC, Combe C. Henoch-Schöenlein purpura with immunoglobulin A nephropathy and abnormal IgA in Wiskott-Aldrich syndrome carrier. Am J Kidney Dis. 1997;29:285–287. doi: 10.1016/s0272-6386(97)90043-3. [DOI] [PubMed] [Google Scholar]
- 7.Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148–1154. doi: 10.1038/sj.ki.5002185. [DOI] [PubMed] [Google Scholar]
- 8.Higgins EA, Siminovitch KA, Zhuang DL, Brockhausen I, Dennis JW. Aberrant O-linked oligosaccharide biosynthesis in lymphocytes and platelets from patients with the Wiskott-Aldrich syndrome. J Biol Chem. 1991;266:6280–6290. [PubMed] [Google Scholar]
- 9.Nguyen DD, Maillard MH, Cotta-de-Almeida V, Mizoguchiv E, Klein C, Fuss I, Nagler C, Mizoguchi A, Bhan AK, Snapper SB. Lymphocyte-dependent and Th2 cytokine-associated colitis in mice deficient in Wiskott-Aldrich syndrome protein. Gastroenterology. 2007;133:1188–1197. doi: 10.1053/j.gastro.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.