

Abstract
Adoptive cell transfer (ACT) of ex vivo activated autologous tumor-reactive T cells is currently one of the most promising approaches for cancer immunotherapy. Recent studies provided some evidence that Th17/Tc17 cells may exhibit potent anti-tumor activity, but the specific mechanisms have not been completely defined. In the present study, we used a murine melanoma lung-metastasis model and tested the therapeutic effects of gp100-specific polarized Tc1 or Tc17 cells combined with autologous BMT after total TBI. BMT combined with ACT of anti-tumor (gp100-specific) Tc17 cells significantly suppressed the growth of established melanoma, whereas Tc1 cells induced long-term tumor regression. After ACT, Tc1 cells maintained their phenotype to produce IFNγ but not IL-17. However, although Tc17 cells largely preserved their ability to produce IL-17, a subset secreted IFNγ or both IFNγ and IL-17, indicating the plasticity of Tc17 cells in vivo. Furthermore, after ACT the Tc17 cells had a long-lived effector T cell phenotype (CD127hi/KLRG-1low) as compared to Tc1 cells. Mechanistically, Tc1 cells mediated anti-tumor immunity primarily through the direct effect of IFNγ on tumor cells. In contrast, despite the fact that some Tc17 cells also secreted IFNγ, Tc17-mediated anti-tumor immunity was independent of the direct effects of IFNγ on the tumor. Nevertheless, IFNγ played a critical role by creating a microenvironment that promoted Tc17-mediated anti-tumor activity. Taken together, these studies demonstrate that both Tc1 and Tc17 cells can mediate effective anti-tumor immunity through distinct effector mechanisms, but Tc1 cells are superior to Tc17 cells in mediating tumor regression.
Introduction
CD4 and CD8 T lymphocytes can be classified into distinct types of effector cells based on their cytokine-secretion profiles after antigen stimulation (1-4) Tc1 cells secrete IFN-γ and kill tumor targets by either perforin- or Fas-mediated mechanisms, whereas Tc2 cells secrete IL-4, IL-5, and IL-10 and kill tumor targets predominantly through the perforin pathway. Tc17 cells secrete IL-17A, IL-17F, IL-21 and IL-22 and also possess killing activity that can result in anti-tumor responses (4, 5). Although the contribution of adoptively transferred Th1 and Tc1 cells in anti-tumor responses has been clearly established, the role of Th17 and Tc17 cells remains controversial (5-7). After skewing primed naïve CD4 T cells towards a Th17 phenotype, IL-17 was shown to induce Th1-type chemokines (8), recruiting effector cells to the tumor microenvironment. Conversely, Th17 can promote IL-6–mediated Stat3 activation, generating a pro-tumorigenic environment (9, 10). One study showed that tumor-specific Th17 cells exhibited stronger therapeutic efficacy than Th1 cells upon adoptive transfer, and were converted into effective IFN-γ producing cells (5) that promoted the expansion, differentiation, and homing of tumor-specific CD8+ T cells into the tumor microenvironment (11). In addition, adoptive transfer of tumor reactive Tc17 cells also reduced the volume of established tumors while differentiating into long-lasting IFN-γ secreting cells (4). Therefore, IL-17 and IFNγ produced by T cells in the tumor microenvironment may determine whether these cytokines negatively or positively may affect tumor growth.
ACT of ex vivo activated autologous tumor-reactive T cells is currently one of the most promising approaches for the treatment of patients with advanced melanoma (12-14). Therapeutic efficiency mediated by ACT is dependent in part on the ability of tumor Ag-specific T cells to persist and to retain their anti-tumor activity in vivo. ACT in combination with non-myeloablative lymphoid depletion has emerged as one of the most effective anti-tumor therapies. The mechanisms involved in this mode of immunotherapy include the elimination of regulatory T cells and the increased cytokine-dependent homeostatic proliferation of effector T cells (12, 15). The improved effectiveness of ACT following a non-myeloablative lymphodepletion provides the rational basis for the evaluation of more intensive conditioning regimens such as a myeloablative regimen in conjunction with BMT. In fact, more recent studies have indicated that hematopoietic stem cells promote the expansion and function of anti-tumor CD8+ T cells after ACT into tumor-bearing hosts (16, 17).
In the present study, we used a murine melanoma model to assess the therapeutic effects of tumor-specific, polarized Tc1 or Tc17 cells combined with autologous BMT after TBI. While ACT with tumor-specific Tc1 or Tc17 cells induced significant regression of established large tumors, Tc1 cells showed higher therapeutic efficacy than Tc17 cells. Furthermore, our results show that Tc1 and Tc17 cells mediated their anti-tumor effects through distinct mechanisms.
Materials and Methods
Animals
C57BL/6 (B6) (H-2b) mice were purchased from the NCI/Charles River program. Pmel-1 mice (B6 background) were purchased from the Jackson Laboratory. All animals were housed in the American Association for Laboratory Animal Care-accredited Animal Resource Center at Moffitt Cancer Center. All experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee.
Tumor cells
The luciferase-transduced B16-F10 cell line, a weakly immunogenic melanoma cell derived from B6 mice, was purchased from Xenogen Corporation (Berkeley, CA). The B16-IFNγRDN tumor cell line expressing high levels of dominant negative interferon-γ receptor (IFNγRDN), which renders the cells unresponsive to the effects of this cytokine, was established as described (18). The EL4 cell line was obtained from the American Type Culture Collection (Manassas, VA).
Spleen and lung cell preparation
Spleens were collected from mice, and single-cell suspensions were prepared, washed twice in PBS, and resuspended in RPMI 1640 (Life Technologies, Grand Island, NY), supplemented with 2 mM pyruvate, 100 units/ml penicillin, 100 mg/ml streptomycin, 10 mM HEPES, and 10% heat-inactivated FCS (Life Technologies). To prepare single-cell suspension of lung parenchyma, lung tissues were minced finely and incubated in RPMI 1640 with 5% FCS, penicillin/streptomycin, 10 mM HEPES, 50 μM 2-ME, 20 mM L-glutamine containing 20 U/ml collagenase D (Sigma, St. Louis, MO) and 1 μg/ml DNAse (Sigma). After incubation for 60 min at 37°C, cells were collected by centrifugation. The cell pellet was suspended in 40% Percoll and layered onto 80% Percoll. The cells at the interphase were then collected for further analysis.
Generation of tumor-specific Tc1 and Tc17 cells
Tc1 cells recognizing hgp100 25–33 were generated using the methods described previously with slight modifications (19). In brief, splenocytes from Pmel-1 transgenic mice were stimulated with the hgp25–33 peptide (1 μg/ml), recombinant mouse IL-12 (10 ng/ml; R&D Systems) and recombinant mouse IFNγ (1000 U/ml R&D Systems). Tc17 cells were generated by methods described previously (4). Splenocytes from Pmel-1 mice were stimulated with the hgp25-33 peptide (1 μg/ml), recombinant human transforming growth factor-β (5 ng/mL; R&D Systems), and anti–IFN-γ antibodies (10 ng/mL; Bioxcell). Three days after peptide stimulation the cell cultures were expanded with recombinant mouse IL-2. Cell phenotypes were confirmed on day 4 by intracellular cytokine staining for IFNγ and IL-17.
Adoptive immunotherapy model
Syngeneic B6 mice were injected i.v. with 5×105 luciferase-transduced B16F10 or IFNγRDN melanoma cells to establish pulmonary metastases. Six days after tumor challenge, mice were conditioned with TBI (1200 cGy in split doses). Bone marrow was flushed from donor femurs and tibias with RPMI 1640 and passed through sterile mesh filters to obtain single-cell suspensions. BM cells were depleted of T cells with anti-Thy1.2 monoclonal antibody plus low-toxicity rabbit complement (C-6 Diagnostics). T-cell depleted BM cells, referred to as TCD-BM, were used for all immunotherapy experiments. On day 7 after tumor implantation, mice received ACT (i.v.) of 2 ×106 gp100-specific T cells (Tc1 or Tc17). Health status of tumor-bearing mice was monitored daily, and tumor growth was measured at specific time points using in vivo bioluminescent imaging. For the IFNγRDN tumor model, anti-tumor effects were evaluated by examination and measurements of tumor masses or by counting the number of tumor nodules in the lungs.
Antibodies and flow cytometry
The following antibodies were used for cell surface staining: anti-CD4–FITC, or -APC (L3T4), anti-CD8α-FITC, -APC, APC-cy7, anti-CD45.1–FITC, or –APC (A20), anti-CD90.1-PE or APC were purchased from eBioscience; anti-CD4-pacific blue (RM4-5) was purchased from BD Biosciences. Detection of biotinylated antibodies was performed using APC-cy7 or APC conjugated to streptavidin (BD Biosciences). Intracellular staining was carried out using anti-IFNγ-PE or Per-cp 5.5 (XMG1.2; BD Biosciences), anti-IL-17-APC (17B7; eBiosciences). Cells were analyzed on a LSR II (BD Biosciences). Data were analyzed using FlowJo (TreeStar) (Treestar, Ashland, OR). For intracellular cytokine staining, splenocytes from recipient mice at the time specified were stimulated in vitro with 50 ng/mL PMA (Sigma-Aldrich, St Louis, MO), 500 ng/mL ionomycin (Sigma-Aldrich) and 1 μL Golgi Plug (BD Biosciences), and incubated at 37°C for 4 to 5 hours before staining as described in our previous work (20).
ELISPOT assays for IFNγ and IL-17 secretion
ELISPOTs were performed according to the manufacturer’s protocol (Mabtech). Briefly, 96-well Multiscreen-IP plates (Millipore) were coated with 10 μg/ml rat anti-mouse IFNγ mAb (AN-18) or IL-17 mAb (TC11-18H10) overnight at 4°C. After washing the plates, freshly isolated splenocytes were incubated with hgp100 peptide (1 μg/ml) at 37°C for 18-20 hours in RPMI 1640 containing 10% FBS. In some ELISPOT assays, peptide-pulsed (10 μg/ml) or un-pulsed EL4 cells were used as APCs. After washing the plates and removing the cells, biotinylated rat anti-mouse IFNγ mAb (R4-6A2) or biotinylated rat anti-mouse IL-17mAb (TC11-8H4.1) was added and incubated for 2 hours at room temperature, followed by addition of streptavidin-HRP for 1–2 hours at room temperature. Spot counting was done with an ELISPOT reader system (Autoimmun Diagnostika). Experiments were performed in duplicate or triplicate wells.
Statistical Analysis
To compare the survival of tumor bearing mice in different groups, the log-rank test was used to determine statistical significance. To compare T-cell expansion or cytokine production, a paired student’s t-test was used. p values less than 0.05 were considered statistically significant.
Results
ACT with Tc1 or Tc17 cells effectively control established melanoma after myeloablative conditioning and syngeneic BMT
It has been reported that both Tc1 and Tc17 cells promote anti-tumor immunity and increase survival of mice with established subcutaneous B16 melanomas (4, 21). Given that the effectiveness of immunotherapy is enhanced by a lymphodepleting regimen and transfer of hematopoietic stem cells (17), we tested the hypothesis that the efficiency of adoptively transferred Tc1 or Tc17 cells would be enhanced by myeloablative TBI and syngeneic BMT. We injected B16-F10 cells i.v. to establish melanoma metastases in the lung. After tumor injection the mice were separated into 2 groups: one group was sublethally irradiated on day 6 and received ACT using polarized pmel-1 Tc1 or Tc17 cells on day 7; the other group was lethally irradiated on day 6 and received Tc1 or Tc17 ACT in combination with syngeneic TCD-BM on day 7. The anti-tumor effects mediated by either Tc1 or Tc17 cells were more evident in mice receiving myeloablative TBI/TCD-BMT as compared to mice treated using the non-myeloablative TBI regimen (Fig. 1A). To understand the mechanism accounting for the enhanced anti-tumor effect after myeloablative TBI/TCD-BMT, we assessed the in vivo persistence of transferred Tc1 or Tc17 cells. The numbers of Tc1 or Tc17 cells recovered from the spleens of mice that received myeloablative TBI/TCD-BMT were markedly higher as compared to mice that received the non-myeloablative TBI regimen (Fig 1B). Because infiltration of effector T cells into the tumors is critical for generating anti-tumor responses (22, 23), we analyzed the cellular composition in the lungs of the treated mice and found that higher numbers of Tc17 or Tc1 pmel-1 cells infiltrated into the lungs of mice treated with myeloablative TCD-TBI as compared to the lungs of mice that received the non-myeloablative TBI regimen (Fig. 1B). Supported by published data from others (15), we reason that the augmented proliferation and tumor infiltration of adoptively transferred pmel-1 Tc1 or Tc17 T cells were likely driven by both lymphodepletion and hematopoietic stem cells.
Figure 1.

Strong anti-tumor effect was mediated by ACT with Tc1 or Tc17 cells in the combination of TBI and BMT. (A) B6 mice were inoculated i.v. with luciferase-transduced B16-F10 melanoma cells. Six days later, tumor-bearing mice received 1200 cGy (split doses). One day after, Tc1 and Tc17 cells generated from Pmel-1/Thy1.1 mice were transferred into tumor-bearing mice together with TCD-BM cells from normal B6 mice. In the non-myeloablative regiment, Tc1 and Tc17 cells were transferred into tumor bearing mice with sublethally irradiation and no BMT. Tumor growth was monitored by in vivo bioluminescent imaging 14 days after ACT. (B) Recipient mice were injected i.v. with 5× 105 B16F10 tumor cells. Seven days later, 2×106 gp100 Ag-specific Tc1 and Tc17 cells (Thy 1.1) in combination with BM (Ly5.1) were adoptively transferred into mice bearing established metastases. Fourteen days later, spleen and lungs from individual animals were harvested, and single cell suspensions were made as described in “Materials and Methods”. Cells were stained with anti-Thy 1.1, anti-CD8, anti-ly5.1. Gates were set on the ly5.1-Thy 1.1+CD8+ T cell populations. Data shown are from a representative experiment of 2 repeating experiments showing the percentages of lung and spleen-derived Thy 1.1+CD8+ T cells.
Characterization of Tc1 and Tc17 cells after ACT using myeloablative TCD-TBI
Because the myeloablative TBI/TCD-BMT more clearly promoted the therapeutic efficacy of ACT (Fig. 1A), we selected this regimen as the platform for the remainder of our studies. Tc1 and Tc17 cells were generated in vitro as described in “Materials and Methods”, and their phenotypes were confirmed prior to ACT (Fig. 2A). To directly compare the proliferation of Tc1 and Tc17 cells in vivo, polarized cells were labeled with CSFE and adoptively transferred into lethally irradiated B16-tumor bearing mice. We observed that almost 100% (99.6 ± 0.8%) Tc1 cells but only 56% (56.3 ± 2.4%) Tc17 cells underwent through more than 3 cell division cycles, indicating that Tc1 cells proliferated substantially faster than Tc17 cells during the first 5 days after cell transfer (Fig. 2B). To monitor anti-tumor responses mediated by Tc1 and Tc17, the frequency of antigen-specific CD8 T cells that produced IFNγ and IL-17 was measured with ELISPOT ex vivo (Fig. 2C and 2D). We found that Tc1 cells had an antigen-specific response exclusively in IFNγ production that was significantly higher than Tc17 cells. In contrast, Tc17 cells had an antigen-specific response in IL-17 as well as IFNγ production. These results indicate that after adoptive transfer Tc1 cells have a stable phenotype whereas some Tc17 cells can switch to a Tc1 phenotype, reflecting the inherent plasticity of Tc17 cells.
Figure 2.

Generation and Tc1 and Tc17 Responses of antigen-specific to tumor antigen stimulation ex vivo. (A) Tc1 and Tc17 cells were generated as described in “Materials and Methods”, and their phenotype in terms of IFNγ and IL-17 production was shown on gated Pmel-1 (Thy1.1+) cells. (B) In a separate experiment, pmel-1 Tc1 and Tc17 cells were labeled with CSFE and transferred into B16 tumor-bearing B6 mice (n = 4). Five days after cell transfer, mice were euthanized and their splenocytes were stained for CD8 and Thy1.1. The CSFE profile is shown on gated live CD8+Thy1.1+ cells for one of four representative mice. (C) A set of B6 mice (n = 3) were inoculated i.v. with luciferase transduced B16 F10 cells and received Tc1 or Tc17 cells combined with BMT. Mice were euthanized on day 14 after treatment. Splenocytes were isolated and processed for cytokine-release ELISPOT assays. Gp100-specific IFNγ- and IL-17-producing cells were detected after co-culture with gp100-pulsed and unpulsed EL4 for 20 hours. Experiments were repeated 2 times with similar results. (D) The graph shows the average number of IFNγ- and IL-17-producing spots from triplicate wells with 1SD as error bars.
Tc1 cells are more effective than Tc17 cells in mediating anti-tumor responses
Using the treatment schedule shown in Figure 3A, we further assessed the therapeutic efficacy of ACT using either Tc1 or Tc17 cells. Mice that received Tc1 ACT completely controlled tumor growth, resulting in long-term tumor free survival (Figs. 3B and 3C). On the other hand, Tc17 ACT, at the same dose was only able to partially control tumor growth and significantly prolonged survival but ultimately all mice succumbed to their disease. These experiments were repeated at least 5 times, with nearly identical results.
Figure 3.

Tc1 cells mediated stronger anti-tumor immunity responses than Tc17 cells. (A) Schema for treatment regimen of B16 melanoma. B6 mice were inoculated i.v. with luciferase-transduced B16-F10 melanoma cells. Six days later, tumor-bearing mice received 1200 cGy (split doses). One day after, Tc1 and Tc17 cells generated from Pmel-1/Thy1.1 mice were transferred into tumor-bearing mice together with TCD-BM cells from normal B6 mice. (B) Percentage survival of tumor bearing mice was presented as pooled data from 2 replicate experiments combined. (C) Tumor growth was monitored by in vivo bioluminescent imaging (n = 4); one representative experiment from a total of 5 experiments is shown.
Anti-tumor effects of Tc1 cells, but not Tc17 cells, depend on intact IFNγ signaling on the tumor
Although CD8 T cells typically kill tumor targets via perforin/granzyme and CD95 ligand-dependent mechanisms, IFNγ produced by these cells can also play an important role in achieving anti-tumor effects (16, 17). Because the Tc1 and some of the Tc17 cells produced IFNγ after adoptive transfer (Figs. 2C and 2D), we investigated whether IFNγ was required for the Tc1- or Tc17-mediated anti-tumor activity in vivo. To this end, we utilized a B16 tumor line transduced with a plasmid encoding a dominant-negative IFNγ receptor (IFNγRDN). The impairment of IFNγR-signaling on IFNγ RDN B16 cells was verified in our previously published work showing that IFNγRDN B16 cells failed to up-regulate MHC-I, MHC-II and PD-L1 molecules upon IFNγ stimulation (18). Using the same schedule as presented above for B16-F10 cells, polarized Tc1 or Tc17 cells were adoptively transferred into B6 mice that had established B16 IFNγRDN lung tumors after myeloablative TBI and syngeneic TCD-BMT. In contrast to the findings using B16-F10 tumor cells (Figs. 3B and 3C), Tc1 cells were not able to achieve eradication of the B16 IFNγRDN tumors, although they were able to prolong the survival of the tumor bearing mice (Fig. 4A). Strikingly, ACT using Tc17 cells mediated complete regressions of the B16 IFNγRDN tumors. These results indicate that the Tc1-mediated anti-tumor responses depended on functional IFNγR on the tumor cells, which are not required for Tc17-mediated anti-tumor responses. Furthermore, the results also suggest that IFNγ signals on the tumor cells limit the therapeutic efficacy of Tc17 cells.
Figure 4.

Requirement of IFNγR on tumor cells for Tc1- or Tc17-mediated anti-tumor responses. (A) B6 mice were inoculated i.v. with IFNγRDN B16 tumor cells. gp100 specific Tc1 and Tc17 cells were administered 7 days after tumor implantation. Overall survival rate was observed. (B) Ag-specific IL-17 cytokine-release was analyzed by an ELISPOT assay using splenocytes from IFNγRDN B16 tumor bearing mice. ELISPOT was performed using several stimulator cells: IFNγ-treated (100 U/mL, 20 hours), and non-treated and IFNγ-treated (100 U/mL, 20 hours) B16, and non-treated B16 IFNγRDN B16 tumor. (C) Ag-specific IFNγ and cytokine-release was analyzed by ELISPOT assays using splenocytes from IFNγRDN B16 tumor bearing mice. ELISPOT was performed using several stimulator cells: IFNγ-treated (100 U/mL, 20 hours), and non-treated B16 and non-treated B16 IFNγRDN B16 tumor.
In a parallel experiment we assessed the effector function of Tc1 and Tc17 cells obtained from mice bearing B16 IFNγRDN tumors after ACT/TCD-BMT. In these experiments we evaluated the ability of the Tc1 and Tc17 cells to recognize B16-F10 and B16 IFNγRDN tumors by IFNγ and IL-17 ELISPOT assays. High numbers of gp100-specific IFNγ-secreting cells, but few or no IL-17-secreting cells, were present in spleens of tumor-bearing mice that received Tc1 cells after stimulation with parental B16-F10 or IFNγRDN tumor cells (Figs. 4B and 4C). Furthermore, the Tc1 responses increased when the parental B16-F10 cells were previously treated with IFNγ, indicating that IFNγ augmented the capacity of the tumor cells and their potential to be recognized by Tc1 cells (Fig. 4C). Consistent with previous observations (Figs. 2C and 2D), a low frequency of IFNγ-secreting cells and high frequency of tumor reactive IL-17-producing cells were detected in the spleens of tumor-bearing mice that received Tc17 cells (Figs. 4B and 4C). Similarly to the Tc1 response, Tc17 responses (measured by IL-17 secretion) were increased by pre-treatment of the parental B16-40 cells with IFNγ (Fig. 4C). Lastly, splenocytes derived from both Tc1 and Tc17 treated mice were effective in recognizing the B16 IFNγRDN tumor cells.
Tc17 mediated anti-tumor activity depends on the effects of IFNγ in the tumor environment
The data presented so far demonstrate that Tc1 cells mediate anti-tumor responses largely through the direct effects of IFNγ on the tumor cells, while IFNγ-signaling on tumor cells was not required for Tc17-mediated anti-tumor responses (Fig. 4A). Because Tc17 cells can produce both IL-17 and IFNγ and either cytokine can promote anti-tumor responses directly or indirectly, we assessed their potential roles by neutralizing either cytokine in vivo. In the B16-F10 parental tumor model, we found that neutralization of IFN-γ negatively affected the anti-tumor activity of Tc17 cells, whereas neutralization of IL-17A did not impair the Tc17-mediated anti-tumor effect (Fig. 5A), indicating that IFN-γ but not IL-17A contributed to Tc17-mediated anti-tumor activity. Because IFNγ-signaling on tumor cells was not required (Fig. 4A), we hypothesize that Tc17-mediatated anti-tumor activity required an indirect effect of IFNγ. To test this hypothesis, we evaluated the impact of neutralization of IFNγ or IL-17A on Tc17-mediated anti-tumor activity against IFNγRDN B16 cells. Similar to parental B16 cells, neutralization of IFNγ dramatically reduced Tc17-mediated anti-tumor activity against IFNγRDN B16 cells, whereas neutralization of IL-17A did not have any effect (Fig. 5B). Given that IFNγRDN B16 cells express 10 fold more IFNγR than parental B16 tumor, we surmise that IFNγ binds to IFNγR on tumor cells creating a microenvironment that promotes Tc17-mediated killing in an IL-17A independent manner.
Figure 5.
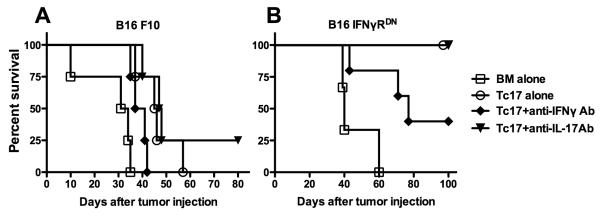
Role of IFN-γ and IL-17 in Tc17-mediated anti-tumor effect. WT C57/B6 mice (n = 4 per group) were given B16 (A) and IFNγRDN B16 tumor (B) i.v. by tail vein. Seven days later, tumor-bearing mice were adoptively transferred with Tc1 and Tc17 cells in the combination of BM. Anti-IFNγ mAb and anti-IL-17 mAb was administered intraperitoneally on days 0 and continuously twice a week for 4 weeks total. Overall survival was monitored. Corresponding groups of untreated tumor-bearing mice served as controls.
Distinct mechanisms of anti-tumor responses medicated Tc1 and Tc17 cells
The data presented so far indicate that Tc1 cells had superior anti-tumor response through an IFNγR-signaling dependent manner. In contrast, Tc17 cells had modest anti-tumor response against WT B16 but superior response against IFNγRDN B16. Because some cytokines play important roles in T-cell mediated tumor progression (14, 24), we measured proliferation and cytokine production of transferred Tc1 and Tc17 cells in the spleen and lung of tumor-bearing recipients. Consistent with ELISPOT data in Figure 3, Tc1 cells expressed higher levels of IFNγ but little IL-17 in B16-bearing mice, whereas Tc17 cells expressed higher levels of IL-17 and low levels of IFNγ, indicating that Tc1 cells had a more stable phenotype than Tc17 cells (Fig. 6A, left). Similar cytokine profiles were detected for transferred Tc1 or Tc17 cells in the recipients with IFNγRDN B16 tumor (Fig. 6A, right). Interestingly, % IFNγ+ (p = 0.07) and % IFNγ+/IL-17+ cells (p = 0.03) in particular among transferred Tc17 cells were significantly increased in the mice with IFNγRDN B16 than those with WT B16 tumor (Fig. 6B).
Figure 6.

Activation and expansion of antigen-specific Tc1 and Tc17 cells in vivo. Normal B6 mice with established WT or IFNγRDN B16 tumor received Tc1 or Tc17 cells in combination with BMT. Four mice were euthanized on day 14 and 28 after ACT. (A) Leukocytes isolated from spleen or lung on day 14 were stained with anti-Thy 1.1, anti-CD8, anti-IFNγ, anti-IL-17 and TNFα. Intracellular cytokine expression was shown on gated Thy 1.1+ cells. Flow plots display 1 of 3-4 mice in each group. (B) Percentages of IFNγ+, /IL-17+, IFNγ+/IL-17+and TNFα+ on gated Thy 1.1+ cells are shown from 3-4 mice per group on day 14 in spleen and lung. (C) Absolute numbers of adoptive transferred Tc1 or Tc17 cells are shown from 3-4 mice per group in spleen and lung on day 14 and 28. The data represent 1 of 3 experiments using similar settings. * p < 0.05, ** p < 0.01, and *** p < 0.001.
The data suggest that enhanced frequency of IFNγ+/IL-17+ cells in the lungs might contribute to the superior activity of Tc17 cells against IFNγRDN B16 tumor, given this double positive population was suggested to be the most functional subset among Th17 cells. TNFα was expressed at low but similar levels by Tc1 and Tc17 cells (Fig. 6B), suggesting that TNFα was not responsible for the differential ability of Tc1 and Tc17 cells in mediating anti-tumor responses against WT or IFNγRDN B16.
To define the underlying mechanisms, we determined the expansion and migration of Tc1 and Tc17 cells by qualifying numbers of transferred cells in tumor-bearing mice. In the spleen, the numbers of Tc17 cells were significantly higher than Tc1 cells regardless of tumor type in day 14 and 28 after cell transfer (Fig. 6C, upper). In the lung, the numbers of Tc1 and Tc17 cells were similar in the recipients with B16 tumor regardless of the time. However, the numbers of Tc17 cells were significantly higher than Tc1 cells in the recipients with IFNγRDN B16 tumor in both time points (Fig. 6C, lower). Moreover, the numbers of Tc17 cells were significantly higher in the recipients with WT tumor than those with IFNγRDN B16 tumor in day 28, which may contribute to the superior activity of Tc17 cells against IFNγRDN B16 tumor (Fig. 3).
Higher levels of T-cell proliferation in spleen were likely contributed to the higher numbers of Tc17 cells accumulated in the lungs as compared with Tc1 cells. However, the ability of T cells in migration is also an important factor for effector T-cell accumulation in lung. To this end, we measured chemokine receptors expressed on the cells present in lung. As expected, Tc1 cells expressed significantly higher levels of CXCR3 than Tc17 cells, whereas Tc17 cells expressed significantly higher levels of CCR6 than Tc1 cells (Figs. 7A and 7B). Because CCR6+ cells preferentially migrate into lung, these data indicate that higher accumulation of Tc17 cells in lung might have also resulted from their stronger ability to migrate. However, Tc17 cells expressed lower levels of CCR6 but higher levels of CXCR3 in the mice with WT B16 than those with IFNγRDN B16 tumor (Figs. 7A and 7B).
Figure 7.

Phenotypes of transferred Tc1 and Tc17 cells in vivo. The experiment was set as in figure 6. Leukocytes isolated from recipient lung on day 14 were stained for surface expression with Thy 1.1, CD8, CXCR3, CCR6 and CD107a. Percentages of CXCR3+ (A), CCR6+ (B), and CD107a+ cells (C) on gated Thy 1.1+ population are shown from 3-4 mice per group. The data represent 1 of 3 experiments using similar settings. * p < 0.05, ** p < 0.01, and *** p < 0.001.
Comparing to quantity, quality or function of T cells is likely more critical for their ability to respond against tumor. Previously it was reported that Tc17 cells have lower killing function than Tc1 cells (4, 21, 25). To compare their cytolytic function, we measured CD107a expression (which correlated with cytolytic activity) on isolated pmel-1 cells from lung tissues. Tc1 cells expressed significantly higher levels of CD107a in the mice with WT B16 (Fig. 7C), in agreement with the published observation that Tc1 cells have strong killing ability compared to Tc17 cells. In the mice with IFNγRDN B16 tumor, the levels of CD107a expression on Tc1 cells were lower than the levels observed on B16 cells, but these levels were similar as those observed on Tc17 cells (Fig. 7C). Reduced killing function of Tc1 cells likely contributed to the impaired ability for Tc1 cells to eradicate IFNγRDN B16 tumor. Although Tc17 cells also expressed low levels of CD107a, they were able to effectively reject IFNγRDN B16 tumor, suggesting that Tc17-medidated anti-tumor response was largely independent of their cytolytic function under the situation.
Considering other molecules also contribute to T-cell function and fitness, we further analyzed the phenotype of Tc1 and Tc17 cells including CD27 (costimulation) CD62L (homing and memory), CD127 (IL-7Rα for maintenance), and KLRG-1 (survival and memory). While CD27 and CD62L expressed similarly on both types of cells, Tc17 cells expressed higher levels of CD127 but lower levels of KLRG-1 than Tc1 cells (Table I). Given that CD127hi and KLRG-1low cells represent long-lived effector T cells, such a phenotype was consistent with better survival of Tc17 cells in vivo. T-cell mediated anti-tumor response may also be attributed to other types of effector cells recruited at the tumor site. We therefore examined the infiltration of neutrophils (Gr-1), monocytes (CD11b) and macrophages (F4-80) in the lung, but found that comparable levels of each population were present in lung tissue of the mice transferred with Tc1 or Tc17 cells (data not shown).
Table I. Phenotype of Tc1 and Tc17 cells after ACT.
| A. Spleen | ||||
|---|---|---|---|---|
| B16 | IFNγ DN B16 | |||
| Tc1 | Tc17 | Tc1 | Tc17 | |
| CD27 | 85.7 ± 8.8 | 85.1 ± 2.4 | 86.3 ± 4.8 | 83.7 ± 2.1 |
| CD62L | 40.6 ± 11.6 | 47.8 ± 2.5 | 42.8 ± 6.7 | 42.6 ± 2.9 |
| CD127 | 29.4 ± 3.3 | 31.7 ± 4.3 | 24.5 ± 3.1 | 32.9 ± 2.4 |
| KLRG1 | 12.8 ± 3.5 | 6.4 ± 1.1 | 13.1 ± 2.9 | 7.6 ± 1.3 |
| B. Lung | ||||
| B16 | IFNγ DN B16 | |||
| Tc1 | Tc17 | Tc1 | Tc17 | |
| CD27 | 67.4 ± 4.3 | 60.8 ± 11.4 | 49.9 ± 7.4 | 59.4 ± 1.2 |
| CD62L | 21.7 ± 7.3 | 23.7 ± 8.5 | 14.3 ± 4.4 | 16.4 ± 0.6 |
| CD127 | 32.8 ± 6.1 | 45.5 ± 2.7 | 30.9 ± 4.5 | 41.1 ± 2.7 |
| KLRG1 | 16.7 ± 2.8 | 9.6 ± 2.0 | 13.1 ± 2.9 | 9.1 ± 2.8 |
The experiment was set us as in figure 6. Leukocytes isolated from recipient lung on day 14 were stained for surface expression of with Thy 1.1, CD27, CD62L, CD127 and KLRG-1. Percentages of CD27+, CD62L+, CD127+, and KLRG-1+ cells on gated Thy 1.1+ population are shown as average ± 1SD from 3-4 mice per group.
Discussion
The use of ACT with tumor specific T cells has shown to be quite effective for the treatment of established melanoma both in animal models as well as in patients. However, eradication of established melanoma has not been consistently achieved when using T cells specific for naturally-processed, tumor-associated Ags (e.g. gp100) (19, 26-30). In the current study, we clearly demonstrate that a myeloablative conditioning plus syngeneic BMT dramatically increased the anti-tumor efficacy of ACT using gp100 specific Tc1 or Tc17 cells against established B16 lung tumors. Using a BMT platform, we further showed that gp100-specific Tc1 cells could eradicate established parental B16 melanoma, and that gp100-spcific Tc17 cells could eradicate established IFNγRDN B16 tumors. To our knowledge, this is the first report showing that the ACT with T cells specific for naturally-processed tumor-associated Ags completely cured established melanomas without the use of IL-2 or vaccination.
To stringently assess the potency of Tc1 and Tc17 cells in anti-tumor responses, we utilized an established lung-metastatic model of B16 melanoma, a poorly immunogenic (31) and a highly aggressive tumor in C57BL/6 mice (32). Analogous to human melanoma, B16 melanoma expresses the mouse homologue (pmel-17) of human gp100 (33), a naturally processed melanoma-associated Ag. We utilized pmel-1 TCR transgenic T cells specific for gp100 to generate Tc1 and Tc17 cells as effector T cells. Consistent with previous reports that ACT with tumor-specific T cells cannot eradicate established tumors (23-27), we observed that either adoptive transfer with pmel-1 Tc1 or Tc17 cells only partially controlled tumor growth in mice that received non-myeloablative conditioning (Fig. 1A). However, the therapeutic efficacy of ACT was significantly augmented using Tc1 or Tc17 cells in mice that received myeloablative conditioning followed by TCD-BMT. We reason that myeloablative conditioning and hematopoeitic stem cells increased the expansion and/or infiltration of transferred T cells, as the numbers of Tc1 and Tc17 cells were increased in the spleen and lung of those treated mice (Fig. 1B).
Using BMT as a platform, Tc1 cells exhibited anti-tumor activity superior to Tc17 cells (Fig. 3B). Effective tumor rejection by adoptively transferred T cells is likely to rely on several direct and indirect immune effector mechanisms. First, both Tc1 and Tc17 effectors may directly eradicate tumor cells through cognate interactions involving perforin/granzyme mediated lytic mechanisms (4, 28). As a result, tumor-associated Ags may be released and presented by host APCs that may enhance host immune responses at sites proximal and distal to tumor growth. Second, release of Tc1 and Tc17 cytokines, such as IFNγ and TNFα, have been shown to directly inhibit tumor cell growth (34-36), as they enhance Ag presentation through up-regulation of MHC-I on both tumors and host APCs (37, 38), and increase the expression of effector molecules by T cells to facilitate anti-tumor responses and tumor rejection (39-41). Third, Tc1 and Tc17 effector cells can induce Ag non-specific inflammatory responses that may indirectly aid in cytolytic and/or cytostatic anti-tumor effects. Local release of cytokines, such as Tc17-derived IFNγ, TNF-α, IL-17 and IL-21 and Tc1-derived IFNγ, have been shown to mediate the selective recruitment and localization of macrophages, NK cells, and granulocytes that may facilitate enhancement of tumor Ag presentation and inhibition of tumor growth (21, 42). In either case, utilization of discrete cytokines produced by Tc1 or Tc17 effector cells can mediate tumor rejection with distinct mechanisms, and potentially affect the efficacy of anti-tumor responses and tumor regression.
Following ACT, Tc1 cells proliferated substantially faster than Tc17 cells during first 5 days after cell transfer (Fig. 2B), but the numbers of Tc17 cells were significantly higher in spleen on days 14 and 28 regardless of tumor types (Fig. 6C). We interpret that Tc1 cells have superior potential to proliferate than Tc17 cells, but Tc17 cells have an advantage to survive as supported by our previously published observation (43). In WT B16 tumor, Tc1 cells were more efficient than Tc17 cells to mount anti-tumor responses against this poorly immunogenic melanoma (Figs. 1 and 2). Because the comparable numbers of Tc1 and Tc17 cells were observed at the tumor site (lung) on days 14 and 28, we reason that the therapeutic efficacy of Tc1 and Tc17 cells against B16 tumor did not result from their differential expansion or infiltration in vivo. Instead, we propose that it is the quality of transferred T cells that dictates the therapeutic efficacy. Tc1 cells are known to have strong cytolytic activity against tumor targets, and the current study demonstrated that IFNγ-signaling through tumor cells was required for Tc1-mediated anti-tumor activity (Fig. 4A). Consistent with recent reports showing that Tc17 cells have much weaker cytolytic activity (4, 21, 25), we also found that Tc1 cells expressed significantly higher levels of CD107a than Tc17 cells (Fig. 7C) Tc17 cells could produce IFNγ after adoptive transfer in vivo, but the amount of IFNγ or the frequency of IFNγ-producing cells was lower than that observed in Tc1 cells. As Tc17-mediated anti-tumor activity was also dependent on IFNγ, we surmise that lower cytolytic activity and IFNγ production by Tc17 cells likely accounted for their inferior therapeutic efficacy against B16 tumor as compared to Tc1 cells.
Strikingly and unexpectedly, we found that Tc17 cells were highly effective against IFNγRDN melanoma (Fig. 4). It appears to be counterintuitive that neutralization of IFNγ not only inhibited Tc17-mediated anti-tumor activity against parental B16 but against IFNγRDN B16 cells as well. Because IFNγRDN B16 cells express 10-fold more dominate-negative receptors that cannot mediate IFNγ signaling, it is clear that Tc17-mediated anti-tumor activity is independent of receptor signaling on tumor cells. However, IFNγRDN B16 cells could absorb significant amount of IFNγ. We surmise that a large number of IFNγ/IFNγR complexes created a microenvironment that promoted Tc17-mediated killing, which was independent of IL-17A since neutralization of this cytokine had no effect (Fig. 5). It is likely that the IFNγ/IFNγR complexes on the IFNγRDN B16 cells serves as a rich source of IFNγ for the tumor microenvironment, which promotes the anti-tumor effect. Although a precise mechanism remains to be further investigated, additional data (Fig. 6 and 7) suggest that abundant IFNγ accumulated at the tumor site might increase the recruitment of Tc17 cells through CXCR3 and the Tc17 activity by enhancing IFNγ+/IL-17+ subset, which has strong anti-tumor activity (44). In addition, Tc17 cells have the phenotype of long-lived effector T cells (CD127hiKLRG-1low), which may also contribute Tc17-mediated anti-tumor response against IFNγRDN tumor. Other potential mechanisms may include that other cytokines produced by Tc17 such as IL-21 and IL-22 could exert indirect anti-tumor response against IFNγRDN tumor by recruiting innate immune cells such as macrophages and neutrophils.
Overall, the current study leads to two novel and important findings. 1) the combination of BMT with TBI and adoptive transfer of tumor specific Tc1 cells can cure established melanoma, which may represent a new strategy to treat patients with metastatic melanoma; 2) local accumulation of IFNγ at the tumor site significantly contributes to T-cell mediated anti-tumor responses independent of IFNγ-signaling by tumor cells, which may have an implication that IFNγ inside the tumor may act as an adjuvant to improve immunotherapy against established tumor.
Acknowledgment
We thank Dr. Jeff Weber for his critical review of this manuscript, and Dr. Hailing Zhang for her assistance in lung pathology. We are grateful for the technical assistance provided by Flow Cytometry and Mouse Core Facility at the Moffitt Cancer Center.
Footnotes
Disclosure of Potential Conflicts of Interest
Authors had no potential conflicts of interest to be disclosed.
This work was supported in part by National Institutes of Heath Grants R01s CA143812, CA118116 and AI082685 to X.-Z.Y
Abbreviations used in this paper: adoptive cell transfer, ACT; bone marrow transplantation, BMT; total body irradiation, TBI; Tc1, type I CD8+ cytotoxic T cells; Tc2, type II CD8+ cytotoxic T cells; Tc17, IL-17-producing CD8+ T cells; TIL, tumor-infiltrating lymphocyte. Th17, IL-17-producing CD4+ T cells; IFNγRDN, dominant-negative IFNγ receptor.
Reference
- 1.Croft M, Carter L, Swain SL, Dutton RW. Generation of polarized antigen-specific CD8 effector populations: reciprocal action of interleukin (IL)-4 and IL-12 in promoting type 2 versus type 1 cytokine profiles. J Exp Med. 1994;180:1715–1728. doi: 10.1084/jem.180.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter LL, Dutton RW. Type 1 and type 2: a fundamental dichotomy for all T-cell subsets. Curr Opin Immunol. 1996;8:336–342. doi: 10.1016/s0952-7915(96)80122-1. [DOI] [PubMed] [Google Scholar]
- 3.Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271–279. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 4.Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, Wrzesinski C, Borman ZA, Muranski P, Restifo NP. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114:596–599. doi: 10.1182/blood-2009-02-203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan C-C, Restifo NP. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ngiow SF, Smyth MJ, Teng MWL. Does IL-17 suppress tumor growth? Blood. 2010;115:2554–2555. doi: 10.1182/blood-2009-11-254607. [DOI] [PubMed] [Google Scholar]
- 8.Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Regulation of the IL-23 and IL-12 Balance by Stat3 Signaling in the Tumor Microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin-Orozco N, Dong C. The IL-17/IL-23 axis of inflammation in cancer: Friend or foe? Curr. Opin. Invest. Drugs. 2009;10:543–549. [PubMed] [Google Scholar]
- 12.Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 14.Yee C, Greenberg P. Modulating T-cell immunity to tumours: new strategies for monitoring T-cell responses. Nat Rev Cancer. 2002;2:409–419. doi: 10.1038/nrc820. [DOI] [PubMed] [Google Scholar]
- 15.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. 2005;17:195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wrzesinski C, Paulos CM, Gattinoni L, Palmer DC, Kaiser A, Yu Z, Rosenberg SA, Restifo NP. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8(+) T cells. J. Clin. Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anasetti C, Mule JJ. To ablate or not to ablate? HSCs in the T cell driver’s seat. J. Clin. Invest. 2007;117:306–310. doi: 10.1172/JCI30973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho H-I, Lee Y-R, Celis E. Interferon gamma limits the effectiveness of melanoma peptide vaccines. Blood. 2011;117:135–144. doi: 10.1182/blood-2010-08-298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishimura F, Dusak JE, Eguchi J, Zhu XM, Gambotto A, Storkus WJ, Okada H. Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: Critical roles of IFN-inducible protein-10. Cancer Res. 2006;66:4478–4487. doi: 10.1158/0008-5472.CAN-05-3825. [DOI] [PubMed] [Google Scholar]
- 20.Yu XZ, Liang YM, Nurieva RI, Guo F, Anasetti C, Dong C. Opposing effects of ICOS on graft-versus-host disease mediated by CD4 and CD8 T cells. J. Immunol. 2006;176:7394–7401. doi: 10.4049/jimmunol.176.12.7394. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Hernandez M. d. l. L., Hamada H, Reome JB, Misra SK, Tighe MP, Dutton RW. Adoptive Transfer of Tumor-Specific Tc17 Effector T Cells Controls the Growth of B16 Melanoma in Mice. J. Immunol. 2010;184:4215–4227. doi: 10.4049/jimmunol.0902995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukai S, Kjaergaard J, Shu S, Plautz GE. Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res. 1999;59:5245–5249. [PubMed] [Google Scholar]
- 23.Young MR, McCloskey G, Wright MA, Pak AS. Increasing infiltration and activation of CD8+ tumor-infiltrating lymphocytes after eliminating immune suppressive granulocyte/macrophage progenitor cells with low doses of interferon gamma plus tumor necrosis factor alpha. Cancer Immunol Immunother. 1994;38:9–15. doi: 10.1007/BF01517164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 25.Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, Hünig T, Mittrücker HW, Brüstle A, Kamradt T, Lohoff M. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol. 2009;39:1716–1725. doi: 10.1002/eji.200939412. [DOI] [PubMed] [Google Scholar]
- 26.Gyobu H, Tsuji T, Suzuki Y, Ohkuri T, Chamoto K, Kuroki M, Miyoshi H, Kawarada Y, Katoh H, Takeshima T, Nishimura T. Generation and targeting of human tumor-specific Tc1 and Th1 cells transduced with a lentivirus containing a chimeric immunoglobulin T-cell receptor. Cancer Res. 2004;64:1490–1495. doi: 10.1158/0008-5472.can-03-2780. [DOI] [PubMed] [Google Scholar]
- 27.Tsuji T, Yasukawa M, Matsuzaki J, Ohkuri T, Chamoto K, Wakita D, Azuma T, Niiya H, Miyoshi H, Kuzushima K, Oka Y, Sugiyama H, Ikeda H, Nishimura T. Generation of tumor-specific, HLA class I-restricted human Th1 and Tc1 cells by cell engineering with tumor peptide-specific T-cell receptor genes. Blood. 2005;106:470–476. doi: 10.1182/blood-2004-09-3663. [DOI] [PubMed] [Google Scholar]
- 28.Sad S, Kagi D, Mosmann TR. Perforin and Fas killing by CD8+ T cells limits their cytokine synthesis and proliferation. J Exp Med. 1996;184:1543–1547. doi: 10.1084/jem.184.4.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Bi XG, Yuan JY, Xu SL, Guo XL, Xiang J. Combined CD4(+) Th1 effect and lymphotactin transgene expression enhance CD8(+) Tc1 tumor localization and therapy. Gene Therapy. 2005;12:999–1010. doi: 10.1038/sj.gt.3302486. [DOI] [PubMed] [Google Scholar]
- 30.Kemp RA, Ronchese F. Tumor-specific Tc1, but not Tc2, cells deliver protective antitumor immunity. J. Immunol. 2001;167:6497–6502. doi: 10.4049/jimmunol.167.11.6497. [DOI] [PubMed] [Google Scholar]
- 31.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poste G, Doll J, Hart IR, Fidler IJ. In vitro selection of murine B16 melanoma variants with enhanced tissue-invasive properties. Cancer Res. 1980;40:1636–1644. [PubMed] [Google Scholar]
- 33.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohm W, Thoma S, Leithauser F, Moller P, Schirmbeck R, Reimann J. T cell-mediated, IFN-gamma-facilitated rejection of murine B16 melanomas. J. Immunol. 1998;161:897–908. [PubMed] [Google Scholar]
- 35.Krasagakis K, Garbe C, Zouboulis CC, Orfanos CE. Growth control of melanoma cells and melanocytes by cytokines. Recent results in cancer research. Fortschritte der Krebsforschung. Progres dans les recherches sur le cancer. 1995;139:169–182. doi: 10.1007/978-3-642-78771-3_12. [DOI] [PubMed] [Google Scholar]
- 36.Pardoll DM. Paracrine cytokine adjuvants in cancer immunotherapy. Annu Rev Immunol. 1995;13:399–415. doi: 10.1146/annurev.iy.13.040195.002151. [DOI] [PubMed] [Google Scholar]
- 37.Ogasawara M, Rosenberg SA. Enhanced expression of HLA molecules and stimulation of autologous human tumor infiltrating lymphocytes following transduction of melanoma cells with gamma-interferon genes. Cancer Res. 1993;53:3561–3568. [PubMed] [Google Scholar]
- 38.Wong LH, Hatzinisiriou I, Devenish RJ, Ralph SJ. IFN-gamma priming up-regulates IFN-stimulated gene factor 3 (ISGF3) components, augmenting responsiveness of IFN-resistant melanoma cells to type I IFNs. J. Immunol. 1998;160:5475–5484. [PubMed] [Google Scholar]
- 39.Barth RJ, Jr., Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991;173:647–658. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morisaki T, Morton DL, Uchiyama A, Yuzuki D, Barth A, Hoon DS. Characterization and augmentation of CD4+ cytotoxic T cell lines against melanoma. Cancer Immunol Immunother:CII. 1994;39:172–178. doi: 10.1007/BF01533383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aruga A, Shu S, Chang AE. Tumor-specific granulocyte/macrophage colony-stimulating factor and interferon gamma secretion is associated with in vivo therapeutic efficacy of activated tumor-draining lymph node cells. Cancer Immunol Immunother: CII. 1995;41:317–324. doi: 10.1007/BF01517220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagoshi M, Goedegebuure PS, Burger UL, Sadanaga N, Chang MP, Eberlein TJ. Successful adoptive cellular immunotherapy is dependent on induction of a host immune response triggered by cytokine (IFN-gamma and granulocyte/macrophage colony-stimulating factor) producing donor tumor-infiltrating lymphocytes. J. Immunol. 1998;160:334–344. [PubMed] [Google Scholar]
- 43.Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, Yu XZ. Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORgammat in mice. Blood. 2011;118:5011–5020. doi: 10.1182/blood-2011-03-340315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tajima M, Wakita D, Satoh T, Kitamura H, Nishimura T. IL-17/IFN-gamma double producing CD8+ T (Tc17/IFN-gamma) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Int Immunol. 2011;23:751–759. doi: 10.1093/intimm/dxr086. [DOI] [PubMed] [Google Scholar]