

Abstract
AID promotes chromosomal translocations by inducing DNA double-strand breaks (DSBs) at immunoglobulin (Ig) genes and oncogenes in G1. RPA is a ssDNA-binding protein that associates with resected DSBs in the S phase and facilitates the assembly of factors involved in homologous repair (HR) such as Rad51. Notably, RPA deposition also marks sites of AID-mediated damage, but its role in Ig gene recombination remains unclear. Here we demonstrate that RPA associates asymmetrically with resected ssDNA in response to lesions created by AID, RAG, or other nucleases. Small amounts of RPA are deposited at AID targets in G1 in an ATM-dependent manner. In contrast, recruitment in S-G2/M is extensive, ATM-independent, and associated with Rad51 accumulation. RPA in S-G2/M increases in NHEJ-deficient lymphocytes, where there is more extensive DNA-end resection. Thus, most RPA recruitment during CSR represents salvage of un-repaired breaks by homology-based pathways during the S-G2/M phases of the cell cycle.
INTRODUCTION
Antigen receptor genes are assembled from variable (V), diversity (D) and joining (J) gene segments by the V(D)J recombinase Rag1 and Rag2 (Fugmann et al., 2000). This process requires generation of DNA DSBs in lymphocyte precursors in the G1-phase of the cell cycle. These targeted DSBs are joined by non-homologous end joining (NHEJ) as revealed by a complete block in Ig and T cell receptor gene assembly in the absence of NHEJ (Jung et al., 2006; Rooney et al., 2004).
Upon antigenic stimulation the Ig genes in B-lymphocytes undergo additional diversification by somatic hypermutation (SHM) and class switch recombination (CSR). CSR is a deletional recombination reaction between highly repetitive switch regions that replaces the heavy chain constant domain Igμ with one of several downstream isotypes Igγ, Igε, or Igα (Honjo et al., 2002; Stavnezer et al., 2008). Similar to V(D)J recombination, NHEJ plays a key role in the resolution of DSBs incurred during CSR (Helmink and Sleckman, 2012).
Both SHM and CSR are initiated by activation induced cytidine deaminase (AID), an enzyme that converts cytidines to uracils at Ig variable genes and switch (S) regions (Maul et al., 2011). dU:dG mismatches are recognized by base-excision and mismatch repair proteins leading to formation of DNA nicks and DSBs which are obligate intermediates in CSR (Stavnezer et al., 2008). In addition to Ig loci, AID can also target a large number of non-Ig genes (Liu et al., 2008; Pasqualucci et al., 1998; Shen et al., 1998; Yamane et al., 2011), including oncogenes that are frequently translocated to Ig in human and mouse B cell tumors (Chiarle et al., 2011; Klein et al., 2011; Kuppers and Dalla-Favera, 2001).
How AID is targeted to Ig and non-Ig loci is still unknown, but transcriptional pausing has been implicated (Peters and Storb, 1996; Rajagopal et al., 2009; Wang et al., 2009). In support of this idea the RNA exosome and RNA polymerase II stalling factor Spt5 appear to be required for AID to access its target genes (Basu et al., 2011; Pavri et al., 2010). Another potential AID co-factor, the ssDNA binding protein RPA, was isolated as part of a biochemical screen for activities that enhance AID hypermutation of in vitro-transcribed substrates (Chaudhuri et al., 2004). Consistent with this idea, RPA associates with Ig and non-Ig AID target genes (Vuong et al., 2009; Yamane et al., 2011) in a manner that is directly proportional to the extent of AID activity (Hakim et al., 2012). However, the precise role of RPA in CSR remains unknown.
In eukaryotic cells RPA forms a complex with ssDNA that is essential for DNA replication, telomere maintenance, DNA recombination, DNA repair, and DNA damage checkpoint activation (Oakley and Patrick, 2010; Wold, 1997). There are at least three ways whereby RPA might impact CSR. First, RPA could help stabilize AID’s ssDNA targets during Ig gene transcription (Chaudhuri et al., 2004). Second, RPA might stimulate long patch base excision repair (DeMott et al., 1998; Ranalli et al., 2002) or mismatch repair (Genschel and Modrich, 2003; Lin et al., 1998), which play critical roles in the processing of Ig gene deamination (Stavnezer et al., 2008). Third, RPA might associate with and stabilize resected ssDNA that cannot easily be repaired by classical NHEJ, thereby facilitating salvage by homology mediated repair pathways (Bothmer et al., 2010; Hasham et al., 2010; Zhang et al., 2010).
RESULTS
RPA accumulates in response to DNA breaks
Deletion of 53BP1 markedly increases RPA recruitment to Ig genes, particularly in the presence of the IgκAID transgene (Figure S1 and (Hakim et al., 2012)). To clarify the nature of RPA recruitment at AID targets, we monitored RPA by ChIP-Seq in the absence of H2AX, a factor that like 53BP1 limits DNA-end resection during intrachromosomal recombination (Bothmer et al., 2010; Helmink et al., 2011). B cells were stimulated with lipopolysaccharide (LPS) and interleukin 4 (IL4) to promote CSR from Igμ to Igγ1 and Igε. Similar to 53BP1−/− B cells we found broad RPA islands centered at recombining Sμ, Sγ1, and to a lesser extent Sε (Figure 1A, lanes I and II). Thus, absence of H2AX enhances RPA deposition at Igh.
Figure 1. RPA recruitment to AID, RAG, and I-SceI-induced breaks.

(A) RPA occupancy at the Igh locus of 53BP1−/− (lane I), H2AX−/− (lane II), and 2AX−/−UNG−/−Msh2−/− (lane III) B cells activated ex-vivo in the presence of LPS+IL-4. Deep-sequencing read densities were normalized to adjust for library size and bin width (reads per million per kb; RPKM). Numbers in parentheses represent average read density in RPKM for the displayed genomic window. (B) TCRα locus showing recruitment of RPA in 53BP1−/− (lane I) and 53BP1+/+ (lane III) control thymocytes. Rag2 occupancy is also included in lane II. (C) Kinetics of RPA recruitment at an I-SceI site engineered at Myc intron 1. Activated B cells were transduced with retroviruses expressing ER-I-SceI and cells were harvested at 0, 0.5, 3, or 24h after tamoxifen treatment.
To ascertain whether RPA recruitment requires DSBs, as opposed to AID recruitment, we performed the same experiment in H2AX−/−UNG−/−Msh2−/− B cells. In this genetic background AID is targeted to and deaminates Ig switch regions normally, but the resulting uracils are not processed to produce DSBs (Maul et al., 2011; Rada et al., 2004; Xue et al., 2006). Notably, RPA signals fell to background levels in the absence of DSBs (Figure 1A, lane III). This finding is consistent with the notion that RPA deposition requires the formation of DSBs, but is independent of DNA deamination and AID targeting.
To determine whether RPA is also associated with other DBSs produced in G1 we examined CD4+CD8+ double-positive thymocytes, which actively undergo RAG mediated TCRα recombination. Consistent with extensive nucleolytic processing of coding ends in 53BP1−/− T cells (Difilippantonio et al., 2008) we found prominent RPA signals in Jα (Figure 1B, lane I), a profile that is reminiscent of RAG2 recruitment to this locus (Figure 1B, lane II, and (Ji et al., 2010)). In contrast to wild type B cells, which accumulate RPA at the Ig locus during CSR, RPA accumulation in T cells required 53BP1 deletion, because ChIP signals were indistinguishable from background in wild type thymocytes (Figure 1B, lane III).
To verify that DSBs are sufficient to stimulate RPA accumulation and to examine the kinetics of recruitment, we induced DNA breaks in 53BP1−/−AID−/−MycI/I B cells by expression of an estrogen inducible I-SceI meganuclease (ER-I-SceI (Robbiani et al., 2008)). RPA recruitment became detectable 3h following induction of ER-I-SceI and persisted for at least 24h (Figure 1C). Taken together the data demonstrate that in 53BP1−/− lymphocytes RPA accumulates rapidly in response to physiological DSBs introduced in G1 (RAGs, AID) as well as nuclease-induced (I-SceI) damage.
RPA associates with resected ssDNA
During DNA repair RPA associates with ssDNA created by end resection (Symington and Gautier, 2011). Resected DNA is typically asymmetric and immunoprecipitated RPA associated with such DNA structures would be expected to reflect that asymmetry. Indeed, when RPA ChIP-Seq libraries from 53BP1−/−Ig AID B cells were resolved into upper (+) and lower (−) DNA strands RPA was asymmetrically distributed around AID target sites. As exemplified by Grap, a non-Ig AID target gene (Klein et al., 2011; Yamane et al., 2011), RPA was enriched on the + strand upstream, and on the - strand downstream of intron 1 (Figure 2A). This distribution is consistent with 5′-3′ DNA-end resection (Figure 2A, schematics). Directionality in RPA recruitment was also observed at Igh and other AID target genes in 53BP1−/− B cells (Figure 2B and not shown). Similar results were also obtained with MycI cells transduced with I-SceI (Figure S2A). In contrast to RPA, PolII ChIP-Seq libraries did not display any obvious strand biases at Grap or other AID target genes (Figure 2B and not shown).
Figure 2. RPA interacts with resected ssDNA downstream of AID or I-PpoI endonuclease.

(A) RPA association with + and − DNA strands at the Grap gene locus from IgκAID-53BP1−/− activated B cells. RPKM values for the specified window (chr11:61,399,129-61,572,079) are provided in parenthesis. (B) RPA bound to Igh locus. As in (A), ChIP-Seq signals were resolved into upper and lower strands. Technical replicates were treated with E. coli ExoI or RecJ exonucleases nuclease prior to deep-sequencing library preparation. (C) PolII binding to + and − strands at the Grap locus. (D) Technical IP replicates from panels A and C samples incubated in the presence of ExoI. (E) Composite diagram showing RPA profiles at 19 I-PpoI mouse genomic sites in 53BP1−/− MEFs transduced with retroviruses expressing the I-PpoI homing endonuclease.
To confirm that RPA binds to resected DNA at AID target genes, we incubated RPA and control PolII immunoprecipitates with E. coli single-strand exonuclease ExoI which digests ssDNA from 3′–5′ before library preparation. While PolII signals were largely unchanged, RPA enrichment at Grap or other AID targets could not be detected after nuclease treatment (Figure 2C–D, and Figure S2B). Although to a lesser extent, treatment of IP DNA with RecJ, a 5′-3′ exonuclease, also diminished RPA signals substantially (Figure 2B and 2D, and Figure S2B). This observation is consistent with the notion that resected, 200bp-sonicated DNA is susceptible to both 5′ and 3′ nucleolytic attack (Figure S2C). On the basis of these findings we conclude that RPA associates with resected DNA in response to AID or nuclease-mediated DNA damage.
In yeast, DNA-end resection can extend up to 10 kb in the absence of a homologous donor or Rad51 (Bishop et al., 1992; Sugawara et al., 1995; Zhu et al., 2008). The extent and dynamics of DNA end resection in mammalian cells however are unknown. Because AID-mediated damage is not site-specific but occurs over a relatively large genomic domain (~1–3kb (Klein et al., 2011; Yamane et al., 2011)), we monitored DNA-end resection in 53BP1−/− mouse embryonic fibroblasts transduced with the I-PpoI endonuclease which recognizes 19 sites in the C57BL/6 genome (San Filippo et al., 2008). RPA recruitment to all 19 I-PpoI sites was strand biased, centered on the DSB, and extended over an area of slightly more than 20kb (Figure 2E). Analogous results were also obtained at Myc upon I-SceI expression in MycI B cells (Figure S2A). We conclude that in mammalian cells deficient in 53BP1, resection occurs for up to 10–15kb on either side of a DSB.
Rad51 recruitment to AID-mediated breaks
RPA has been studied primarily in the context of HR where it recruits Rad51, a recombinase that forms the presynaptic nucleoprotein filament required for strand invasion (Ogawa et al., 1993; Sung and Robberson, 1995). To determine whether Rad51 also associates with resected DNA damaged by AID we performed Rad51 ChIP-Seq on 53BP1−/−Ig AID B cells. We found extensive Rad51 recruitment at Igh (Figure 3A). Analogous to RPA, the Rad51 ChIP signal was centered at recombining switch domains and displayed a marked DNA strand bias (Figure 3A). In addition, Rad51 was also found at AID off-target genes, such as Mir155 and Cd83 (Figure 3B). Additional examples are provided in Figure S3. At these sites, the domain separating the strand-specific Rad51 islands overlapped with hotspots of AID-induced chromosomal translocations (Figure 3B), as determined by TC-Seq (Klein et al., 2011). Moreover, the relative amount of Rad51 deposition per off-target gene (TSS +/− 2kb) was directly proportional to its translocation frequency (Spearman ρ = 0.65, Figure 3C). This result is consistent with the notion that, as described for RPA (Hakim et al., 2012), the extent of Rad51 occupancy is a function of AID activity. In contrast, genes not associated with RPA displayed background levels of Rad51 (Figure 3D and not shown). We conclude that in the absence of 53BP1, RPA and Rad51 associate with sites of AID-mediated damage at Ig and non-Ig genes.
Figure 3. Rad51 recruitment to AID on- and off-targets.

(A) Rad51 accumulation on the upper (+) and lower (−) strand at the Igh locus of IgκAID-53BP1−/− activated B cells. (B) Rad51 (blue), RPA (red), and translocation profiles at Mir155 and Cd83 genes in chromosomes 16 and 13 respectively from IgκAID-53BP1−/− B cells. (C) Chromosomal translocations (TSS +/− 2kb) involving Igh or Myc at Rad51-recruiting genes. Chromosomes 12 and 15 carrying the I-SceI sites were excluded from the analysis. Correlation between the two datasets is calculated using Spearman’s ρ. (D) Rad51 levels (TSS +/− 2kb) at RPA+ and RPA− genes in IgκAID-53BP1−/− B cells.
RPA recruitment occurs mostly in S-G2/M
Recruitment of RPA and Rad51 to resected DNA might occur in G1 during NHEJ repair of V(D)J or CSR DSBs. Alternatively, RPA could be recruited in S and G2/M phases of the cell cycle as part of a salvage mechanism for unrepaired DSBs. To address this question we purified H2AX−/− lymphocytes in the G1, S, and G2/M phases of the cell cycle and performed RPA ChIP-Seq. Consistent with limited DNA-end resection during NHEJ (Bothmer et al., 2010; Helmink et al., 2011), G1 cells displayed localized RPA deposition at the Igh locus (Figure 4A) in a manner that overlapped with sites of AID activity (Hakim et al., 2012; Klein et al., 2011). In contrast, lymphocytes from S and G2/M phases recruited far greater amounts of RPA (Figure 4A). Analogous results were obtained using 53BP1−/− B cells (see below). Thus, RPA recruitment occurs in both G1 and S-G2/M, with the majority of the signal accumulating in cells that progress to S-G2/M.
Figure 4. RPA and γ H2AX recruitment during cell cycle.

(A) RPA accumulation in H2AX−/− activated B cells at the Igh locus during the cell cycle. Samples were stained with Hoechst dye and sorted into G1, S, and G2/M-phased cells. RPKM values for the specified genomic windows are provided in parenthesis. (B) Extent of H2AX phosphorylation (γH2AX) at the Igh locus in wild type (G1, lane I; S, lane II), H2AX−/− (G1, lane III), or 53BP1−/− (G1, lane IV; S, lane V) activated B cells. (C) RPA (G1, G2/M) and γH2AX (G2/M) deposition at Igh in cells from H2AX−/− or 53BP1−/− respectively. ChIP-Seq data was split into + and − strands.
Our results suggested that in end joining deficient cells, a significant fraction of CSR lesions produced in G1 persist into S and G2/M, where they are processed for HR. To explore this possibility we monitored Igh breaks in cycling cells by ChIP for the phosphorylated histone H2AX (γH2AX), which accumulates at sites of DSBs (Rogakou et al., 1999). In wild type B cells in G1, γH2AX was primarily associated with Igh (1.40 RPKM, Figure 4B lane I), and these signals decreased in S phase cells to levels found in H2AX−/− controls (Figure 4B, lanes II and III). These results support the idea that most CSR breaks are resolved before DNA replication (Hasham et al., 2012; Petersen et al., 2001; Schrader et al., 2007; Sharbeen et al., 2012). In contrast, 53BP1−/− B cells showed similar levels of γH2AX accumulation in G1 and in the S phase (Figure 4B, lane V), indicating that in end joining deficient cells a significant fraction of CSR breaks persist beyond G1, even in the presence of intact ATM and p53 (Callen et al., 2007). These data indicate that un-repaired AID-mediated DNA damage does not efficiently activate cell cycle checkpoints in the absence of 53BP1 or H2AX.
As expected based on results with unsorted samples (Figure S2A), RPA displayed strand biases at Igh in all cell cycle stages (Figure 4C and not shown). In contrast, γH2AX signals were not asymmetric around S domains (Figure 4C), indicating that this chromatin mark, unlike RPA, is mostly associated with unresected double-strand lesions. The persistence of γH2AX signals in G2/M-phased cells is consistent with the presence of frequent chromosome 12 breaks in NHEJ deficient metaphases (Bothmer et al., 2011; Bunting et al., 2010; Gostissa et al., 2011).
ATM is required for resection in G1 but not S-G2/M
ATM is required for DNA end resection during CSR, V(D)J recombination and in cells treated with clastogenic agents (Bothmer et al., 2011; Bothmer et al., 2010; Helmink et al., 2011; Jazayeri et al., 2006). To examine the role of ATM in RPA recruitment during CSR we treated LPS+IL4 activated B cells with the small molecule ATM inhibitor KU-55933 (ATMi) and performed RPA ChIP-Seq on purified G1 and S-G2/M-phase cells. ATMi-treated G1 phase cells showed only background RPA signals at non-Ig AID targets in the absence of 53BP1. In addition, RPA was 3-fold lower at Igh in ATMi-treated cells relative to controls (P = 0.02, Figure 5B, n = 3). In contrast, activated B cells treated with ATR inhibitor (ATR45 (Charrier et al., 2011; Schoppy et al., 2012)) did not show this effect (Figure 5B) which is consistent with the observation that ATR is activated only after significant resection has occurred (Brown and Baltimore, 2003; Pellicioli et al., 2001; Shiotani and Zou, 2009; Zou and Elledge, 2003). Furthermore, ATR inhibitor treatment did not affect the extent of resection in S-G2/M (Morin et al., 2008).
Figure 5. ATM is required for G1 but not S-G2/M resection.

(A) RPA accumulation at Pim1 and IL4Rα loci from 53BP1−/− activated B cells that were either treated (lower two panels) or not treated (upper panels) with the ATM inhibitor KU-55933. Samples were sorted into G1 or S/G2/M phased cells using the Hoechst dye 33342. Numbers in parenthesis represent RPKM values within the specified genomic windows. (B) RPA accumulation at Igμ and Igγ1 loci in WT, ATMi-, or ATRi-treated B cells. Values represent the RPKM ratio between G1 and S-G2/M-phased cells. (C–D) RPA recruitment to the TCRα in thymocytes (C) or Igh in activated B cells (D) from 53BP1−/− (upper) or 53BP1−/−ATM−/− (lower) mice. (E) RPA and Rad51 recruitment to activated B cells with an intact NHEJ: IgκAID transgenics, AID+/+, and AID−/−.
To confirm a role for ATM in initiating resection in G1 we examined 53BP1−/− and 53BP1−/−ATM−/− thymocytes. The analysis showed a substantial reduction in RPA deposition at the TCRα locus in the absence of ATM (Figure 5C). Consistent with ATMi results, RPA signals at Igh in unfractionated B cells undergoing CSR from the same mice were not affected (Figure 5D). Taken together these findings demonstrate that ATM is required to promote initiation of RPA recruitment to resected DNA in G1- but is not required for its recruitment in S-G2/M-phased lymphocytes.
We have shown that in NHEJ deficient cells (H2AX−/− or 53BP1−/−), a fraction of DNA breaks that are normally repaired in G1 become substrates for homologous-mediated repair in S/G2/M. To investigate whether this also occurs under physiological conditions we measured Rad51 and RPA occupancy in activated wild type B cells undergoing CSR. We found strand-biased recruitment of Rad51 and RPA at Igμ and Igγ1 (Figure 5E), consistent with 5′-3′ resection activity in wild type cells. Resection required AID activity because RPA signals were increased in the presence of IgκAID and were indistinguishable from background in AID−/− lymphocytes (Figure 5E). As expected, the extent of resection in wild type cells was reduced relative to H2AX−/− or 53BP1−/− (compare RPKM values to Figures 5D and 2B for instance). We thus conclude that recombining Ig genes are resected under physiological conditions, arguing that non-homologous and homologous repair pathways contribute to repair during CSR.
DISCUSSION
Our results show that RPA is recruited to resected DNA DSBs produced by AID, RAGs, and site-specific endonucleases. RPA accumulation begins within 3 hours in the G1 phase of the cell cycle and unrepaired lesions persist into the S and G2/M phases where HR or other salvage repair mechanisms resolve DSBs. While our findings do not exclude the possibility that RPA can enhance AID activity (Chaudhuri et al., 2004), we can conclude that RPA recruitment is not essential for Ig gene deamination by AID.
RPA recruitment to DNA damage is asymmetric around DSBs, displaying a strong bias for the + DNA strand 5′, and the − strand 3′ of the break. This profile is best explained by the 5′-3′ directionality of DNA end resection which we find spreads 10–15Kb away from the site of the break in both directions as previously described in yeast (Bishop et al., 1992; Sugawara et al., 1995; Zhu et al., 2008). Consistent with this idea, exonuclease treatment of immunoprecipitated DNA, a procedure that specifically removes ssDNA 3′ tracks, ablates RPA ChIP signals. In addition, Rad51, which is required for strand invasion during HR, mirrors RPA deposition. The striking overlap between RPA and Rad51 profiles is consistent with the idea that RPA accumulation on resected DNA in S-G2/M facilitates HR ((New et al., 1998), Figure 6).
Figure 6. DNA-end resection during CSR.
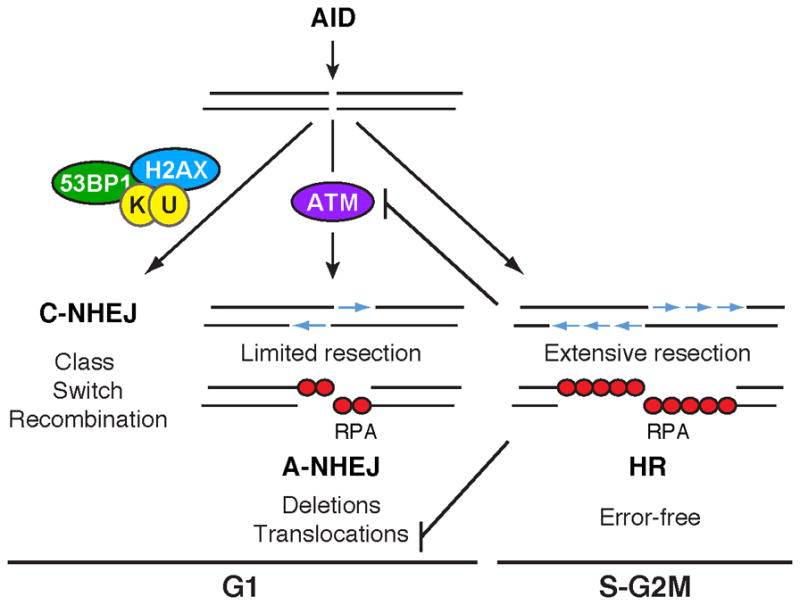
Model showing how AID-mediated breaks at Igh are either processed by C-NHEJ proteins into efficient switch recombination, or are resected by ATM-dependent A-NHEJ in G1 or by the HR in S-G2/M, leading to RPA recruitment and repair. Based on previous work (Shiotani and Zou, 2009), extensive resection in connection with HR is expected to inhibit ATM activity and potentially reduce the formation of chromosomal translocations via A-NHEJ.
DNA end resection has been primarily studied in yeast, where the Sae2 (CtIP) nuclease and the MRX (MRN) complex initiate the trimming phase, and the Exo1 and the RecQ helicase Sgs1 extend the resected tracks (Mimitou and Symington, 2008; Symington and Gautier, 2011; Zhu et al., 2008). The mechanistic details of end resection in mammalian cells are less clear, but both CtIP and ATM appear to play a role. During V(D)J recombination, depletion of CtIP in H2AX−/− cells protects DSBs from end resection (Helmink et al., 2011). In like manner, inhibition of ATM in 53BP1−/− B cells protects CSR breaks from degradation (Bothmer et al., 2010). Our results confirm a role for ATM in the resection of V(D)J and CSR lesions, but notably, only in the G1 phase of the cell cycle. Thus, an ATM-independent pathway controls resection at postreplicative stages.
The accumulation of RPA at resected AID target genes in the S-G2/M phases of the cell cycle suggest that some AID-induced DSBs are carried over from G1 to S where they would be preferentially repaired by HR. Consistent with this model, B cells deficient in the HR factor XRCC2 accumulate unrepaired AID-mediated breaks at postreplicative stages of the cell cycle (Hasham et al., 2010; Hasham et al., 2012). However, the mechanism that allows these lesions to persist beyond G1 was not resolved. ATM is an essential mediator of the G1-S phase checkpoint, and when it is deleted DSBs persist through cell cycle phase transitions leading to chromosome breaks and genome instability (Bredemeyer et al., 2006; Callen et al., 2007; Derheimer and Kastan, 2010). While it is conceivable that the relative low number of AID-mediated DSBs per cell might be insufficient to activate the checkpoint, this scenario seems unlikely because ATM can be activated by only a few breaks (Bakkenist and Kastan, 2003; Chen et al., 2000; Petersen et al., 2001). Moreover, AID activity engages ATM as indicated by formation of γH2AX foci on recombining Igh loci (Petersen et al., 2001). Cells that resolve these breaks by NHEJ extinguish ATM and progress. In contrast, DSBs that fail to resolve would be expected to undergo further processing, including end resection and RPA recruitment, which can suppress ATM signaling and abort the G1-S checkpoint (Callen et al., 2007; Shiotani and Zou, 2009). Thus, RPA accumulation in G1 may help avert the G1-S checkpoint and facilitate homology-mediated repair by enabling progression of cells carrying unrepaired breaks from G1 into the S phase.
What is the role of homology-mediated DNA repair during CSR? AID initiates CSR by introducing multiple DSBs in highly repetitive switch regions, which vary in length from 1 to ~10 Kb (Honjo et al., 2002). NHEJ repair of paired DSBs in heterologous switch regions results in productive CSR. However, ~20% of the time productive recombination fails, and DSBs are re-joined by an alternative form of non-homologous end joining (A-NHEJ) that promotes intra-switch deletions (Bottaro et al., 1998; Dudley et al., 2002; Gu et al., 1993; Hummel et al., 1987; Reina-San-Martin et al., 2003; Winter et al., 1987). We propose that this salvage mechanism is mediated by S region microhomology uncovered by DNA-end resection (Bothmer et al., 2011; Bothmer et al., 2010) and RPA recruitment (Figure 6). The presence of localized RPA at Igμ and Igγ1 in the G1 phase of the cell cycle is consistent with this idea. Whether A-NHEJ is also active at Igh in S-G2M remains to be determined. Our results also imply that DNA breaks not process by the canonical or alternative NHEJ pathways would be further resected in S-G2M and repaired by HR using the second, intact Igh allele (Figure 6). The physiologic advantage of engaging error-free HR during CSR is that switch regions thus repaired would be available for subsequent recombination attempts in the next G1 phase. In addition, HR should be able to counteract AID-induced mutations and limit the formation of chromosomal translocations.
MATERIALS AND METHODS
Mice
Ig AID (Robbiani et al., 2009); UNG−/− (Endres et al., 2004); ATM−/− (Barlow et al., 1996); AID−/− (Muramatsu et al., 2000), 53BP1−/− (Ward et al., 2003), MycI (Robbiani et al., 2008); Msh2−/− (Reitmair et al., 1995); and H2AX−/− (Bassing et al., 2003) mice were previously described. All experiments were in accordance with protocols approved by the NIAMS and Rockefeller Institutional Animal Care and Use Committee.
ChIP-seq
Ex-vivo cultured B-cells or thymocytes were fixed with 1% formaldehyde (Sigma) for 10 minutes at 37°C. The fixation was qu enched by addition of glycine (Sigma) at a final concentration of 125mM. Twenty million fixed cells were washed with PBS and then re-suspended in 1ml of RIPA buffer (10mM Tris pH7.6, 1mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 1× Complete Mini EDTA free proteinase inhibitor (Roche)) or stored at −80°C until further processing. The sonication was performed on S2 sonicator (Covaris) at duty cycle 20%, intensity 5, cycle/burst 200 for 30 minutes. Ten μg of anti-RPA32 (EMD, NA19L), anti-RAD51 (Santa Cruz, H-92) or anti-γH2AX (Epitomics, 2212-1) was incubated with 40 μl of Dynabeads Protein A for 40 minutes at room temperature. The antibody-bound beads were added to 500 μl of sonicated chromatin, incubated at 4°C overnigh t and washed twice with RIPA buffer, twice with RIPA buffer containing 0.3M NaCl, twice with LiCl buffer (0.25M LiCl, 0.5% Igepal-630, 0.5% sodium deoxycholate), once with TE pH8 plus 0.2% Triton X-100, and once with TE pH8. Crosslinking was reversed by incubating the beads at 65°C for 4 hours with 0. 3% SDS and 1 mg/ml Proteinase K. ChIP DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. The DNA was subsequently blunt-ended with End-It DNA end repair kit (Epicentre) and A-tailed with Taq DNA polymerase (Invitrogen) in the presence of 200mM of dATP for 40 minutes at 70°C. The sample was purified by phenol-chloroform extraction after each reaction. Illumina compatible adaptors (Illumina or Bioo Scientific) were then ligated with T4 DNA ligase (Enzymatics) and the reaction was purified once with AMpure XP magnetic beads (Beckman Coulter). Samples were PCR amplified for 18-cycles with KAPA HiFi DNA polymerase mix (KAPA biosystems). The amplicon was run on a 2% agarose gel and size-selected at 200–300 bp. Thirty-six or 50 bp of sequencing data were acquired on GAII or HiSeq2000 (Illumina). For nuclease treatment, ChIP DNA was incubated for 2 hours with 20 units of E. coli ExoI (New England Biolabs) at 37°C or 30 units of RecJf nuclease (New England Biolabs) at 37°C for 2 hours prior to the blunt-end ligation step. How ssDNA is amplified and detected during the Illumina library protocol has been recently addressed by two independent studies (Croucher et al., 2009; Khil et al., 2012). In brief, ssDNA is rendered double-stranded by intermolecular or intramolecular (hairpin) priming, facilitated by DNA microhomologies (Croucher et al., 2009; Khil et al., 2012). Table S1 contains a list of all ChIP-Seq experiments as they appear in the manuscript figures. Biological replicates or comparable experiments that further support the results presented in each panel are also provided.
Cell culture and retroviral infection
For I-SceI assays, CD43− resting B cells were isolated from spleens, stimulated, and infected as previously described (Bothmer et al., 2011). The pMX-IRES-GFP based retrovirus encoding I-SceI was previously described (Robbiani et al., 2008). The new pMX-ER-I-SceI was generated by fusing in frame the optimized ligand-binding domain of the human estrogen receptor (ERT2) with I-SceI. For I-PpoI assays, immortalized 53BP1−/− MEFs were transduced with the pMY retroviral vector encoding BFP-I-PpoI fusion gene. Following spin-infection MEFs were re-seeded until they reached ~70% confluency, at which time cells were trypsinized for 5′ at 37°C, and trypsin was quenched with complete media containing 1.25% formaldehyde for 10′. Following incubation a final concentration of 125mM glycine was added to stop crosslinking.
Flow Cytometry and Cell Sorting
I-SceI-GFP B cells and BFP-I-PpoI MEFs were sorted on a FACSAriaIII (Becton Dickinson) or Moflo (Beckman Coulter). For cell cycle sorting, B cells were cultured for 72 hours with LPS, IL4, and anti-Cd180 antibody and incubated for an additional for 45 minutes in the presence of 10 μg/ml of Hoechst 33342 (Invitrogen). Cells were then fixed the same conditions used for ChIP-seq sample preparation. Samples were the resuspended in HBSS buffer containing Hoechst 33342 and G1, S, or G2/M phase cells were sorted with Influx or AriaIII machines (BD Biosciences) using 355nm or 375nm lasers respectively.
Supplementary Material
RPA and Rad51 bind asymmetrically to recombining immunoglobulin genes
Ig gene DNA breaks are resected as B cells enter the S-G2M cell cycle stages
ATM regulates localized DNA-end resection in G1
DNA-end resection in S-G2M is extensive and ATM independent 3
Acknowledgments
We thank all members of the Casellas and Nussenzweig labs for helpful discussions, Matthias F. Muellenbeck, Jim Simone, Jeff Lay, and Gustavo Gutierrez for technical assistance. The work was supported in part by the intramural program of NIAMS at the NIH. This study utilized the high-performance computational capabilities of the Helix/Biowulf Systems at the National Institutes of Health, Bethesda, MD (http://helix.nih.gov). Deep-sequencing data is available at GEO under accession number GSE42451.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DK, Park D, Xu L, Kleckner N. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottaro A, Young F, Chen J, Serwe M, Sablitzky F, Alt FW. Deletion of the IgH intronic enhancer and associated matrix-attachment regions decreases, but does not abolish, class switching at the mu locus. Int Immunol. 1998;10:799–806. doi: 10.1093/intimm/10.6.799. [DOI] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, Mortimore M, O’Donnell ME, Pinder JL, Reaper PM, et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem. 2011;54:2320–2330. doi: 10.1021/jm101488z. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, et al. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Fookes MC, Perkins TT, Turner DJ, Marguerat SB, Keane T, Quail MA, He M, Assefa S, Bahler J, et al. A simple method for directional transcriptome sequencing using Illumina technology. Nucleic Acids Res. 2009;37:e148. doi: 10.1093/nar/gkp811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMott MS, Zigman S, Bambara RA. Replication protein A stimulates long patch DNA base excision repair. J Biol Chem. 1998;273:27492–27498. doi: 10.1074/jbc.273.42.27492. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–3681. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Manis JP, Zarrin AA, Kaylor L, Tian M, Alt FW. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc Natl Acad Sci U S A. 2002;99:9984–9989. doi: 10.1073/pnas.152333499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, et al. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annual Review of Immunology. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Mol Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–350. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol. 2010;11:820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Snow KJ, Donghia NM, Branca JA, Lessard MD, Stavnezer J, Shopland LS, Mills KD. Activation-Induced Cytidine Deaminase-Initiated Off-Target DNA Breaks Are Detected and Resolved during S Phase. J Immunol. 2012 doi: 10.4049/jimmunol.1200414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2011;469:245–249. doi: 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T, Kinoshita K, Muramatsu M. MOLECULAR MECHANISM OF CLASS SWITCH RECOMBINATION: Linkage with Somatic Hypermutation. Annu Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- Hummel M, Berry JK, Dunnick W. Switch region content of hybridomas: the two spleen cell Igh loci tend to rearrange to the same isotype. J Immunol. 1987;138:3539–3548. [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, Petukhova GV. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res. 2012;22:957–965. doi: 10.1101/gr.130583.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–5594. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- Lin YL, Shivji MK, Chen C, Kolodner R, Wood RD, Dutta A. The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J Biol Chem. 1998;273:1453–1461. doi: 10.1074/jbc.273.3.1453. [DOI] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM, 3rd, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. [see comments] Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- Oakley GG, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci. 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Yu X, Shinohara A, Egelman EH. Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science. 1993;259:1896–1899. doi: 10.1126/science.8456314. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, Chaganti RS, Klein U, Kuppers R, Rajewsky K, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci U S A. 1998;95:11816–11821. doi: 10.1073/pnas.95.20.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, San-Martin BR, Barreto V, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranalli TA, DeMott MS, Bambara RA. Mechanism underlying replication protein a stimulation of DNA ligase I. J Biol Chem. 2002;277:1719–1727. doi: 10.1074/jbc.M109053200. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, Waterhouse P, Mittrucker HW, Wakeham A, Liu B, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- Robbiani DF, Bothmer A, Callen E, Reina San-Martin B, Dorsett Y, difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, et al. Activation Induced Deaminase is Required for the Chromosomal Translocations in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–131. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, Fernandez-Capetillo O, Diehl JA, Brown EJ. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest. 2012;122:241–252. doi: 10.1172/JCI58928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- Sharbeen G, Yee CW, Smith AL, Jolly CJ. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med. 2012;209:965–974. doi: 10.1084/jem.20112379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–1752. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ivanov EL, Fishman-Lobell J, Ray BL, Wu X, Haber JE. DNA structure-dependent requirements for yeast RAD genes in gene conversion. Nature. 1995;373:84–86. doi: 10.1038/373084a0. [DOI] [PubMed] [Google Scholar]
- Sung P, Robberson DL. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter E, Krawinkel U, Radbruch A. Directed Ig class switch recombination in activated murine B cells. Embo J. 1987;6:1663–1671. doi: 10.1002/j.1460-2075.1987.tb02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gostissa M, Hildebrand DG, Becker MS, Boboila C, Chiarle R, Lewis S, Alt FW. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol. 2010;106:93–133. doi: 10.1016/S0065-2776(10)06004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.