

Abstract
Type I interferons are induced during microbial infections and have well characterized anti-viral activities. TRAF3 is a signaling molecule crucial for type I IFN production and therefore represents a potential target for disarming immune responses. Chlamydia pneumoniae is a human pathogen which primarily infects respiratory epithelial cells where onset of symptoms takes several weeks, and the course of infection is protracted. C. pneumoniae has also been associated with a variety of chronic inflammatory conditions. Thus, typical C. pneumoniae infections of humans are consistent with an impairment in inflammatory responses to the microorganism. We demonstrate that infection of epithelial cells with C. pneumoniae does not lead to IFNβ production. Instead, infected cells are prevented from activating IRF3. This effect is mediated by C. pneumoniae-dependent degradation of TRAF3, which is independent of a functional proteasome. Hence it is likely that C. pneumoniae express a unique protease targeting TRAF3-dependent immune effector mechanisms.
Introduction
The respiratory epithelium represents a major site for entry of pathogenic microorganisms. Therefore the epithelial cell lining of the respiratory tract plays a pivotal role in innate immune defense against bacterial and viral pathogens. Innate immune recognition of microbial products is an essential component of this defense and is mediated by germline-encoded pattern recognition receptors (PRRs). PRRs detect pathogen-associated molecular patterns (PAMPs) that signal the presence of a foreign microorganism to the host Toll-like receptors. These receptors which are members of the tumor necrosis factor receptor (TNFR) family, represent the best characterized class of PRRs. TLRs are localized in the cytoplasmic or endosomal membranes, and out of the ten functional TLRs identified in humans, human bronchial epithelial cells express functional TLR1-6 (1). Ligation of a TLR initiates a cascade of signaling pathways that proceed in either a MyD88-dependent (TLR1, 2, 4, 5, and 6) or TRIF-dependent (TLR3 and TLR4) manner. MyD88-dependent pathways drive induction of various inflammatory cytokines, whereas TRIF-dependent pathways are responsible for the induction of type I interferons (IFNs) in addition to inflammatory cytokines (2). Another group of PRRs represent RNA helicases, retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), which recognize viral dsRNA in the cytoplasm and lead to the production of IFNα/β (3–5).
Transduction of various signals including those initiated from all PRRs requires participation of TNFR-associated factors (TRAFs). TRAFs are intracellular signaling molecules that serve as both adaptor proteins and E3 ubiquitin ligases. TRAF1 and TRAF2 are constitutively associated and ubiquitously expressed together with TRAF3 and TRAF6 in most cell types, whereas expression of functional TRAF5 is mainly restricted to immune cells (6). K-63 linked autoubiquitination of TRAF proteins is essential for the assembly of downstream signaling effectors. The engagement of TLR3 or TLR4 leads to TRIF-dependent K63-linked ubiquitination of TRAF3. This ubiquitinated TRAF3 is crucial for downstream activation of kinases TBK1 and IKKε that catalyze the phosphorylation of interferon regulatory factor 3 (IRF3) and subsequent induction of IFNβ (7–9). K63-linked ubiquitination of TRAF3 also plays a role in transduction pathways initiated from RIG-I and MDA5 by binding directly to activated mitochondrial anti-viral signaling protein (MAVS) (10, 11). Thus, TRAF3 represents a key signaling molecule in multiple transduction pathways, and deficiency in this adapter molecule impairs IFNα/IFNβ induction by TLR3, TLR4, TLR7/8, TLR9 (8, 9), RIG-I and MDA5 (10). Besides IRF3, type I interferons are also regulated by the transcription factor IRF7, that is activated upon stimulation of endosomal TLR7/8 and TLR9 in immune cells, leading to the generation of primarily IFNα (10). Although the importance of type I IFNs in the innate immune response to viral infections is well characterized, their role during the course of bacterial infections is less clear.
Chlamydia pneumoniae is a gram-negative, obligate intracellular bacterium that infects mucosal surfaces of the human respiratory tract causing pneumonia, bronchitis, pharyngitis and sinusitis. Epidemiological data suggest that most people are infected and re-infected throughout life (12). Therefore, C. pneumoniae represents an invader commonly encountered by respiratory defenses. The pathogen has also been associated with a variety of chronic diseases such as reactive arthritis, sarcoidosis, asthma, COPD (chronic obstructive pulmonary disease), multiple sclerosis, Alzheimer disease and atherosclerosis (13). However, the role of the pathogen in the course of chronic diseases is unknown. C. pneumoniae undergoes a developmental cycle in which two functionally and morphologically distinct cell types are recognized. The infectious cell type, which is specialized for extracellular survival and transmission, is termed the elementary body (EB), and the intracellular, vegetative cell type is called the reticulate body (RB). Upon internalization of C. pneumoniae by a host cell, the bacterium proceeds with a developmental cycle which occurs entirely within a membrane bound vesicle termed an inclusion. The chlamydial inclusion does not fuse or interact with endosomes or lysosomes during productive growth of the microorganism within an epithelial cell (14–16). Conversely, C. pneumoniae cannot escape the endosomal/lysosomal fusion upon invasion of an immune cell such as a macrophage where the pathogen is destined for degradation (17).
In this study we demonstrate that C. pneumoniae has evolved a unique mechanism to disarm essential aspects of innate immunity. Unlike other chlamydial species, infection of epithelial cells with C. pneumoniae does not lead to IFNβ production. Moreover, the microorganism actively suppresses type I interferon induction since infection interferes with stimulation of cells with poly(I:C) (polyinosinic-polycytidylic acid), the synthetic analog of dsRNA. Finally, we provide systematic evidence that C. pneumoniae targets TRAF3 for degradation. Therefore, C. pneumoniae infection blocks type I IFN induction by preventing phosphorylation and nuclear translocation of IRF3.
Material and Methods
Cell culture and organisms
C. pneumoniae, AR-39 purchased from American Type Culture Collection (Manassas, VA) was propagated in HeLa cells and purified using MD-76R as previously described (18). HeLa 229 and A549 cells were also obtained from American Type Culture Collection. Sub-confluent monolayers of epithelial cells were infected with C. pneumoniae, AR-39 at an indicated multiplicity of infection (MOI) by incubation with chlamydial inoculum in a 37 C incubator for 2 h with occasional agitation. Chlamydia-infected and uninfected cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) for HeLa cells and F12K for A549 cells supplemented with 10% FBS (Sigma, St. Louis, MO) and in the presence of 1 μg/ml cycloheximide (Sigma) for the first 30 hrs. No antibiotics were present in the culture medium after 30 h post infection (p.i.). For Fig. 1A, crude C. pneumoniae inoculum was prepared as described by Maass et al. (19) and HeLa cells were infected at an MOI of 3 by centrifugation at 800 × g for 30 min (20). In experiments where human IFNβ was quantified, uninfected and chlamydiae-infected cells were incubated in the absence of antibiotics. For inhibition of chlamydial protein synthesis, C. pneumoniae infected cells were incubated in the presence of 200 μg/ml chloramphenicol (Sigma). Growth and MD-76R purification of C. trachomatis, serovars L2 and D was performed as previously described (21).
FIGURE 1.
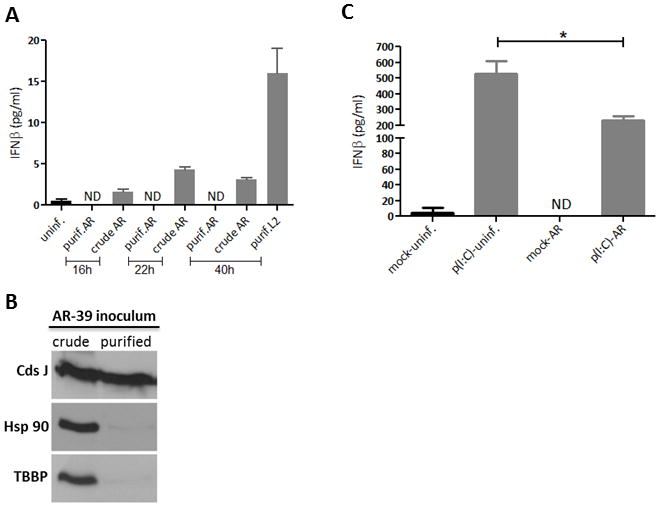
C. pneumoniae does not induce IFNβ. (A) HeLa cells were infected with either crude or MD-76R purified C. pneumoniae, AR-39 at an MOI of 3, and IFNβ released into the culture media was quantified by ELISA. Uninfected and cells infected with purified C. trachomatis, L2 (MOI of 1) were included as negative and positive controls, respectively. ND, none detected. (B) Immunoblot analysis of crude and purified C. pneumoniae inoculum probed with anti-Hsp90 and anti-TATA box binding protein (TBBP) antibodies. Total amount of chlamydial protein was controlled by probing with anti-CdsJ antibodies. (C) Uninfected and A549 cells infected with C. pneumoniae (MOI of 0.5) were either mock treated or transfected at 45 h p.i. with 0.5 μg/ml of p(I:C) for additional 20 h. IFNβ in cell culture media was measured using ELISA. Asterisk indicates significant difference (P < 0.05) as determined by Student’s t-test. ND, none detected.
Antibodies and other reagents
The antibodies recognizing the FLAG tag (M2) and human actin were purchased from Sigma. The anti-TRAF3 specific antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and LifeSpan BioSciences (Seattle, WA). The anti-phospho-IRF3 (Ser396) and anti-TATA box binding protein (TBBP) antibodies were purchased from Millipore (Temecula, CA), anti-IRF3 from Santa Cruz Biotechnology and anti-MAVS/VISA antibody from Bethyl Laboratories (Montgomery, TX). Genus-specific anti-CdsJ antibodies were previously described (22). Anti-Hsp90 and anti-phospho-IκBα (Ser32 and Ser36) mAb were obtained from BD Biosciences (San Jose, CA). Polyinosinic-polycytidylic acid [poly(I:C)] was purchased from Sigma. Lactacystin and MG132 were obtained from EMD Chemicals (Billerica, MA).
Transfections
Uninfected and C. pneumoniae infected HeLa cells were transfected at 45 h p.i. with either HA-TRAF3 or FLAG-TRAF2 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The HA-TRAF3 and FLAG-TRAF2 constructs were kindly provided by Dr. E. W. Harhaj and Dr. S. C. Sun. The pUNO-hIPS1 vector was purchased from InvivoGen (San Diego, CA) and co-transfections of uninfected and chlamydiae-infected HeLa cells with HA-TRAF3 and pUNO-hIPS1 were performed as described above. Transfected cell cultures were incubated in RPMI 1640 supplemented with 5% FBS for an additional 19 h. For induction experiments, uninfected and C. pneumoniae or C. trachomatis infected HeLa cells were transfected with poly(I:C) for indicated periods of time using Lipofectamine 2000. Mock treated cells were incubated in the presence of Lipofectamine 2000 without poly(I:C).
Immunoblotting assays and immunoprecipitation
Protein samples obtained from whole cell lysates of uninfected and C. pneumoniae or C. trachomatis infected cultures were harvested with 8M Urea (23) and concentrated by addition of trichloroacetic acid (Sigma) to 10% (v/v). Collected protein pellets were analyzed by SDS-PAGE electrophoresis followed by immunoblot analysis using appropriate primary antibodies and peroxidase conjugated secondary antibody (Sigma). Nuclear fractions of uninfected and chlamydiae-infected HeLa cells were obtained through stepwise separation of membrane, cytoplasmic and nuclear fractions using Subcellular Protein Fractionation Kit according to the manufacturer’s instructions (Pierce, Rockford, IL). Polypeptide bands were visualized by development with ECL Plus Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ) according to the manufacturer’s instructions.
C. pneumoniae and uninfected HeLa cells co-transfected with HA-TRAF3 and pUNO-hIPS1 were lysed in RIPA buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% sodium deoxycholate and protease inhibitors (Roche, Indianapolis, IN). Pre-cleared cell lysates were incubated with anti-HA antibodies followed by incubation with 50% slurry of protein A-sepharose (Sigma). The protein A-bound immunocomplexes were subjected to Western blot analysis and probed with anti-MAVS antibodies as described above.
Co-incubation of host and chlamydial cell lysates
Uninfected HeLa cells transfected with HA-TRAF3 were harvested into Tris-buffered saline (20 mM Tris [pH 7.4], 150 mM NaCl) (24) and gently lysed using a Par Cell Disruption Bomb (Parr Instrument Company, Moline, IL) according to the manufacturer’s instructions. Density gradient purified C. pneumoniae were lysed in Tris-buffered saline containing 1% NP40. Different amounts of chlamydial lysate were co-incubated with a constant amount of HeLa cell lysate for 1 h at 37°C as previously described (24). Protein samples were prepared as described above.
ELISA
Culture media of uninfected, C. pneumoniae, and C. trachomatis infected HeLa cells were collected at different time points post infection, centrifuged at 20,000 × g for 10 min and IFNβ was measured in supernatants using ELISA kit (PBL, Piscataway, NJ) according to the manufacturer’s instructions. Uninfected or C. pneumoniae infected A549 cells at 48 h p.i. were either mock treated or transfected with 0.5 μg/ml of poly(I:C) as described above. Culture media were collected 20 h post transfection and levels of IFNβ were quantified by ELISA. Student t-test was used to analyze data.
Results
Infection of epithelial cells with C. pneumoniae does not induce IFNβ
IFNβ response elicited during productive infection of epithelial cells with C. pneumoniae has not been demonstrated, Buss et al. reported that in human endothelial cells C. pneumoniae induces production of IFNβ via a MAVS-dependent IRF3 and IRF7 activation during first 16 h post infection (20). However, in that study endothelial cells were infected with C. pneumoniae inoculum that had not undergone purification through diatrizoate meglumine plus diatrizoate sodium, formally known as Renografin described by Caldwell et al. (21). Instead a crude C. pneumoniae preparation was used (20). Renografin purification of chlamydiae is routinely employed for all chlamydial species as it removes significant amounts of cellular debris from lysed eukaryotic cells as well as RBs, which are released with EBs during sonication of infected cells at the end of chlamydial cycle. We therefore compared IFNβ production during infection of HeLa cells with MD-76R (diatrizoate meglumine plus diatrizoate sodium) purified and crude C. pneumoniae inoculum using ELISA assays. Whilst IFNβ secretion was not detected in culture media obtained from cells infected with purified C. pneumoniae at 16, 22 or 40 h post infection (p.i.), the cytokine was clearly detected in media of cultures infected with crude inoculum (Fig. 1A). MD-76R purified C. trachomatis, L2 at 40 h p.i. was included as a positive control. It is highly likely that the crude chlamydial inoculum contains ample amounts of cellular debris, including nucleic acid, which might be responsible for activation of host cells. Indeed, unlike purified C. pneumoniae, the crude inoculum contains high levels of the eukaryotic cytoplasmic protein Hsp90 and nuclear TATA box binding protein, TBBP (Fig. 1B). The lack of IFNβ production in cells infected with purified C. pneumoniae was further confirmed in A549 lung epithelial cells at 65 h p.i. (Fig. 1C). Moreover, in C. pneumoniae infected cells transfected with poly(I:C), the production of IFNβ detected in the culture medium was inhibited by more than 50% when compared to uninfected poly(I:C) stimulated cells (Fig. 1C). The percent reduction in levels of IFNβ corresponded to the number of infected cells as approximately 50%–60% of cultures contained chlamydial inclusions (data not shown). These data demonstrate that infection of epithelial cells with purified, intact C. pneumoniae does not stimulate IFNβ production at any time post infection, and suggests that the microorganism actively impairs generation of IFNβ in infected cultures.
C. pneumoniae prevents IRF3 activation in infected cells
We further confirmed that C. pneumoniae inhibits IFNβ production in infected HeLa cells where the infection of cells at multiplicity of infection (MOI) of 1 is easily and consistently achievable. Stimulation of uninfected cells with poly(I:C) for 4 h caused significant cytotoxicity which was detected by the presence of a large number of rounding cells compared to the mock treated cells. This cytotoxic effect of poly(I:C) appeared to be greatly reduced in cultures infected with C. pneumoniae for 64 h (Fig. 2A). In epithelial cells, the induction of IFNβ is initiated upon phosphorylation and nuclear translocation of the transcription factor IRF3. Therefore we examined the activation status of IRF3 in Chlamydia infected cultures. As expected, the Ser-396 specific phosphorylation of IRF3 was detected in uninfected cells transfected with poly(I:C) but not in quiescent cells. Interestingly, IRF3 phosphorylation was not detected in C. pneumoniae infected cultures even in cells stimulated with poly(I:C) (Fig. 2B). These data were further confirmed by probing of nuclear extracts for IRF3. Nuclear translocation of IRF3 was detected in uninfected cells and cells infected with C. trachomatis, L2 at 24 h p.i. but not in C. pneumoniae infected cells at 64 h p.i. upon induction with poly(I:C) (Fig. 2C). C. trachomatis was included as a positive control since infection of cultures with this chlamydial species induces expression of IFNβ (25). C. trachomatis infected cells were harvested at 24 h p.i. due to differences in chlamydial growth rate.
FIGURE 2.
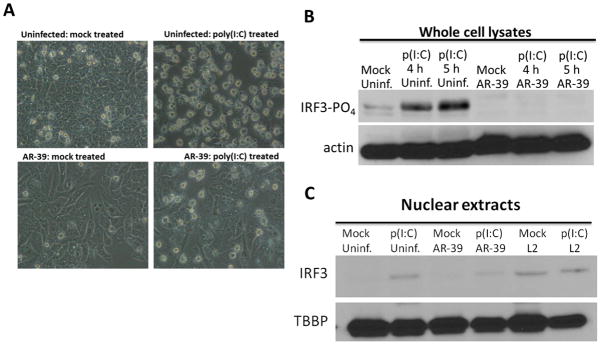
C. pneumoniae does not activate IRF3 and protects infected cells from detrimental effects of poly(I:C). (A) Phase-contrast microscopic images of uninfected and C. pneumoniae infected HeLa cells were either mock treated or transfected with 4 μg/ml of p(I:C) for 4 h. Micrographs were acquired using Olympus CX41microscope. (B) Immunoblot analysis of uninfected and infected HeLa cells, treated as above for 4 h or 5 h, and probed with anti-phospho-IRF3 specific antibody. The amount of protein loaded per sample was controlled by probing with anti-actin antibodies. (C) C. pneumoniae (64 h p.i.) or C. trachomatis (24 h p.i.) infected and uninfected cultures were either mock treated or transfected with 4 μg/ml of poly(I:C) for 5 h and subjected to subcellular fractionation. Immunoblot of the nuclear fraction was probed with anti-IRF3 antibody. Anti-TATA box binding protein (TBBP) was employed as a loading control.
C. pneumoniae targets host TRAF3
In epithelial cells transfected with poly(I:C), the signaling cascade leading to IRF3 activation is primarily initiated from MDA5, which recognizes this dsRNA analog (26). Ligation of MDA5 to dsRNA leads to the recruitment of mitochondrial anti-viral signaling protein (MAVS) followed by activation of TRAF3 through direct association with MAVS (10, 27). Formation of TRAF3 dependent K63-linked polyubiquitin chains in induced cells is essential for activation of TBK1-IKKε kinases that phosphorylate IRF3. Conversely, in cells treated with poly(I:C) in the absence of a transfecting reagent, the dsRNA enters the cell via endocytosis and initiates the TLR3 signaling pathway leading to the activation of IRF3 and NFκB (reviewed in (28). Interestingly, unlike with C. trachomatis, infection of cultures with C. pneumoniae in HeLa cells treated with poly(I:C) for 1 h in the absence of a transfecting reagent prevented phosphorylation of the cytoplasmic NFκB inhibitor, IκBα, indicating that the pathogen is also capable of inhibiting signaling initiated from TLR3 (Fig. 3A). The transduction pathways initiated from both MDA5 and TLR3 converge at one critical signaling molecule, TRAF3. Indeed, in C. pneumoniae infected and uninfected cells co-transfected with HA-TRAF3 and pUNO-hIPS1, TRAF3 was immunoprecipitated together with MAVS from uninfected cells but not from C. pneumoniae infected cultures. The co-precipitation of TRAF3 and MAVS was detected in Chlamydia-infected cells treated with chloramphenicol (CMP), indicating that bacterial protein synthesis is required to prevent MAVS binding to TRAF3 (Fig. S1). We therefore analyzed TRAF3 levels during C. pneumoniae infection. In A549 cells which contain detectable amounts of endogenous TRAF3, the infection with C. pneumoniae resulted in lower levels of the signaling molecule as early as 24 h p.i. when compared to uninfected cells. This decrease in TRAF3 was detected throughout the time course of infection (Fig. 3B). These results indicate that C. pneumoniae targets host TRAF3 during infection.
FIGURE 3.
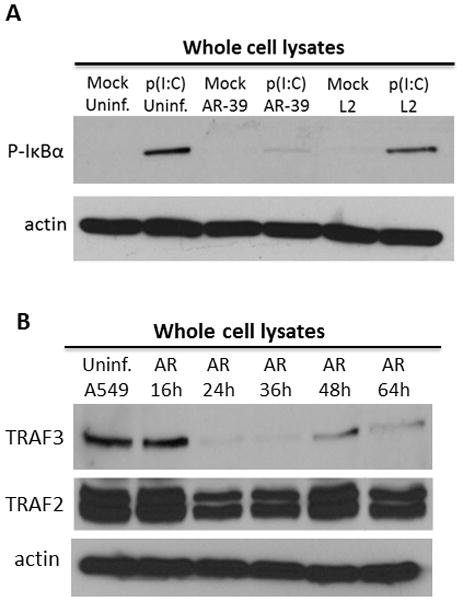
C. pneumoniae decreases TRAF3 in infected cells. (A) Uninfected and C. pneumoniae or C. trachomatis infected HeLa cells were either mock treated or treated with 2 μg/ml of poly(I:C) for 1 h in the absence of a transfecting reagent. Immunoblots of cell lysates were probed with anti-S32 and S36 phospho-specific IκBα mAb. Amount of protein loaded per sample was controlled by probing with anti-actin antibodies. (B) Uninfected A549 cells or cells infected with C. pneumoniae were harvested at 16, 24, 36, 48 and 64 h p.i. and probed with anti-TRAF3 or TRAF2 antibodies. Probing with anti-actin mAb was used as a loading control.
C. pneumoniae degrades TRAF3 in epithelial cells
The possibility that C. pneumoniae may degrade TRAF3 was investigated in uninfected and chlamydiae infected HeLa cells ectopically expressing HA-TRAF3. It has been demonstrated that overexpression of TRAF3 does not activate any known transduction pathway (29). Whole cell lysates were harvested at 64 h p.i. for C. pneumoniae infected cells and at 24 h p.i. for C. trachomatis, L2 infected cells and probed with TRAF3-specific antibody. Converse to uninfected and C. trachomatis-infected cells, this signaling molecule was not readily detectable in C. pneumoniae infected cultures (Fig. 4A), raising the possibility that TRAF3 is degraded. We performed transcriptional analysis of TRAF2, TRAF3, and TRAF5 in uninfected and C. pneumoniae infected HeLa cells to rule out the possibility of alterations at the transcriptional level. Indeed, the mRNA message of all the analyzed genes, including TRAF3, was not lower than in uninfected cultures (Fig. S2). Furthermore, we examined the overexpression of TRAF3 in C. trachomatis, serovar D infected cells and the levels of the signaling molecule remained unaltered (Fig. S3). The apparent TRAF3 degradation was therefore a C. pneumoniae-specific phenomenon requiring bacterial translation (Fig. 4A). The levels of TRAF3 degradation correlated with the number of cells infected with C. pneumoniae as more TRAF3 was detected at a lower MOI (Fig. 4B). Conversely, the ectopic expression of TRAF2 in HeLa cells was unaffected by C. pneumoniae or C. trachomatis (Fig. 4C).
FIGURE 4.
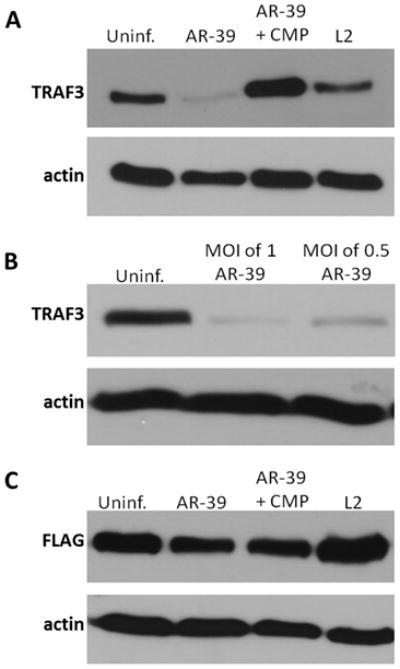
C. pneumoniae degrades host TRAF3. (A) Whole cell lysates of uninfected and C. pneumoniae infected at 64 h p.i., or C. trachomatis infected at 24 h p.i. HeLa cells transfected with HA-TRAF3. Immunoblots were probed with anti-TRAF3 antibody. (B) C. pneumoniae cultures infected at an MOI of 1 or 0.5 and uninfected cells were transfected with HA-TRAF3. Resolved material was probed with anti-TRAF3 mAb. (C) Uninfected and chlamydiae infected HeLa cells transfected with Flag-TRAF2 were probed with anti-Flag mAb. Immunoblots probed with anti-actin were used as a loading control.
Treatment of infected cells with protease inhibitors does not prevent C. pneumoniae induced TRAF3 degradation
TRAF3 is an adaptor protein that also acts as an E3 ubiquitin ligase, a function essential for K63-linked autoubiquitination (30), whereas K48-linked ubiquitylation of TRAF3 marks this signaling molecule for proteasome-dependent degradation (31, 32). Interestingly, only K63-linked polyubiquitin chains are stimulated directly by TRAF3 whereas other cytoplasmic E3 ligases execute K48-linked ubiquitination (30, 33). It was recently demonstrated that TRAF3 degradation in A549 cells is mediated by the E3 ligase, Triad3A, and is prevented by the proteasome inhibitors Lactacystin and MG132 (33). Therefore, we investigated whether the degradation of TRAF3 during infection of epithelial cells with C. pneumoniae depends on the host proteasome. Uninfected and C. pneumoniae infected HeLa cells ectopically expressing TRAF3 were treated with Lactacystin for 15 hours. None of the Lactacystin concentrations tested prevented TRAF3 degradation in C. pneumoniae infected cells (Fig. 5A). We were unable to exploit the alternative proteasome inhibitor MG132 in this assay as treatment of cultures with MG132 for 15 h resulted in toxicity and cell death (data not shown). Instead, we employed a cell-free lysates approach (24) to further investigate the mechanism of TRAF3 degradation. 5×106 uninfected HeLa cells expressing TRAF3 were lysed using a Parr Cell Disruption Bomb, and the cell lysates were co-incubated for 1 h with different amounts of lysates prepared from purified C. pneumoniae. Even in the presence of MG132, chlamydial lysates were sufficient to decrease the abundance of TRAF3 (Fig. 5B). These data are consistent with changes in TRAF3 abundance not being manifested at the transcriptional level. More directly, they suggest that the protease targeting TRAF3 is chlamydial since degradation of TRAF3 is not impaired by two commonly used inhibitors of the eukaryotic 26S proteasome, and C. pneumoniae lysates are sufficient to yield reduced detection of TRAF3.
FIGURE 5.
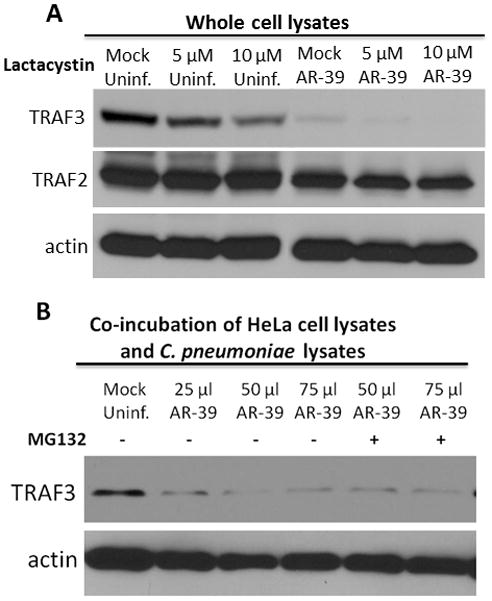
Degradation of TRAF3 is not inhibited by Lactacysitin or MG132. (A) Uninfected HeLa cells and cells infected with C. pneumoniae were transfected with HA-TRAF3 and treated with different concentrations of Lactacystin for 15 h. Immunoblots were probed with anti-TRAF3 and anti-TRAF2 antibodies. (B) Uninfected cells transfected with HA-TRAF3 were disrupted using a Parr Cell Disruption Bomb (1500 psi/5 min). HeLa cell lysates were co-incubated with various amounts of C. pneumoniae lysates in the presence or absence of 20 μM MG132. The level of TRAF3 degradation was detected by probing immunoblots with anti-TRAF3 mAb. Probing with anti-actin was used as a loading control.
Discussion
Type I interferons are pleiotropic cytokines secreted by a variety of cells and are known to act in an autocrine as well as paracrine manner. Type I IFNs are primarily comprised of multiple IFNα subtypes and a single IFNβ and represent an essential component of the innate immune response to viral infections in humans. Secreted interferons mediate anti-microbial activities by binding the IFNα/β receptor (IFNAR) leading to heterodimerization of the signal transduction activator 1 (STAT1) and STAT2. The heterodimer forms a complex with interferon regulatory factor 9 (IRF9) and is translocated into the nucleus where it interacts with the interferon-stimulated response element (ISRE). This ultimately drives the induction of transcription of type I interferon responsive genes. The importance of type I IFNs in innate response to numerous viruses is well established. Although type I interferons are induced, the role of this cytokine during the course of bacterial infections is less defined.
TRAF3 is a key signaling molecule involved in multiple pathways that lead to generation of type I IFNs and is essential for the innate immune response to a broad range of infections. K-63 linked autoubiquitination of TRAF3 is crucial for activation of IRF3 upon induction of cells with numerous stimuli. However, the signaling molecule also plays an important role as a negative regulator of various transduction pathways (31). For instance, in quiescent cells TRAF3 binds to the NFκB inducing kinase (NIK). Upon induction of the non-canonical NFκB pathway, TRAF3 undergoes non self-mediated K48-linked ubiquitination and proteosomal degradation releasing NIK, which results in phosphorylation of IKKα and p100 (NFκB2) processing (34). Furthermore, degradative ubiquitination of TRAF3 is also required for MyD88-dependent MAPK activation (30). Although C. pneumoniae inhibits induction of IFNβ upon MDA5 and TLR3 engagement (Fig. 6), it is highly likely that by targeting TRAF3, the pathogen modulates a great number of transduction pathways within its eukaryotic host.
FIGURE 6.
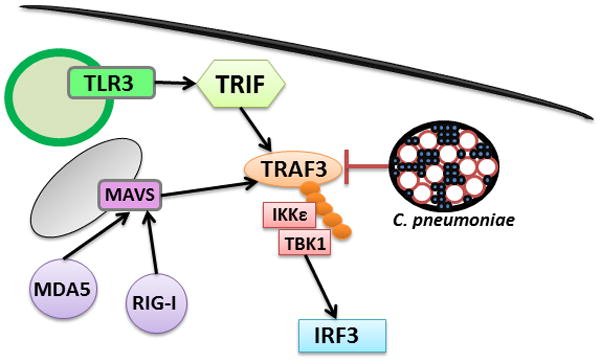
C. pneumoniae inhibits multiple signaling pathways by degradation of TRAF3. Upon stimulation of either MAVS- or TRIF-dependent signaling pathways, C. pneumoniae interferes with the signaling cascade by targeting host TRAF3. This prevents transduction of the inducing signal to IRF3, thereby suppressing the IFNβ response.
The majority of data describing chlamydial infections and IFN production comes from in vitro and in vivo studies with Chlamydia muridarum. C. muridarum is a murine pathogen that has been established as a mouse model of genital infection as it shares many aspects of acute genital infection with C. trachomatis in women (35). The production of type I IFNs in mice infected with C. muridarum appeared to be more beneficial to the microorganism than to the host. IFNAR−/− mice were able to resolve genital infection faster and displayed reduced pathology in the oviduct when compared to the WT mice (36). In C. muridarum infected murine macrophages, the expression of IFNβ was induced in MyD88-dependent manner, yet the survival of chlamydiae in MyD88−/− macrophages was comparable to chlamydial survival in macrophages obtained from WT mice (37). Conversely, IFNβ production in mouse oviduct epithelial cells was primarily TLR3 dependent i.e. MyD88-independent (38). The production of IFNβ has also been described in McCoy cells infected with C. trachomatis, serovar L2 (39), and Lad et al. detected significant up-regulation of IFNβ gene expression in C. trachomatis infected HeLa cells (25). Unlike with C. muridarum, no difference in chlamydial burden was detected in lungs of WT, IFNAR−/− and IRF3−/− mice infected with C. pneumoniae (40). A great number of studies have described diverse effects of C. pneumoniae on various cell types in vitro, including the production of IFNβ in endothelial cells (20). However, it is absolutely essential to clearly distinguish whether the observed effects are caused directly by the pathogen or by cellular debris which may enter the host cell together with the bacterium. Furthermore, it is important to differentiate between immune responses to productive chlamydial growth and chlamydial degradation when C. pneumoniae cannot avoid fusion with the lysosomal pathway within an infected cell (17).
In this study we have demonstrated that during productive infection of human epithelial cells with purified, intact C. pneumoniae, the pathogen does not induce production of IFNβ. Furthermore, C. pneumoniae induces species-specific degradation of the host signaling molecule TRAF3 thereby preventing activation of IFR3. The impaired transduction pathway initiated from MDA5 was detected in epithelial cell lines such as HeLa, A549, but also in HEK 293 cells (data not shown), all of which support C. pneumoniae growth and the levels of inhibition closely corresponded to the number of infected cells. Chlamydial protein synthesis was required for all of the events caused by the bacterium. In addition, the treatment of C. pneumoniae infected cells with the proteasomal inhibitors, Lactacystin and MG132, did not prevent TRAF3 degradation and co-incubation of chlamydial and host cell lysates resulted in degradation of the signaling molecule, suggesting that C. pneumoniae utilizes a species-specific protease that targets TRAF3. Although chlamydiae reside within membrane-bound inclusions during their intracellular growth, insertion of chlamydial proteins into the inclusion membrane as well as secretion of chlamydial effectors into the host cytoplasm are essential for successful exploitation of the eukaryotic cell. Chlamydiae utilize sophisticated strategies to evade host immune responses. C. trachomatis and C. pneumoniae secrete a serine protease designated chlamydial protease/proteasome like activity factor (CPAF) into the host cytosol. CPAF is responsible for degradation of several eukaryotic components but it primarily targets the regulatory factor X5 (RFX5) and upstream stimulation factor 1 (USF-1), both of which are required for MHC antigen expression (41). Moreover, chlamydiae employ a Tail-specific protease (Tsp) that cleaves the p65/RelA subunit of NFκB1 into 40 kDa and ~22 kDa fragments, preventing NFκB activation during chlamydial infection (24). Although the target specificity of these two chlamydial proteases has been recently questioned since CPAF and Tsp failed to cleave their respective targets in 8M Urea (23), we have detected the degradation of TRAF3 even in the presence of 8M Urea (Figs. 3 and 4), which suggests that targeting of the signaling molecule is not an artifact of post lysis degradation. Our data represent a critical shift in understanding C. pneumoniae pathogenesis. Whilst it is postulated that the pathogen elicits numerous pro-inflammatory responses by host cells, we propose that during productive infection with purified C. pneumoniae, the bacteria actively modify and impair the host immune system, which prevents or significantly delays chlamydial recognition and efficient elimination. Indeed, C. pneumoniae seems to utilize species-specific strategies which are comparatively more efficient than those employed by C. trachomatis, in order to avoid or delay recognition by the host innate immunity. It has been previously established that C. pneumoniae intercepts the signaling pathway from IL-17R in IL-17 stimulated epithelial cells by sequestering the adaptor molecule Act1to the chlamydial inclusion membrane (42). The degradation of TRAF3 represents another unique tactic of C. pneumoniae that may significantly contribute to protracted or asymptomatic respiratory infections and/or chronic infections associated with this pathogen.
Supplementary Material
Acknowledgments
This work was supported by the Public Health Service Grant AI072126 from the National Institutes of Health to K.A.F.
We thank Drs. H. Betts-Hampikian, K. Mueller and N. Shembade for critical review of the manuscript. We would also like to thank Dr. E. Harhaj for FLAG-TRAF2 and Dr. S. C. Sun for HA-TRAF3 construct.
Footnotes
Disclosures
The authors have no financial conflict of interest.
References
- 1.Mayer AK, Muehmer M, Mages J, Gueinzius K, Hess C, Heeg K, Bals R, Lang R, Dalpke AH. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J Immunol. 2007;178:3134–3142. doi: 10.4049/jimmunol.178.5.3134. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 5.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11:457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 8.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 9.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 10.Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paz S, Vilasco M, Werden SJ, Arguello M, Joseph-Pillai D, Zhao T, Nguyen TL, Sun Q, Meurs EF, Lin R, Hiscott J. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011;21:895–910. doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo CC, Jackson LA, Campbell LA, Grayston JT. Chlamydia pneumoniae (TWAR) Clinical microbiology reviews. 1995;8:451–461. doi: 10.1128/cmr.8.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell LA, Kuo CC, Grayston JT. Chlamydia pneumoniae and cardiovascular disease. Emerg Infect Dis. 1998;4:571–579. doi: 10.3201/eid0404.980407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schramm N, Wyrick PB. Cytoskeletal requirements in Chlamydia trachomatis infection of host cells. Infect Immun. 1995;63:324–332. doi: 10.1128/iai.63.1.324-332.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf K, Hackstadt T. Sphingomyelin trafficking in Chlamydia pneumoniae-infected cells. Cell Microbiol. 2001;3:145–152. doi: 10.1046/j.1462-5822.2001.00098.x. [DOI] [PubMed] [Google Scholar]
- 17.Wolf K, Fischer E, Hackstadt T. Degradation of Chlamydia pneumoniae by peripheral blood monocytic cells. Infect Immun. 2005;73:4560–4570. doi: 10.1128/IAI.73.8.4560-4570.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf K, Fischer E, Hackstadt T. Ultrastructural analysis of developmental events in Chlamydia pneumoniae-infected cells. Infect Immun. 2000;68:2379–2385. doi: 10.1128/iai.68.4.2379-2385.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maass M, Bartels C, Engel PM, Mamat U, Sievers HH. Endovascular presence of viable Chlamydia pneumoniae is a common phenomenon in coronary artery disease. Journal of the American College of Cardiology. 1998;31:827–832. doi: 10.1016/s0735-1097(98)00016-3. [DOI] [PubMed] [Google Scholar]
- 20.Buss C, Opitz B, Hocke AC, Lippmann J, van Laak V, Hippenstiel S, Krull M, Suttorp N, Eitel J. Essential role of mitochondrial antiviral signaling, IFN regulatory factor (IRF)3, and IRF7 in Chlamydophila pneumoniae-mediated IFN-beta response and control of bacterial replication in human endothelial cells. J Immunol. 2010;184:3072–3078. doi: 10.4049/jimmunol.0902947. [DOI] [PubMed] [Google Scholar]
- 21.Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–1176. doi: 10.1128/iai.31.3.1161-1176.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fields KA, Mead DJ, Dooley CA, Hackstadt T. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Molecular microbiology. 2003;48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen AL, Johnson KA, Lee JK, Sutterlin C, Tan M. CPAF: A Chlamydial Protease in Search of an Authentic Substrate. PLoS pathogens. 2012;8:e1002842. doi: 10.1371/journal.ppat.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lad SP, Li J, da Silva Correia J, Pan Q, Gadwal S, Ulevitch RJ, Li E. Cleavage of p65/RelA of the NF-kappaB pathway by Chlamydia. Proc Natl Acad Sci U S A. 2007;104:2933–2938. doi: 10.1073/pnas.0608393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lad SP, Fukuda EY, Li J, de la Maza LM, Li E. Up-regulation of the JAK/STAT1 signal pathway during Chlamydia trachomatis infection. J Immunol. 2005;174:7186–7193. doi: 10.4049/jimmunol.174.11.7186. [DOI] [PubMed] [Google Scholar]
- 26.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 27.Tang ED, Wang CY. MAVS self-association mediates antiviral innate immune signaling. J Virol. 2009;83:3420–3428. doi: 10.1128/JVI.02623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clinical microbiology reviews. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 30.Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11:70–75. doi: 10.1038/ni.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuzawa A, Tseng PH, Vallabhapurapu S, Luo JL, Zhang W, Wang H, Vignali DA, Gallagher E, Karin M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS pathogens. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farris CM, Morrison RP. Vaccination against Chlamydia genital infection utilizing the murine C. muridarum model. Infect Immun. 2011;79:986–996. doi: 10.1128/IAI.00881-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagarajan UM, Prantner D, Sikes JD, Andrews CW, Jr, Goodwin AM, Nagarajan S, Darville T. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun. 2008;76:4642–4648. doi: 10.1128/IAI.00629-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagarajan UM, Ojcius DM, Stahl L, Rank RG, Darville T. Chlamydia trachomatis induces expression of IFN-gamma-inducible protein 10 and IFN-beta independent of TLR2 and TLR4, but largely dependent on MyD88. J Immunol. 2005;175:450–460. doi: 10.4049/jimmunol.175.1.450. [DOI] [PubMed] [Google Scholar]
- 38.Derbigny WA, Johnson RM, Toomey KS, Ofner S, Jayarapu K. The Chlamydia muridarum-induced IFN-beta response is TLR3-dependent in murine oviduct epithelial cells. J Immunol. 2010;185:6689–6697. doi: 10.4049/jimmunol.1001548. [DOI] [PubMed] [Google Scholar]
- 39.Devitt A, Lund PA, Morris AG, Pearce JH. Induction of alpha/beta interferon and dependent nitric oxide synthesis during Chlamydia trachomatis infection of McCoy cells in the absence of exogenous cytokine. Infect Immun. 1996;64:3951–3956. doi: 10.1128/iai.64.10.3951-3956.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothfuchs AG, Trumstedt C, Mattei F, Schiavoni G, Hidmark A, Wigzell H, Rottenberg ME. STAT1 regulates IFN-alpha beta- and IFN-gamma-dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J Immunol. 2006;176:6982–6990. doi: 10.4049/jimmunol.176.11.6982. [DOI] [PubMed] [Google Scholar]
- 41.Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf K, Plano GV, Fields KA. A protein secreted by the respiratory pathogen Chlamydia pneumoniae impairs IL-17 signalling via interaction with human Act1. Cell Microbiol. 2009;11:769–779. doi: 10.1111/j.1462-5822.2009.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.