

Abstract
Transposon mutagenesis and single-gene deletion are two methods applied in genome-wide gene knockout in bacteria 1,2. Although transposon mutagenesis is less time consuming, less costly, and does not require completed genome information, there are two weaknesses in this method: (1) the possibility of a disparate mutants in the mixed mutant library that counter-selects mutants with decreased competition; and (2) the possibility of partial gene inactivation whereby genes do not entirely lose their function following the insertion of a transposon. Single-gene deletion analysis may compensate for the drawbacks associated with transposon mutagenesis. To improve the efficiency of genome-wide single gene deletion, we attempt to establish a high-throughput technique for genome-wide single gene deletion using Streptococcus sanguinis as a model organism. Each gene deletion construct in S. sanguinis genome is designed to comprise 1-kb upstream of the targeted gene, the aphA-3 gene, encoding kanamycin resistance protein, and 1-kb downstream of the targeted gene. Three sets of primers F1/R1, F2/R2, and F3/R3, respectively, are designed and synthesized in a 96-well plate format for PCR-amplifications of those three components of each deletion construct. Primers R1 and F3 contain 25-bp sequences that are complementary to regions of the aphA-3 gene at their 5' end. A large scale PCR amplification of the aphA-3 gene is performed once for creating all single-gene deletion constructs. The promoter of aphA-3 gene is initially excluded to minimize the potential polar effect of kanamycin cassette. To create the gene deletion constructs, high-throughput PCR amplification and purification are performed in a 96-well plate format. A linear recombinant PCR amplicon for each gene deletion will be made up through four PCR reactions using high-fidelity DNA polymerase. The initial exponential growth phase of S. sanguinis cultured in Todd Hewitt broth supplemented with 2.5% inactivated horse serum is used to increase competence for the transformation of PCR-recombinant constructs. Under this condition, up to 20% of S. sanguinis cells can be transformed using ~50 ng of DNA. Based on this approach, 2,048 mutants with single-gene deletion were ultimately obtained from the 2,270 genes in S. sanguinis excluding four gene ORFs contained entirely within other ORFs in S. sanguinis SK36 and 218 potential essential genes. The technique on creating gene deletion constructs is high throughput and could be easy to use in genome-wide single gene deletions for any transformable bacteria.
Keywords: Genetics, Issue 69, Microbiology, Molecular Biology, Biomedical Engineering, Genomics, Streptococcus sanguinis, Streptococcus, Genome-wide gene deletions, genes, High-throughput, PCR
Protocol
1. Primer Design
Primers are designed using in house scripts based on the S. sanguinis SK36 genome sequence. Three sets of primers, F1/R1, F2/R2, and F3/R3 are designed for amplification of the 1-kb upstream sequence of the target gene, the aphA-3 gene encoding kanamycin resistance (Kmr) protein3 and the 1-kb downstream sequence of the target gene, respectively (Figure 1). Among these primers, F1 and R3 are designed using ePrimer3 in the EMBOSS suite of programs (http://emboss.sourceforge.net/apps/cvs/emboss/apps/index.html) to amplify flanking regions upstream or downstream of the target gene. Gene specific primers, R1 and F3, are designed based upon the 5' and 3' sequences of the target gene. R1 and F3 primers contain 25-bp adaptor sequences at their 5' end that are complementary to the aphA-3 gene. The melting temperatures of each primer were designed to be as close as possible to 60 °C to enable use of a uniform annealing temperature for all PCR reactions in 96-well format.
F1, R1, F3 and R3 primers are synthesized in 96-well plates based on a gene order. Each plate contains one type of primers in the same gene order. Dilute the primers in 96-well working primer plate to a final concentration of 10 μM for PCR amplification using a multichannel pipette.
2. High-throughput PCR Amplification and Purification
To high-throughput amplify 1-kb upstream or downstream of the target S. sanguinis genes, assemble a PCR cocktail mixture on ice in a 15-ml conical tube containing 1640 μl ddH2O, 250 μl 10xHigh Fidelity PCR Buffer, 200 μl of 10 mM dNTP mixture, 100 μl of 50 mM MgSO4, 100 μl of 10 ng/ μl S. sanguinis SK36 genomic DNA and 10 μl Platinum Taq DNA polymerase high fidelity and transfer 23 μl of the mixture into each well on 96-well PCR plate using a multichannel pipette. Transfer 1 μl of each F1 and R1 (or F3 and R3) from the 10 μM working primer plates to the PCR plate using multichannel pipette. Seal the PCR plate and perform amplification at 94 °C for 1 min, and 30 cycles of 94 °C for 30 sec, 54 °C for 30 sec and 68 °C for 1.5 min.
Prepare 1% agarose gel containing ethidium bromide with 48 wells fitting multichannel pipette loading. Mix 4 μl PCR product with 1 μl of 5xDNA loading buffer on 96-well plate and load the samples on agarose gel using a 10-μl multichannel pipette. Run electrophoresis at 135 V for 30 min. Examine the band on gel under UVP documentation and analysis system.
Purify the PCR products by PureLink 96 PCR purification kit using centrifugation according to the manufacturer's instruction. For eluting DNA, add 40 μl sterile ddH2O to the well of the binding plate.
Randomly pick several purified amplicons on the plate to examine the DNA concentrations using NanoDrop spectrophotometer. Adjust the concentration of amplicons on plate to ~10 ng/μl.
A plasmid containing Kmr cassette is digested using EcoRI as PCR template. The digested plasmid is purified by QIAquick PCR purification kit and the linear plasmid DNA is adjusted to final DNA concentration 10 ng/μl. Assemble 25 μl mixture for Kmr cassette amplicon including 16.4 μl ddH2O, 2.5 μl 10xHigh Fidelity PCR Buffer, 2 μl of 10 mM dNTP mixture, 1 μl of 50 mM Mg SO4, 1 μl of 10 μM F2, 1 μl of 10 μM R2, 1 μl linear plasmid DNA and 0.1 μl Platinum Taq DNA polymerase high fidelity. Perform PCR amplification at 94 °C for 1 min, and 30 cycles of 94 °C for 30 sec, 55 °C for 30 sec and 68 °C for 1 min. Examine the PCR product by gel electrophoresis on 1% agarose. Pool a bulk of the Kmr cassette amplicon from 10 individual PCR purified by QIAquick PCR purification kit. Adjust the amplicon concentration to 10 ng/μl.
To obtain the final linear recombinant PCR amplicon, three PCR amplicons are combined with each 1 μl (in nearly equal-molar amounts) in one PCR plate well as the PCR template. Other PCR components is added including 14.4 μl ddH2O, 2.5 μl 10xHigh Fidelity PCR Buffer, 2 μl of 10 mM dNTP mixture, 1 μl of 50 mM Mg SO4, 1 μl of 10 μM F1, 1 μl of 10 μM R3, 0.1 μl Platinum Taq DNA polymerase high fidelity. PCR amplification is performed at 94 °C for 2 min, 30 cycles of 94 °C for 30 sec 55 °C for 30 sec and 68 °C for 3.5 min, and finally 68 °C for 4 min.
Examine the PCR amplicons on agarose gel. Purify and quantify the PCR amplicons as described above. Freeze the recombined PCR amplicons at -20 °C.
3. Competent Cells Preparation
Todd Hewitt broth is prepared, adjusted pH to 7.6 using 10 N NaOH, heated to boil and then cooled to room temperature and sterilized using 0.22 μm polystyrene filter4. Add 300 μl heat-inactivated horse sera (final concentration to 2.5%) to 11.7 ml Todd Hewitt broth in 15-ml conical tube to make up TH+HS medium. Aliquot TH+HS medium into 2 ml and 10 ml in tubes.
Inoculate 5 μl of stock S. sanguinis SK36 frozen at -80 °C into 2 ml TH+HS medium and incubate the culture overnight with capping tightly at 37 °C, accompanied by pre-incubating the 10 ml-TH+HS tube.
After overnight, transfer 50 μl culture into 10 ml TH+HS and incubate the tube at 37 °C for 3 hr (corresponding to OD660 of 0.07-0.08), immediately using for transformation.
4. Cell Transformation and Antibiotic Selection
Add 2 μl of 70 ng S. sanguinis SK36 competence stimulating peptide (CSP) and 2 μl linear recombinant PCR amplicon (~50 ng) to Eppendorf tubes on 96-well block and pre-warm them at 37 °C. Transfer 330 μl of 3 hr-incubated SK36 culture into each tube. Incubate at 37 °C for 1 hr. Replace DNA with sterile ddH2O as control.
Place the block on ice and spread 100 μl of each transformation on brain heart infusion (BHI) agar plate with 500 μg/ml kanamycin. Incubate the plates at 37 °C for 2 d under microaerobic conditions.
5. Mutant Confirmation and Storage
For each replacement mutant, randomly pick up 2 individual colonies, inoculate the colony into 5 ml BHI containing 500 μg/ml kanamycin and incubate the inoculum microaerobically overnight at 37 °C. Cryopreserve each culture in 30% glycerol at -80 °C
To examine whether the mutant containing the expected gene replacement, perform colony PCR for each mutant using F1 and R3 primers in 96-well PCR plate. About 1-μl overnight culture from individual colony is used as the DNA template in 25-μl PCR-amplification reaction. PCR is performed at 94 °C for 5 min, 35 cycles of 94 °C for 30 sec, 55 °C for 30 sec and 68 °C for 3.5 min, and finally 68 °C for 4 min.
To precisely identify double-band mutants or contaminant PCR amplicon, examine the PCR amplicon by electrophoresis for 4 hr on >12 cm long 2% agarose gel with ethidium bromide staining. Under this agarose gel electrophoresis condition, any amplicons with ≥ 100 bp difference were clearly identified. When bands resulting from amplification of the Kmr cassette and the wild-type gene are anticipated to differ by < 100 bp, an internal T1 primer is used to determine whether a wild-type gene can be detected by PCR.
To further confirm the deletion, the amplicons are purified by PureLink 96 PCR purification kit and sequenced using the P1 primer which binds to the Kmr cassette. Keep only correct mutants confirmed by sequencing.
Representative Results
After PCR amplification using primers F1 and R1, and F3 and R3, approximately 1-kb upstream and downstream of each S. sanguinis gene were obtained in 96-well format, respectively (Figure 2A). Under our PCR conditions and using designed primers, a specific product was amplified from S. sanguinis genomic DNA in each PCR reaction. This result indicated the primers were highly specific to the targets of S. sanguinis. Through PCR re-amplification using primers F1 and R3 and three amplicons as DNA templates, linear recombinant PCR constructs (~3 kb) were high-throughput created on 96-well plate, composed of a region upstream of the each target gene, a kanamycin cassette, and a region downstream of each target gene in that order (Figure 2B). The recombinant PCR constructs were then transformed into S. sanguinis SK36 and selected by kanamycin on BHI agar plate. For the majority of transformations, over 1000 colonies were obtained on each plate following a microaerobic 2 d-incubation. When these colonies were PCR-amplified using F1 and R3, a single DNA band corresponding to the expected mutant size (~3 kb) was observed by gel electrophoresis (Figure 2C). When attempting to delete potential essential genes, no colonies could be obtained in the majority of transformations. For deletion of some essential genes, however, colonies were still obtained following transformation. After PCR amplification of these colonies using F1/R3, two DNA bands corresponding to the expected mutant size and the wild type size appeared as revealed by gel electrophoresis (Figure 2C). For some mutants, the expected mutant size and the wild type size of PCR amplicon were too close to be separated by long agarose gel. In this case, an internal primer T1 was designed based upon the wild-type gene sequence. PCR was performed using the internal T1 primer and F1 primer (Figure 2D). The wild-type SK36 was used as a positive control strain for this PCR. If a band with the expected size was amplified from the mutant using the internal primer, the gene was identified as essential gene (double-band mutant) since the presence of kanamycin cassette in the mutant was confirmed previously by DNA sequencing using the P1 primer. Based on this protocol, we eventually collected 2048 non-essential S. sanguinis gene mutants excluding those four ORFs contained entirely within other ORFs (Table 1). We identified 218 genes which are essential for S. sanguinis survival under the experimental conditions.
| Class | Open-reading frames (ORF) |
| Total genes* | 2270 |
| Targeted genes | 2266 |
| Nonessential (promoterless aphA-3) | 1973 |
| Nonessential (promoter aphA-3) | 75 |
| Essential (no transformant) | 60 |
| Essential (double-band) | 158 |
*Four ORFs contained entirely within other ORFs.
Table 1. S. sanguinis gene mutant summary.
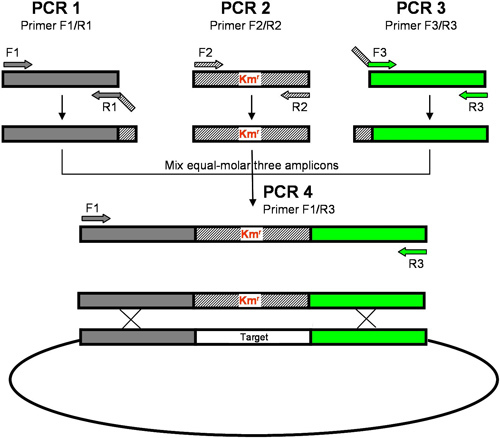 Figure 1. Recombinant PCR. Three sets of primers (F1/R1, F2/R2 and F3/R3) are designed to amplify the upstream sequence, a drug resistance gene (Kmr) and the downstream sequence, respectively. Both 5' ends of R1 and F3 primers contain sequences complementary with the cassette of an antibiotic resistant gene. A final PCR recombinant DNA product containing the antibiotic selection marker flanked with S. sanguinis genomic DNA is produced using F1/R3. The recombinant DNA will be integrated into S. sanguinis genome via double cross-over recombination.
Figure 1. Recombinant PCR. Three sets of primers (F1/R1, F2/R2 and F3/R3) are designed to amplify the upstream sequence, a drug resistance gene (Kmr) and the downstream sequence, respectively. Both 5' ends of R1 and F3 primers contain sequences complementary with the cassette of an antibiotic resistant gene. A final PCR recombinant DNA product containing the antibiotic selection marker flanked with S. sanguinis genomic DNA is produced using F1/R3. The recombinant DNA will be integrated into S. sanguinis genome via double cross-over recombination.
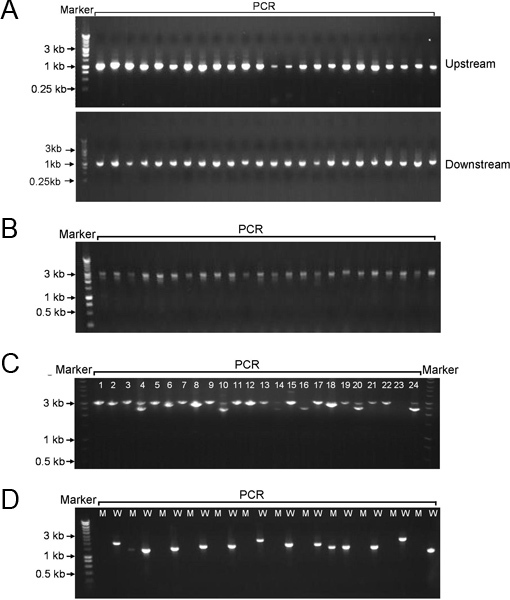 Figure 2. Representative gel electrophoresis of PCR amplicons on agarose. A, 1-kb PCR amplicons of upstream or downstream of the target S. sanguinis genes. B, the PCR recombinant DNAs composed of upstream and downstream of the target genes and promoterless aphA-3. C, colony-PCR amplicons from the transformation of S. sanguinis with PCR recombinant DNAs; double bands at lane 4, 10, 14, 15, 20 and 24. D, PCR amplification of wild-type and mutant colonies using primers F1/T1; M, mutant; W, wild-type.
Figure 2. Representative gel electrophoresis of PCR amplicons on agarose. A, 1-kb PCR amplicons of upstream or downstream of the target S. sanguinis genes. B, the PCR recombinant DNAs composed of upstream and downstream of the target genes and promoterless aphA-3. C, colony-PCR amplicons from the transformation of S. sanguinis with PCR recombinant DNAs; double bands at lane 4, 10, 14, 15, 20 and 24. D, PCR amplification of wild-type and mutant colonies using primers F1/T1; M, mutant; W, wild-type.
Discussion
To minimize the possible polar effects of the replacement of one targeted gene with exogenous antibiotics gene on neighboring genes, two steps are taken while initial primer designing. For most of deleted genes, the primers R1 and F3 are designed to delete the coding region from 6 bp following the start codon to 30 bp prior to the stop codon. The last 30 base pairs are retained to protect potential ribosomal binding site used by the adjacent downstream gene. The retained region between two neighboring genes is extended to 100 bp when they were located in opposite orientations and the N-termini of both proteins is close to each other to prevent deleting a potential promoter region. The promoter of aphA-3 gene is excluded in the recombinant construct for gene deletion5. A ribosomal binding site is introduced into the aphA-3 gene for translation. To test our strategy, we randomly constructed 10 and then 96 allelic exchange mutants. We obtained 10 and 93 mutants, respectively, on kanamycin selection plates, suggesting that the promoter-less aphA-3 gene was expressed well in most cases. We then designed and synthesized the primer for promoter-less aphA-3 gene for all gene deletions in the S. sanguinis genome. However, we found some genes in S. sanguinis were low expressed or not expressed which will likely affect the expression of the promoterless aphA-3 gene and hence the kanamycin selection. To solve this issue, the promoter of aphA-3 gene from the plasmid was introduced into the Kmr cassette. A bulk of the promoter Kmr cassette amplicon was obtained from 10 individual PCR using primers F2p/R2. Except that new R1 promoter primers R1p were designed for complement with the promoter Kmr cassette amplicon, all other construction steps were the same as for promoter-less construction.
To improve the efficiency of homologous recombination, we select long (1 kb) flanking sequences upstream and downstream of the S. sanguinis target gene in making gene-deletion constructs. The flanking sequence size was limited to 1 kb on the basis of two points: (1) the DNA fragment length (~3 kb) allowed ready PCR amplification of the aphA-3 gene (~1 kb) plus 2 kb of flanking sequences using high-fidelity DNA polymerase; and (2) the feasibility of sequencing flanking regions by ABI sequencing method from both ends (~1 kb). High numbers of the transformants in the most of S. sanguinis gene deletions indicated 1 kb upstream and downstream sequences, respectively, were enough to knockout the target genes.
To successful connect the upstream and downstream of the target gene with the two ends of aphA-3 gene in PCR recombinant amplification, primers R1, F2, R2 and F3 will be added at their 5' ends an extra sequence that is complementary with the connecting end of the other side in many bacterial gene deletion cases6,7. Preliminary studies showed that the extra sequence could be reduced to 25 bp for this recombinant PCR protocol. In this protocol, we eliminated the extra sequences of primers F2 and R2 in aphA-3 gene and added 25-bp extra sequence to the 5'-end of R1 and F3 in upstream and downstream of the S. sanguinis target gene. This design will decrease the length and amount of primers and hence reduce the cost and the PCR times to improve the efficiency of single-gene deletion. In our laboratory, this design has also been successfully demonstrated for gene deletions in other microbes such as Streptococcus mutans and Streptococcus pneumoniae, and Porphyromonas gingivalis.
In this protocol, the specificity of PCR primers is critical. The primer melting temperature was high (> 58 °C), the primer size was long (>21 bp), and the primer binding site was unique in the genome of S. sanguinis. This design produced single gene-specific products in nearly each of our PCR amplifications. A single gene-specific product from PCR amplification is critical for the last PCR amplification. Otherwise, the final PCR amplification that results in the formation of the final recombinant composed of each gene's upstream and downstream regions and antibiotic gene cassette, respectively, would be difficult.
Cell-density dependence of transformation has been demonstrated in many streptococci such as S. sanguinis8, S. mutans9and S. pneumoniae10. When cell density of S. sanguinis reached an OD660 of 0.07-0.08 (corresponding to ~5x106 CFU/ml) in TH+HS medium, the highest transformation efficiency of S. sanguinis with a linear recombinant PCR amplicon was obtained and up to 20% of S. sanguinis cells could be transformed. After completion of genome-wide gene deletions in S. sanguinis, we found that the number of the transformants for deletion of some nonessential genes was low (~10). There is no doubt that high transformation efficiency of bacteria is important to fulfill all of nonessential gene deletions in the genome-wide gene knockouts.
Except for recombinant construct creation by PCR, there are also other techniques for single gene inactivation. For example, suicide plasmid creation has been extensively applies to inactivate bacterial genes on a small scale. However, this technique has not been used for genome-wide single gene inactivation because large-scale plasmid creation is much time-consuming and laborious, and some bacteria need to be created specific suicide plasmid. Although marker-less gene deletion may avoid the possible polar effect of the antibiotic resistance gene, traditional marker-less gene deletion method is more time-consuming and difficult to inactivate a gene than the suicide plasmid method. In Escherichia coli, genome-wide mark-less gene deletions have been obtained through replacement with antibiotic resistance gene and then elimination of the resistance cassette6. However, this E. coli strain requires the phage λ Red recombinase. Before gene-deletion transformation, the Red helper plasmid pKD46 should be introduced into E. coli11. In our technique, we eliminated the promoter of aphA-3 resistance gene in the vast majority of gene-deleted mutants and designed the two-ends of aphA-3 ORF to follow the expression structure of the gene-deleted region, to avoid the polar effect issue of the resistance cassette. Up to now, we have not found the polar effect issue after examining the function of many mutants created by this technology. A genome-wide and clear set of essential genes acquired in S. sanguinis using this technique12 also imply low possibility of the polar effect of antibiotic cassette in the system. We have demonstrated other promoter-less antibiotic resistance genes, such as erythromycin and chloramphenicol resistance genes, could be applied in this technique (data not shown). In addition, this technique does not need suicide plasmid and reconstructed host. Overall, this technique is high throughput to create genome-wide gene deletion constructs and could be easy to perform in genome-wide single gene mutation of a bacterium.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by grants R01DE018138 from the National Institutes of Health (PX) and in part, by Virginia Commonwealth University Presidential Research Incentive Program (PRIP) 144602-3 (PX). We thank Drs. Lei Chen, Yuetan Dou and Xiaojing Wang for assisting with the construction of genome wide mutants. We also thank the DNA Core Facility at Virginia Commonwealth University for DNA sequencing.
References
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, et al. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu-Cuot P, Courvalin P. Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3'5"-aminoglycoside phosphotransferase type III. Gene. 1983;23:331–341. doi: 10.1016/0378-1119(83)90022-7. [DOI] [PubMed] [Google Scholar]
- Paik S, et al. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect. Immun. 2005;73:6064–6074. doi: 10.1128/IAI.73.9.6064-6074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukomski S, et al. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect. Immun. 2000;68:535–542. doi: 10.1128/iai.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Berardinis V. A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol. Syst. Biol. 2008;4:174. doi: 10.1038/msb.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AM, et al. Physiological and molecular characterization of genetic competence in Streptococcus sanguinis. Mol. Oral. Microbiol. 2011;26:99–116. doi: 10.1111/j.2041-1014.2011.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry D, Kuramitsu HK. Genetic transformation of Streptococcus mutans Infect. Immun. 1981;32:1295–1297. doi: 10.1128/iai.32.3.1295-1297.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakula R, Walczak W. On the nature of competence of transformable streptococci. J. Gen. Microbiol. 1963;31:125–133. doi: 10.1099/00221287-31-1-125. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, et al. Genome-wide essential gene identification in Streptococcus sanguinis. Sci. Rep. 2011;1 doi: 10.1038/srep00125. [DOI] [PMC free article] [PubMed] [Google Scholar]