

Abstract
Kidney cancer is not a single disease; it is made up of a number of different types of cancer that occur in the kidney. Each of these different types of kidney cancer can have a different histology, have a different clinical course, can respond differently to therapy and is caused by a different gene. Kidney cancer is essentially a metabolic disease; each of the known genes for kidney cancer, VHL, MET, FLCN, TSC1, TSC2, TFE3, TFEB, MITF, fumarate hydratase (FH), succinate dehydrogenase B (SDHB), succinate dehydrogenase D (SDHD), and PTEN genes is involved in the cells ability to sense oxygen, iron, nutrients or energy. Understanding the metabolic basis of kidney cancer will hopefully provide the foundation for the development of effective forms of therapy for this disease.
Keywords: Cancer Metabolism, Renal Cell Carcinoma (RCC), Hereditary Kidney Cancer, Cancer Metabolism Targeted Therapy, Warburg Effect
1: An Introduction to Kidney Cancer
It is now known that kidney cancer is not a single uniform disease; it is in fact a number of different and specific types of cancers that can occur within the kidney. Each of these different types of kidney cancer can be characterized by differing histologies, different clinical courses, differing responses to a number of varied therapies and association with alterations to different tumor suppressor genes or oncogenes. Currently there are at least twelve different genes associated with the development of kidney cancer, the VHL, MET, FLCN, TSC1, TSC2, TFE3, TFEB, MITF, fumarate hydratase (FH), succinate dehydrogenase B (SDHB), succinate dehydrogenase D (SDHD), and PTEN genes. Each one of these genes is involved in the regulation of the ancient and essential mechanisms involving the single cell's ability to respond to nutrient deprivation in the surrounding environment and alter its metabolism accordingly. Thus, these gene pathways are involved in the cell's ability to respond to changes in oxygen, iron, nutrients or energy which might limit growth and advantageous alterations that can overcome this and promote growth are intrinsically useful in tumorigenesis. Understanding the metabolic basis of cancer of the kidney will hopefully provide the foundation for the development of novel therapeutic approaches targeting the metabolic basis of kidney cancer. (Suggested position for Figure 1)
Figure 1.

Kidney cancer is not a single disease; it is made up of a number of different and specific types of cancers that can occur within the kidney. Each of these different types of kidney cancer can be characterized by differing histologies, different clinical courses, differing responses to a number of varied therapies and association with alterations to different genes. (TBD*: to be determined.)
2: Hereditary Kidney Cancer
Much of what we know about the genetic basis of kidney cancer was learned from the study of inherited forms of kidney cancer. There are a number of familial forms of kidney cancer, including von Hippel-Lindau (VHL), Hereditary Papillary Renal Carcinoma (HPRC), Birt-Hogg-Dubé (BHD), Hereditary Leiomyomatosis Renal Cell Carcinoma (HLRCC), Succinate Dehydrogenase Renal Cell Carcinoma (SDH-RCC), Tuberous Sclerosis (TS), and Cowden's Disease.(1, 2) All these syndromes are associated with the inheritance of single mutant copy of a gene that imparts are greatly heighted risk of developing different types of kidney cancer, along with additional clinical features in most cases. Identification of the associated genes and study of their function has highlighted the metabolic nature of kidney cancer and given important insights into the genetics of non-familial, sporadic kidney cancer.
3: Von Hippel-Lindau (VHL): Clear Cell Kidney Cancer
Von Hippel-Lindau (VHL) is a hereditary kidney cancer syndrome in which affected individuals are at risk for the development of tumors in a number of organs, including the kidneys.(3) It represents a well studied form of inherited cancer risk syndrome, which has additionally provided invaluable insight into the study of non-familial, sporadic kidney cancer.
Clinical Presentation of VHL syndrome
Retinal angiomas
Affected individuals in VHL families are at risk for the development of bilateral, multifocal retinal angiomas. These retinal lesions are made up of very hypervascular angiomas that, while being benign, can be very destructive and can cause blindness if not diagnosed and treated early. It is recommended that patients from families affected with VHL undergo genetic testing early and have regular retinal examinations. Early intervention can often be of significant benefit in preserving visual fields. Sadly, we have managed a large number of patients who were not diagnosed and treated early in life, who lost their vision as a result of these late detected retinal angiomas.(4)
Central Nervous System (CNS) Hemangioblastomas
Patients affected with VHL are at risk for the development of cerebellar and spinal hemangioblastomas. These can be early onset and can occur throughout the spine and cerebellum. Occasionally a patient may also develop a hemangioblastoma in the frontal cortex or along the optic nerve. While these CNS hemangioblastomas are benign, they can cause significant morbidity, including paralysis. Surgical management is often recommended when patients develop symptoms or if an impending ventricular obstruction is detected.(3, 5)
Endolymphatic Sac Tumors (ELST)
Patients affected with VHL are at risk for the development of tumors in the inner ear, the endolymphatic sac. These tumors are low grade papillary tumors which rarely metastasize. Endolymphatic sac tumors, which occur in approximately twelve percent of VHL patients, can be associated with disequilibrium and hearing loss and are treated by surgical resection.(6)
Epididymal Cystadenomas
Affected male VHL patients are at risk for the development of bilateral, benign cystic adenomas of the epididymis. These lesions are found by physical examination and/or ultrasound in fifty five percent of affected male patients. The benign course of these lesions favors conservative management.(7)
Pancreatic Neuroendocrine Tumors (PNET)
Patients affected with VHL are at risk for the development of pancreatic neuroendocrine tumors and cysts.(8, 9) Pancreatic neuroendocrine tumors can spread; in a series of 108 VHL patients with PNETs, nine were found to have metastatic disease.(9) Tumors larger than 3 cm are more likely to metastasize, and clinical management involves observation until the tumors reach a certain size, at which time surgical intervention is recommended.
Kidney Cancer: Clear Cell Type
Patients affected with VHL are at risk for the development of renal cysts and bilateral, multifocal kidney cancer. These tumors are always clear cell histologic type.(10) It is estimated that patients with VHL are at risk for the development of up to 600 independent clear cell kidney tumors and 1300 cysts per kidney during their lifetime.(11) Extensive study of VHL-associated kidney cancers growth and metastasis rates has refined the clinical management to active surveillance until the largest kidney tumor reaches three centimeters in size, at which time surgical intervention is recommended.(12-14)
Genetic Analysis of VHL syndrome and the VHL Gene
Genetic linkage analysis was performed on numerous VHL families that localized and subsequently identify the gene for von Hippel-Lindau syndrome (VHL) on the short arm of chromosome 3 at 3p21.4.(15, 16) Germline mutations in the VHL gene have been found in nearly 100% of VHL families.(17) In order to determine if mutation of the VHL gene was also associated with non-inherited forms of kidney cancer, VHL gene mutation analysis was performed in tumors from patients with sporadic, non-hereditary kidney cancer. This resulted in the fundamentally important discovery that VHL gene mutation or methylation was found in nearly 90% of tumors from patients with sporadic clear cell kidney cancer.(18, 19)
Function Analysis of the VHL Gene Protein Product: Oxygen and Iron sensing
The product of the VHL gene, the VHL protein, makes a complex with elongin C, elongin B, CUL2 and Rbx1 (20-23) to target the Hypoxia Inducible Factor 1 alpha (HIF1α) and HIF2α for ubiquitin mediated degradation.(24-26) When there are normal amounts oxygen and iron available to the cell, the prolyl hydroxylase enzymes (PHDs) can actively transfer hydroxyl groups onto two specific proline residues in the oxygen dependent domain of the hypoxia inducible factor protein and this enables the VHL complex to bind and ubiquinate HIF for degradation in the proteosome.(24-26) If there is not enough oxygen or iron present within the cell the PHD enzymes lose their activity, consequently the HIFα subunits are not targeted for degradation and become stable and activate a response to counteract and adapt to the environment conditions. HIF1α and HIF2α are transcription factors which regulate the activity of a number of downstream genes important in dealing with low oxygen/iron levels that are also extremely important to the growth and maintenance of a cancer, such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), epidermal growth factor (EGF) and the glucose transporter, GLUT1. Activation of the HIF pathway is a natural response to low oxygen/iron that is shutdown again once normal oxygen levels are present, but when the VHL gene is mutated the VHL complex cannot bind and degrade HIF, even in normoxia. Therefore the HIFα subunits are continuously stable, they accumulate and transcription of downstream genes such as VEGF, PDGF, EGF and GLUT1 is greatly and stably upregulated. This occurs in the VHL kidney tumors due to loss of the one remaining wild-type copy of VHL resulting in the production of inactive or no VHL protein and in sporadic kidney tumors by the loss of activity from both wild-type genes, acting as a classical tumor suppressor gene. This also provides a factor for the earlier onset of kidney tumors in VHL patients compared to sporadic cases as a single gene alteration is required opposed to a double hit.
VHL Gene Pathway Targeted Therapy in Clear Cell Kidney Cancer
The indepth knowledge of the actions of the VHL pathway and the results of its deregulation have provided the foundation for the development of a number of targeted therapeutic agents for patients with advanced clear cell kidney cancer. The first trial to target the VHL pathway in clear cell kidney cancer involved the use of bevacizumab, a neutralizing antibody against VEGF, in patients with metastatic clear cell kidney cancer. In this randomized, double blind phase 2 trial, bevacizumab was shown to significantly prolong the time to progression of disease in patients with advanced clear cell kidney cancer.(27) Sunitinib, an agent which targets the VEGF and PDGF receptors has been shown to have a thirty one percent partial response rate in patients with advanced kidney cancer and an increased survival time over treatment with interferon α.(28) Sorafenib, which also targets the VEGF and PDGF receptors, has been shown to have a 10% partial response rate and an increase of progression free survival to 5.5 months versus 2.8 months for placebo.(29) Temsirolimus, which targets the amounts HIFα via down regulation of its translation through down regulating the mTOR pathway, has been shown to have an 8.6% response rate and an increase in progession free survival (3.8 months versus 1.9 months) and an increase in overall survival (10.9 months versus 7.3 months) versus interferon α.(30) Everolimus, which also targets HIF translation through the mTOR pathway, has been shown to increase progression free survival to 4 months (everolimus) versus 1.9 months (placebo).(31) Pazopanib, an agent which targets the VEGF receptors (VEGFR1, VEGFR2 and VEGFR3) has been shown to have a 30% response rate (versus 3% placebo) in patients with advanced kidney cancer with a median duration of response over 1 year.(32)
4: Hereditary Papillary Renal Carcinoma (HPRC): Type 1 Papillary Kidney Cancer
Hereditary papillary renal carcinoma (HPRC) is an autosomal dominant hereditary cancer syndrome in which affected individuals are at risk for the development of bilateral, multifocal type 1 papillary kidney cancer.(33, 34) It is estimated that patients affected with HPRC are at risk for the development of up to 1,100 tumors per kidney throughout their lifetime.(35) Clinical management, like in patients with VHL-associated kidney tumors, involves active surveillance until the largest tumor reaches the 3 cm threshold. At this time, surgical intervention is recommended.(13)
Genetic Analysis of HPRC and Therapeutic Targeting of the MET Gene: Nutrient sensing
Genetic linkage analysis was performed in HPRC families and the proto-oncogene, MET, was identified as the HPRC gene.(36) Activating mutations of the MET gene are found in the germline of patients affected with HPRC.(36) Within most assessed tumors, there is amplification of the mutated copy of the MET gene, presumably to produce a threshold level of over-activated protein product of the MET gene within the kidney cell. An early onset form of HPRC was observed in two apparently unrelated families carrying a specific MET V1101I germline mutation.(37)
MET encodes the cell surface receptor for the growth factor, hepatocyte growth factor (HGF). Growth factor-dependent activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway increases cell surface expression of nutrient transporters, resulting in increased uptake of glucose, amino acids and other nutrients, and increased growth and proliferation through the up regulation of the PI3K-AKT and PI3K-RAS-Erk pathways. Additionally, nutrient-stimulated HGF/MET signaling induces phosphorylation of LKB1 on Ser428 through the RAS-Erk1/2-p90RSK pathway in a manner that results in uncoupling it with its low energy sensing partner, 5'AMP –activated protein kinase (AMPK).(38) If this occurred in an uncontrolled manner it would inhibit AMPK activation in the presence of low energy levels and implicates HGF/MET activation in deregulation of the LKB1-AMPK-mTOR nutrient and energy sensing pathway. (2, 39) Therefore normal growth patterns can be controlled by the growth factor receptor activity levels and the surrounding nutrient levels, whereas the mutated constitutively active HGF/MET can drive uncontrolled growth irrelevant of the surrounding environmental conditions and overcome the negative regulation of AMPK.
A clinical trial has been conducted evaluating the role of a small molecule inhibitor of both HGF/MET and VEGFR2/KDR in patients with papillary kidney cancer either with or without germline MET mutations. A partial response to therapy has been seen in patients with papillary kidney cancer, and results in patients with or without germline MET mutation is currently being analyzed.(40)
5: Birt-Hogg-Dubé (BHD): Chromophobe Kidney Cancer
Birt-Hogg-Dubé is an autosomal dominant, hereditary cancer syndrome in which affected individuals are at risk for the development of cutaneous fibrofolliculomas (41), pulmonary cysts (42) and kidney cancer.(43) BHD-associated kidney cancers can be multifocal, bilateral and they can metastasize. Patients affected with BHD are at risk for the development of chromophobe, hybrid oncocytic, and clear cell kidney cancer and oncocytoma.(44) Like VHL and HPRC, BHD-associated kidney cancers are managed with active surveillance; when the largest tumor reaches the three centimeter threshold, surgical intervention is recommended.
Genetic Analysis of BHD and Functional Analysis of the FLCN gene: Nutrient and Energy Sensing Pathway
Genetic linkage analysis in families identified FLCN as the BHD syndrome associated gene.(45) Germline mutations of FLCN have been detected in a high percentage of BHD families and FLCN functions as a tumor suppressor gene.(42, 46) When tumors from BHD patients were analyzed for alteration of the second allele of FLCN, sequence alterations or LOH of the somatic copy of FLCN were detected in seventy percent.(47)
When FLCN was initially identified, it was a novel gene of unknown function. Subsequently, it has been shown that the product of the FLCN gene, Folliculin or FLCN, forms a complex with protein products of two novel genes, Folliculin Interacting Protein 1 (FNIP1) and FNIP2, which can bind the γ subunit of AMPK.(48, 49) As mentioned before, AMPK is a critical energy and nutrient sensor in the cell that responds to low ATP levels compared to AMP levels and low nutrient levels by down regulating cell growth, lipid and protein metabolism. When Folliculin is inactivated in either murine or human tumors it is observed that both aspects of the mTOR pathway, mTORC1 and mTORC2, are activated and both are normally down regulated by AMPK signaling.(48, 50) Furthermore, increased TFE3 transcriptional activity was found in Folliculin deficient kidney cancer cells and this was associated with increased TFE3 nuclear localization.(51) TFE3 is a member of the MITF/TFE transcription factor family and TFE3 gene fusions have been found in kidney tumors with Xp11.2 translocations. The importance of this will be further elucidated in the following section.
In order to evaluate the role of agents targeting the FLCN pathway, a FLCN kidney-specific knockout model was developed. These animals, which died at 30 days of age of renal failure, developed large cystic kidneys with focal areas of neoplastic growth. When the FLCN-knockout animals were treated with rapamycin, which specifically targets mTORC1, there was a significant reduction in kidney size and the animal's survival time was doubled.(52)
6: MiT Transcription Factor Associated Kidney Cancer
The MiT family of transcription factors includes TFE3, TFEB, MITF, and TFEC, a family of transcription factors that share a highly homologous basic-helix-loop-helix-leucine zipper DNA binding and dimerization domain. These proteins can produce both hetero- and homodimers and bind identical DNA response elements, suggesting that they may have common downstream targets and a degree of functional redundancy. In tumors, these genes are mainly over expressed due to somatic translocations that create active fusion proteins with MiT transcription factor activity, but not their normal regulation. Importantly, translocation associated kidney cancer represents approximately 15% of the kidney cancers in patients under 45 years of age.(53)
TFE3 Associated Kidney Cancer
Studies of kidney cancers characterized by (X;1)(p11.2,q21.2) translocations (54, 55) led to the identification of a fusion protein involving the X chromosome gene, TFE3.(55-57) Genes shown to be fused to TFE3 in TFE3 associated kidney cancer include CLTC in t(X;17)(p11;q23) NONO in inv(X)(p11.2;q12), ASPSCR1 in t(X;17)(p11.2;q25), SFPQ in t(X;1)(p11.2;p23) and PRCC in t(X;1)(p11.2;q21).(53, 58) The diagnosis is made by TFE3 staining or by a dual color FISH assay.(59)
TFE3 associated kidney cancer is now recognized as a distinct and often very aggressive type of alveolar/papillary kidney cancer that tends to occur in younger individuals (median age 24 years).(60, 61) The disease is seen more often in females than in males and has a worse prognosis in older males. Xp11 translocation kidney cancers have been shown to have increased expression of phosphorylated S6, a marker of elevated mTOR pathway activation, and increased levels of stable HIF1α.(58)
TFEB Associated Kidney Cancer
TFEB associated kidney cancer is a novel, recently described form of pediatric kidney cancer with a distinctive alveolar morphological pattern characterized by a t(6:11)(p21.1;q12) translocation that stained positively for the HMB45 melanocyte marker.(62, 63) In 2003 Davis, et al. cloned an Alpha-TFEB fusion in renal tumors harboring the t(6:11)(p21:q13) chromosome translocation. Histologic similarity between TFE3 and TFEB associated kidney cancer suggests that common MiT family targets may be associated with tumorigenesis, in accordance with theory that similar genes would be affected by both transcription factors.(63)
MITF Associated Kidney Cancer
Since we reported that the t(X;1) (p11.2;q21.2) translocation in alveolar/papillary kidney cancer fuses PRCC to the TFE3 transcription factor in 1996 (55), we have searched in vain for a hereditary variant of a MiT associated kidney cancer involving either TFE3 or TFEB. However, a 2010 study by Maubec, et al. investigating the co-existence kidney cancer and melanoma and the familial clustering of melanoma supported the finding that there is a genetic predisposition underlying the association between kidney cancer and melanoma.(63, 64) These and other findings, including the finding that both melanoma and TFEB kidney cancer express HMB45, led Bertolotto et al., to perform a very elegant study which they identified a specific MITF germline mutation/variant (pE318K) that was significantly enriched for in patients with either melanoma or kidney cancer or a combination of both.(63, 65) The MITF transcription factor variant was found to upregulate a number of genes important in kidney cancer tumorigenesis, including MET and HIF1α.(65, 66)
Therapy of MiF Transcription Factor Family Associated Kidney Cancers
The most successful strategies to date for targeting the MiF transcription factor associated kidney cancers (TFE3 and TFEB associated kidney cancer) have involved approaches targeting the known up regulation of the HIF1α or mTOR pathways. In the largest therapeutic series to date, Malouf, et al. reported three of eleven patients treated with sunitinib had a partial response to therapy, seven of eight patients treated with sorafenib had stable disease and one patient receiving mTOR inhibitors had a partial response while six had stable disease.(67)
7: Tuberous Sclerosis Complex: Regulation of the mTOR Pathway
The tuberous sclerosis complex (TSC) is an autosomal dominant disorder in which affected individuals are at risk for the development of manifestations involving multiple organs, including cutaneous angiofribroma, pulmonary lymphangiomyomatosis and renal tumors.(68) Although angiomyolipoma is the most common type of renal tumor found in TSC patients, other types of tumors have been identified, including clear cell kidney cancer.(69)
TSC1/TSC2 Gene Pathway
Tuberous sclerosis complex is characterized by germline mutation of either the TSC1 or TSC2 genes, the protein products of which are essential parts of the LKB1/AMPK/TSC/mTOR nutrient and energy sensing pathway and both genes act as tumor suppressors.(70, 71) TSC2 encodes the tuberin protein and TSC1 encodes the hamartin protein, which combine to form a heterodimer that acts as a GTPase-activating protein complex on Rheb, a Ras-family GTPase that in turn inhibits mTOR activity. Thus the levels of active TSC protein complex directly controls the activity of the mTOR pathway and is itself controlled AMPK and AKT in opposing manners. TSC2 loss has been shown to result in accumulation of HIF1α and increased expression of HIF-responsive genes, including vascular endothelial growth factor (VEGF) and treatment of TSC2-deficient cells with rapamycin normalizes HIF levels, indicating that TSC2 regulates HIF translation by inhibiting mTOR activity.(70)
Targeting the TSC Pathway
In an effort to target the TSC pathway, patients with angiomyolipoma who were affected with tuberous sclerosis complex were treated with sirolimus, a rapamycin analogue which targets mTORC1. Although sirolimus was found to induce regression of the renal angiomyolipomas, most of the kidney tumors that regressed on therapy tended to increase in size after therapy was stopped.(72) However, this study demonstrated that targeting the LKB1/AMPK/TSC2/mTOR pathway can have a notable effect on TSC-deficient renal tumors.
8: Cowden Syndrome: PTEN and the Regulation of the mTOR Pathway
Cowden syndrome is an autosomal dominant disorder that results from germline mutation of the PTEN gene, in which affected individuals are at risk for manifestations in a number of organs, including tumors of the breast, thyroid, endometrium and kidney.(73-75) The protein product of the PTEN gene, PTEN, is a phosphatase that catalyzes the conversion of PIP3 (phosphatidylinositol 3,4,5 triphosphate) to PIP2 (phosphatidylinositol 4,5 biphosphate). In response to growth factor receptor stimulation, intracellular PIP3 rises, leading to activation of a number of effectors, including AKT/mTOR pathway, then PTEN lowers the levels of PIP3 to abrogate the response. In a PTEN deficient tumor, increased levels of PIP3 remain constant and continuous activation of AKT occurs, which phosphorylates the TSC complex leading to its decreased TSC2 and the up regulation of mTOR pathway.(71)
Targeting the PTEN Deficient AKT/mTOR Pathway
In pre-clinical PTEN-deficient models a therapeutic approach has been developed to target the mTOR pathway with rapamycin. Treatment of an epithelial-specific deletion of Pten in mice has been shown to promote the rapid regression of advanced mucocutaneous lesions. When the drug was administered before disease manifestation, rapamycin halted the development of Cowden's disease-like lesions, prolonging survival.(76) Clinical trials are currently underway evaluating the role of rapamycin in the treatment of patients with PTEN-deficient Cowden's syndrome.
9: Tricarboxylic Acid Mutation Kidney Cancers: Exemplifying the Warburg Effect
In the 1920's Otto Warburg proposed that a basic characteristic of cancer would be that it is characterized by aerobic glycolysis.(77, 78) Subsequently, this has been shown to be true to a greater and lesser extent in nearly all general cancer types, including kidney cancer. There are two types of inherited kidney cancer that are unique examples of the Warburg effect in cancer driven by specific gene mutation; fumarate hydratase-deficient kidney cancer and succinate dehydrogenase deficient kidney cancer.
Hereditary Leiomyomatosis Renal Cell Carcinoma
Hereditary Leiomyomatosis Renal Cell Carcinoma (HLRCC) is a hereditary cancer syndrome in which affected individuals are at risk for the development of cutaneous and uterine leiomyomas and kidney cancer.(79, 80) The cutaneous leiomyomas, which can be painful, are often multiple and can occur on the arms or trunk and may appear on only one side of the body. Up to 90% of affected females develop early onset uterine leiomyomas, many of whom have had hysterectomies in their twenties.(81) Patients affected with HLRCC are at risk of developing a very aggressive form of kidney cancer, where tumors can be solitary, multifocal and/or bilateral. The histologic pattern, which is characterized by orangophilic nucleoli and a unique perinucleolar halo with H&E staining, can be papillary, tubulopapillary or solid.(82) Whereas VHL, HPRC or BHD tumors rarely spread when the tumors are smaller than 3 cm, it is important to note that HLRCC kidney tumors can metastasize quickly, even when the primary tumor is still relatively small.(83) Therefore, clinical management of HLRCC-associated kidney cancer does not include active surveillance; when a kidney tumor is detected in an HLRCC patient, immediate surgical intervention is recommended.
Genetic Analysis of HLRCC and the Fumarate Hydratase Gene
Hereditary Leiomyomatosis Renal Cell Carcinoma is characterized by germline mutation of the fumarate hydratase (FH) gene.(84) In the North American HLRCC families, FH mutation was detected in nearly ninety percent.(81) In fumarate hydratase (FH) deficient cells, including FH-deficient kidney cancer cells, fumarate levels accumulate to excessive levels. These increased fumarate levels inhibit the activity of the HIF prolyl hydroxylases, resulting in accumulation of HIFα and upregulation of HIF target genes such as VEGF and GLUT1, providing a VHL-independent mechanism for dysregulation of HIFα degradation in fumarate hydratase-deficient kidney cancer.(81, 85, 86)
Fumarate Hydratase Deficiency and the AMPK Pathway
Fumarate hydratase-deficient kidney cancer is characterized by aerobic glycolysis. The FH-deficient kidney cancer cell lines do not respire normally; they take up nearly no oxygen and produce a large amount of lactate. As neither the Krebs cycle nor the electron transport chain are functioning normally, these cells are very dependent on glucose transport and glycolysis for energy production.(86, 87) Fumarate hydratase-deficient kidney cancer cells have decreased activated phospho-AMPK and decreased production of AMPKα and AMPKβ. This decreased level of AMPK results in increased fatty acid synthesis and increased activation of the mTOR pathway. Treatment of FH-deficient kidney cancer with metformin increased AMPK levels, a known function of the drug, and significantly inhibited invasion in an in-vitro model.(86) A potential approach to therapy for this most aggressive form of kidney cancer could involve targeting AMPK as well as inhibiting glucose uptake by targeting the vasculature. A clinical trial is currently underway evaluating the role of bevacizumab and erlotinib in patients with advanced HLRCC-associated kidney cancer.
Succinate Dehydrogenase Deficient Kidney Cancer (SDH-RCC)
Germline mutations of three succinate dehydrogenase genes, SDHB, SDHD and SDHC, have been found to be associated with familial paraganglioma/pheochromocytoma.(88-90) Patients affected with these hereditary syndromes are at risk for the development of bilateral, multifocal pheochromocytomas and paragangliomas. Patients with familial renal carcinoma with or without pheochromocytoma/paraganglioma have been characterized with germline mutation of SDHB and SDHD genes.(91-94) SDH-RCC can be a very aggressive form of kidney cancer and is another example of the Warburg effect in cancer. Thus, the current suggested clinical management for SDH-RCC is the same as for HLRCC, where immediate surgical intervention is recommended.
SDH-deficient cells have been shown to have increased levels of succinate and these increased succinate, similar to the increased levels of fumarate, can inhibit the prolyl hydroxylases and lead to increased levels of HIF.(85) Targeting the metabolic basis of SDH-deficient kidney cancer could lead to approaches that would target the vasculature to decrease glucose uptake.
10: Conclusion - Kidney Cancer is a Metabolic Disease
Kidney cancer is fundamentally a metabolic disease. The known genes for kidney cancer, VHL, MET, FLCN, MITF, TFE3, TFEB, TSC1, TSC2, PTEN, FH, SDHB and SDHD are involved in the cell's ability to sense oxygen, iron, nutrients, and, particularly in the TCA cycle enzymes, energy. Although significant progress has been made targeting, we still have a long way to go. Most patients treated with the approved drugs targeting the VHL gene pathway, such as sunitinib, sorafenib, bevacuzimab, temsirolimus and everolimus, eventually fail therapy and their disease progresses. Hopefully targeting the metabolic basis of kidney cancer will provides the opportunity to develop effective forms of therapy for all patients with this disease.
Figure 2.

Patients affected with von Hippel-Lindau are at risk for the development of bilateral, multifocal (A,B) clear cell kidney cancer (C) and have germline mutation of the VHL gene (D). From Linehan, et al.(1)
Figure 3.

Patients affected with Hereditary Papillary Renal Carcinoma (HPRC) at risk for the development of bilateral, multifocal (A,B) type 1 papillary kidney cancer. HPRC is a highly penetrant autosomal dominant hereditary cancer syndrome (D). From Linehan, et al.(1)
Figure 4.

Patients affected with Birt-Hogg-Dubé (BHD) are at risk for the development if bilateral, multifocal kidney cancer (left upper and left lower panels) with chromophobe (upper right panel), hybrid oncocytic (right middle panel) and oncocytoma (right lower panel). From Linehan, et al.(1)
Figure 5.
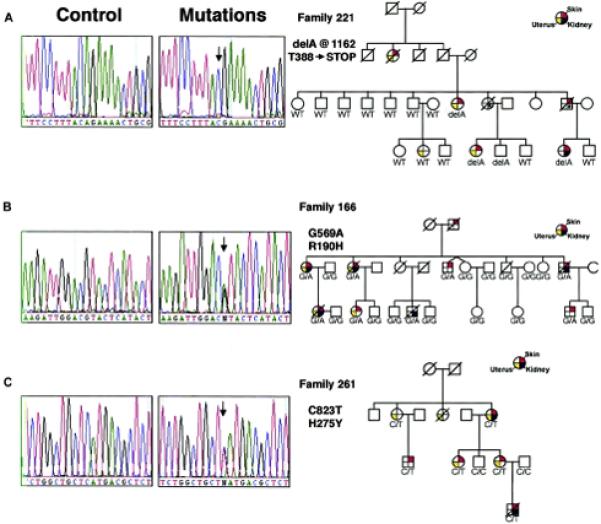
BHD is a highly penetrant autosomal dominant hereditary cancer syndrome characterized by germline mutation of the FLCN gene. From Toro, et al. (81).
Figure 6.
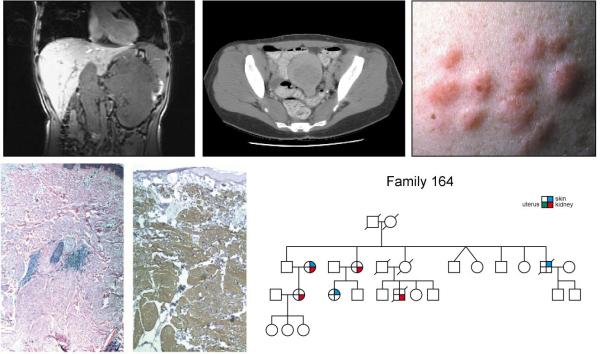
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC) is an autosomal dominant hereditary cancer syndrome (right lower panel) in which affected individuals are at risk for the development cutaneous and uterine leiomyomas (upper middle and upper right panels) and an aggressive form of kidney cancer (upper left panel). HLRCC is characterized by germline mutation of the Krebs cycle enzyme gene, fumarate hydratase. From Linehan, et al.(1)
Figure 7.

Kidney cancer is essentially a metabolic disease. Each of the genes known to be associated with the development of kidney cancer, VHL, MET, FLCN, FH, SDHB, SDHD, TSC2, TSC1, TFE3, TFEB, MITF and PTEN, is involved in the cell's ability to sense oxygen, iron, nutrients or energy. Adapted from Linehan, et al. (2)
Acknowledgement
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The authors thanks Georgia Shaw for the outstanding editorial and graphics support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003;170:2163–72. doi: 10.1097/01.ju.0000096060.92397.ed. [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature Reviews Urology. 2010;7:277–85. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lonser RR, Glenn GM, Walther MM, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 4.Wong WT, Agron E, Coleman HR, Reed GF, Csaky K, Peterson J, et al. Genotype-phenotype correlation in von Hippel-Lindau disease with retinal angiomatosis. Arch Ophthalmol. 2007;125:239–45. doi: 10.1001/archopht.125.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filling-Katz MR, Choyke PL, Oldfield E, Charnas L, Patronas NJ, Glenn GM, et al. Central nervous system involvement in von Hippel Lindau disease. Neurology. 1991;41:41–6. doi: 10.1212/wnl.41.1.41. [DOI] [PubMed] [Google Scholar]
- 6.Choo D, Shotland L, Mastroianni M, Glenn G, van WC, Linehan WM, et al. Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg. 2004;100:480–7. doi: 10.3171/jns.2004.100.3.0480. [DOI] [PubMed] [Google Scholar]
- 7.Choyke PL, Glenn GM, Wagner JP, Lubensky IA, Thakore K, Zbar B, et al. Epididymal cystadenomas in von Hippel-Lindau disease. Urol. 1997;49:926–31. doi: 10.1016/s0090-4295(97)00074-5. [DOI] [PubMed] [Google Scholar]
- 8.Libutti SK, Choyke PL, Bartlett DL, Vargas H, Walther M, Lubensky I, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery. 1998;124:1153–9. doi: 10.1067/msy.1998.91823. [DOI] [PubMed] [Google Scholar]
- 9.Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine tumors (PNETs). Surgery. 2007;142:814–8. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poston CD, Jaffe GS, Lubensky IA, Solomon D, Zbar B, Linehan WM, et al. Characterization of the renal pathology of a familial form of renal cell carcinoma associated with von Hippel-Lindau disease: clinical and molecular genetic implications. J Urol. 1995;153:22–6. doi: 10.1097/00005392-199501000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol. 1995;154:2010–4. [PubMed] [Google Scholar]
- 12.Walther MM, Choyke PL, Glenn G, Lyne JC, Rayford W, Venzon D, et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161:1475–9. doi: 10.1016/s0022-5347(05)68930-6. [DOI] [PubMed] [Google Scholar]
- 13.Herring JC, Enquist EG, Chernoff A, Linehan WM, Choyke PL, Walther MM. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: 10-year experience. J Urol. 2001;165:777–81. [PubMed] [Google Scholar]
- 14.Duffey BG, Choyke PL, Glenn GM, Grubb RL, Venzon D, Linehan WM, et al. The Relationship Between Renal Tumor Size and Metastases in Patients with von Hippel-Lindau Disease. J Urol. 2004;172:63–5. doi: 10.1097/01.ju.0000132127.79974.3f. [DOI] [PubMed] [Google Scholar]
- 15.Hosoe S, Brauch H, Latif F, Glenn GM, Daniel L, Bale S, et al. Localization of the von Hippel-Lindau disease gene to a small region of chromosome 3. Genom. 1990;8:634–40. doi: 10.1016/0888-7543(90)90249-t. [DOI] [PubMed] [Google Scholar]
- 16.Latif F, Tory K, Gnarra JR, Yao M, Duh F-M, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 17.Stolle C, Glenn GM, Zbar B, Humphrey JS, Choyke P, Walther MM, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 18.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore LE, Nickerson ML, Brennan P, Toro JR, Jaeger E, Rinsky J, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: Associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet. 2011;7:1–13. doi: 10.1371/journal.pgen.1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan DR, Pause A, Burgess WH, Aso T, Chen DY, Garrett KP, et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science. 1995;269:1402–6. doi: 10.1126/science.7660122. [DOI] [PubMed] [Google Scholar]
- 21.Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94:2156–61. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science. 1995;269:1444–6. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 23.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–61. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 24.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 25.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–7. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 26.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 27.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 29.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 30.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 31.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:1432–9. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 32.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J Clin Oncol. 2010;28:1061–8. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 33.Zbar B, Tory K, Merino MJ, Schmidt LS, Glenn GM, Choyke P, et al. Hereditary papillary renal cell carcinoma. J Urol. 1994;151:561–6. doi: 10.1016/s0022-5347(17)35015-2. [DOI] [PubMed] [Google Scholar]
- 34.Lubensky IA, Schmidt LS, Zhuang Z, Weirich G, Pack S, Zambrano N, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–26. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ornstein DK, Lubensky IA, Venzon D, Zbar B, Linehan WM, Walther MM. Prevalence of microscopic tumors in normal appearing renal parenchyma of patients with hereditary papillary renal cancer. J Urol. 2000;163:431–3. [PubMed] [Google Scholar]
- 36.Schmidt LS, Duh FM, Chen F, Kishida T, Glenn GM, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genetics. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt LS, Nickerson ML, Angeloni D, Glenn GM, Walther MM, Albert PS, et al. Early onset Hereditary Papillary Renal Carcinoma: germline missense mutations in the tyrosine kinase domain of the Met proto-oncogene. J Urol. 2004;172:1256–61. doi: 10.1097/01.ju.0000139583.63354.e0. [DOI] [PubMed] [Google Scholar]
- 38.Esteve-Puig R, Canals F, Colome N, Merlino G, Recio JA. Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. PLoS ONE. 2009;4:e4771. doi: 10.1371/journal.pone.0004771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srinivasan R, Choueiri TK, Vaishampayan U, Rosenberg JE, Stein MN, Logan T, et al. A phase II study of the dual MET/VEGFR2 inhibitor XL880 in patients (pts) with papillary renal carcinoma (PRC). Journal of Clinical Oncology. 2008;(20 Suppl):5103. May. [Google Scholar]
- 41.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674–7. [PubMed] [Google Scholar]
- 42.Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, Toure O, et al. Lung Cysts, Spontaneous Pneumothrorax and Genetic Associations in 89 Families with Birt-Hogg-Dub&eacute; Syndrome. Am J Respir Crit Care Med. 2007;175:1044–53. doi: 10.1164/rccm.200610-1483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zbar B, Alvord WG, Glenn GM, Turner M, Pavlovich CP, Schmidt LS, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393–400. [PubMed] [Google Scholar]
- 44.Pavlovich CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dub&eacute; syndrome. Am J Surg Pathol. 2002;26:1542–52. doi: 10.1097/00000478-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn GM, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–64. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 46.Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke CD, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321–31. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vocke CD, Yang Y, Pavlovich CP, Schmidt LS, Nickerson ML, Torres-Cabala CA, et al. High Frequency of Somatic Frameshift BHD Gene Mutations in Birt-Hogg-Dube-Associated Renal Tumors. J Natl Cancer Inst. 2005;97:931–5. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 48.Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552–7. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415:60–7. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. 2009;106:18722–7. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong SB, Oh H, Valera VA, Baba M, Schmidt LS, Linehan WM. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS ONE. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, et al. Kidney-targeted Birt-Hogg-Dub&eacute; gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140–54. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malouf GG, Camparo P, Molinie V, Dedet G, Oudard S, Schleiermacher G, et al. Transcription factor E3 and transcription factor EB renal cell carcinomas: clinical features, biological behavior and prognostic factors. J Urol. 2011;185:24–9. doi: 10.1016/j.juro.2010.08.092. [DOI] [PubMed] [Google Scholar]
- 54.Shipley JM, Birdsall S, Clark J, Crew J, Gill S, Linehan WM, et al. Mapping the X chromosome breakpoint in two papillary renal cell carcinoma cell lines with a t(X;1)(p11.2;q21.2) and the first report of a female case. Cytogenet Cell Genet. 1995;71:280–4. doi: 10.1159/000134127. [DOI] [PubMed] [Google Scholar]
- 55.Sidhar SK, Clark J, Gill S, Hamoudi R, Crew J, Gwilliam R, et al. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses anovel gene PRCC to the TFE3 tanscription factor gene. Hum Mol Genet. 1996;5:1333–8. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 56.Weterman MA, Wilbrink M, Janssen I, Janssen HA, Van Den BE, Fisher SE, et al. Molecular cloning of the papillary renal cell carcinoma-associated translocation (X;1)(p11;q21) breakpoint. Cytogenet Cell Genet. 1996;75:2–6. doi: 10.1159/000134444. [DOI] [PubMed] [Google Scholar]
- 57.Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, Smedley D, et al. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene. 1997;15:2233–9. doi: 10.1038/sj.onc.1201394. [DOI] [PubMed] [Google Scholar]
- 58.Argani P, Hicks J, De Marzo AM, Albadine R, Illei PB, Ladanyi M, et al. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol. 2010;34:1295–303. doi: 10.1097/PAS.0b013e3181e8ce5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong M, De AP, Osborne L, Keane-Tarchichi M, Goldfischer M, Edelmann L, et al. Dual-color, break-apart FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of Xp11 translocation renal cell carcinoma and alveolar soft part sarcoma. Am J Surg Pathol. 2010;34:757–66. doi: 10.1097/PAS.0b013e3181dd577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Argani P, Antonescu CR, Couturier J, Fournet JC, Sciot R, Debiec-Rychter M, et al. PRCC-TFE3 renal carcinomas: morphologic, immunohistochemical, ultrastructural, and molecular analysis of an entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol. 2002;26:1553–66. doi: 10.1097/00000478-200212000-00003. [DOI] [PubMed] [Google Scholar]
- 61.Armah HB, Parwani AV. Xp11.2 translocation renal cell carcinoma. Arch Pathol Lab Med. 2010;134:124–9. doi: 10.5858/2008-0391-RSR.1. [DOI] [PubMed] [Google Scholar]
- 62.Argani P, Hawkins A, Griffin CA, Goldstein JD, Haas M, Beckwith JB, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol. 2001;158:2089–96. doi: 10.1016/S0002-9440(10)64680-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A. 2003;100:6051–6. doi: 10.1073/pnas.0931430100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maubec E, Chaudru V, Mohamdi H, Grange F, Patard JJ, Dalle S, et al. Characteristics of the coexistence of melanoma and renal cell carcinoma. Cancer. 2010;116:5716–24. doi: 10.1002/cncr.25562. [DOI] [PubMed] [Google Scholar]
- 65.Bertolotto C, Lesueur F, Giuliano S, Strub T, de LM, Bille K, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011 doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 66.Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- 67.Malouf GG, Camparo P, Oudard S, Schleiermacher G, Theodore C, Rustine A, et al. Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol. 2010;21:1834–8. doi: 10.1093/annonc/mdq029. [DOI] [PubMed] [Google Scholar]
- 68.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 69.Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol. 1996;149:1–8. [PMC free article] [PubMed] [Google Scholar]
- 70.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4:147–58. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 71.Brugarolas J, Kaelin WG., Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 2004;6:7–10. doi: 10.1016/j.ccr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 72.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007;34:428–34. doi: 10.1053/j.seminoncol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 74.Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16:1289–300. doi: 10.1038/ejhg.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 76.Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden's disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68:7066–72. doi: 10.1158/0008-5472.CAN-08-0922. [DOI] [PubMed] [Google Scholar]
- 77.Warburg O, Posener K, Negelein E. Metabolism of the Carcinoma Cell. Biochem Z. 1924;152:309–44. [Google Scholar]
- 78.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 79.Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98:3387–2. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zbar B, Glenn GM, Lubensky IA, Choyke P, Magnusson G, Bergerheim U, et al. Hereditary papillary renal cell carcinoma: Clinical studies in 10 families. J Urol. 1995;153:907–12. [PubMed] [Google Scholar]
- 81.Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95–106. doi: 10.1086/376435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merino MJ, Torres-Cabala C, Pinto PA, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–85. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 83.Grubb RL, III, Franks ME, Toro J, Middelton L, Choyke L, Fowler S, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177:2074–80. doi: 10.1016/j.juro.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 84.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 85.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 86.Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases metabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–27. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet. 2010;196:45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 89.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–70. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 90.Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nature Reviews Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- 91.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–51. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 92.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–9. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–2. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 94.Henderson A, Douglas F, Perros P, Morgan C, Maher ER. SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam Cancer. 2009;8:257–60. doi: 10.1007/s10689-009-9234-z. [DOI] [PubMed] [Google Scholar]