

Abstract
CD4+ Foxp3+ Tregs are an independent cell lineage and their developmental progression during thymic development depends on IL-2R signaling. However, the role of IL-2R signaling during thymic Treg development remains only partially understood. The current study assessed the contribution of IL-2 in the expansion and functional programming of developing Tregs. In the absence of IL-2Rβ signaling, predominately CD4+ CD25- Foxp3lo T cells were found and these cells exhibited somewhat lower expression of the proliferative marker Ki67. These immature Tregs, which represent products of failed development, were also found in normal mice and characterized by markedly lower expression of several Treg functional molecules. IL-2R signaling, therefore, is required for the progression, functional programming, and expansion of Tregs during thymic development. An IL-2R signaling mutant that lowers STAT5 activation readily supported Treg functional programming but Treg proliferation remained somewhat impaired. The requirement for IL-2 during thymic Treg expansion was best illustrated in mixed chimeras where the Tregs with mutant IL-2Rs were forced to compete with WT Tregs during their development. Tregs with impaired IL-2R signaling were more prevalent in the thymus than spleen in these competitive experiments. The general effectiveness of mutant IL-2Rs to support thymic Treg development is partially accounted for by a heighten capacity of thymic Tregs to respond to IL-2. Overall our data support a model where limiting IL-2R signaling is amplified by thymic Tregs to readily support their development and functional programming whereas these same conditions are not sufficient to support peripheral Treg homeostasis.
Keywords: T regulatory cells, autoimmunity, thymic development, IL-2R
Introduction
Natural CD4+ Foxp3+ regulatory T cells (Tregs) are a distinct T cell lineage that develop in the thymus and are essential to prevent self-reactivity by suppressing peripheral autoreactive T cells that escape thymic negative selection. IL-2/IL-2R signaling is essential and non-redundant for thymus Treg development and for peripheral Treg homeostasis and competitive fitness (1-6). The central role of IL-2 in Treg biology is exemplified by the uncontrolled hyper-activation of T cells and lethal autoimmunity associated with IL-2-, IL-2Rα- (CD25), and IL-2Rβ- (CD122) deficient mice that typically die by 4-12 week of age (7-10). Importantly, restoring Tregs in IL-2 or IL-2R-deficient mice is sufficient to completely prevent this autoimmunity (11), directly linking defective Tregs as the main reason for autoimmunity in the absence of IL-2R signaling. The absence of IL-2R signaling in mice results in the development of only CD4+ Foxp3lo CD25- T cells that may represent immature Tregs (12, 13). However, besides defects in Tregs, IL-2/IL-2R-deficient mice do not effectively program CD8+ T memory responses (14) . Thus, the lack of IL-2 also tempers autoreactive T cells that contribute to somewhat delayed lethal autoimmunity when compared to Foxp3-deficient mice.
IL-2 contributes to thymic Treg development through activation of STAT5 (15). Low levels of IL-2 selectively compartmentalized with the thymic microenvironment are sufficient to drive thymic Treg development (16). Indeed, weak IL-2R signaling generated by mutant IL-2Rs, where key cytoplasmic tyrosine residues of IL-2Rβ were replaced with phenylalanine, readily supports thymic Treg development (17). Thus, IL-2 promotes Treg development through a low IL-2R signaling threshold, providing a mechanism to yield Tregs even when IL-2 is limiting. Current models are consistent with a two-step developmental process where TCR signals during thymic selection process lead to induction of CD25 on developing CD4+ CD8- thymocytes and IL-2 provides an instructive signal to these precursor cells to upregulate both Foxp3 and CD25 into mature Tregs (18, 19).
Genome-wide expression analysis comparing peripheral Tregs bearing WT vs. impaired IL-2R revealed an over representation of differentially expressed genes that function for cell growth and death (5, 17), implicating IL-2 as a key regulator of Treg homeostasis. Consistent with this idea, IL-2 favors the expansion of peripheral Tregs that express low levels of the pro-apoptotic molecule Bim (20). Furthermore, the Treg lineage cells from IL-2-/- mice that do not express Bim showed an increase in peripheral CD4+ Foxp3+ T cells such that these mice now contained a normal percentage and number of peripheral CD4+ Foxp3+ T cells, further linking IL-2 in regulation of peripheral Treg numbers. However, these Bim-/- IL-2-/- mice still exhibited severe autoimmunity due to impaired Treg function (21). Overall these and other findings indicate that IL-2R signaling exerts a strong influence on peripheral Treg homeostasis and function that in part is reflected by IL-2R-dependent STAT5 regulation of Foxp3 expression (22-26).
Although the involvement of IL-2 in peripheral Tregs is relatively well studied, comparatively little is known concerning the contribution of IL-2R signaling during thymic Treg development. The above findings for peripheral Tregs raise the possibility that the reduction of Tregs associated with IL-2/IL-2R-deficiency might reflect impaired IL-2-dependent expansion and/or survival of developing thymic Tregs. However, IL-2 may more broadly instruct Tregs as they develop and may be critical to program Treg function. The current study investigated these possibilities and showed that in the absence of IL-2Rβ signaling, the major defect in thymic Tregs was related to impaired development of the Treg functional program. Furthermore, competitive in vivo experiments revealed that low IL-2R signaling is much more efficient in promoting thymic Treg development than maintaining peripheral Treg homeostasis.
Materials and Methods
Mice
C57BL/6 (B6) mice were obtained from the Jackson Laboratory. B6 Rag2-/- γc-/- mice were purchased from Taconic Farms. Congenic CD45.1, congenic Thy1.1 and TCRα-/- B6 mice, all on the C57BL/6 genetic background, were bred and maintained in the animal facility at the University of Miami. C57BL/6 IL-2Rβ-/- (2Rβ-/-), and T cell-specific transgenic B6 IL-2RβWT (Y0) and IL-2RβY341,395,498 (Y3) on the IL-2Rβ-/- genetic background were previously described (8, 17). Foxp3/RFP reporter mice were kindly provided by R. Flavell (Yale University). All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Miami.
Treg suppression of autoimmunity in IL-2Rβ-/- mice
Neonatal IL-2Rβ-/- mice (1-3 day old) received purified CD45.1-congenic CD4+ T cells (2-3 × 106) using anti-CD4 magnetic beads (Miltenyi) by i.v. injection to the superfacial vein. Such treatment results in life-long engraftment of donor Tregs and prevents autoimmunity associated with IL-2Rβ-/- mice (11). These mice are referred to as “cured” 2Rβ-/- in this report.
Antibodies and FACS
Single cell suspensions were prepared for flow cytometry as previously described (3). Typically 300,000 events were collected during analysis. Intracellular staining of Foxp3 was performed according to the manufactures’ instructions (eBioscience). Bcl2, Ki67 and CTLA4 (Biolegend) were usually stained in combination with Foxp3, using the protocol for Foxp3 intracellular staining. Surface mAbs to CD4, CD8, CD45.2, and Thy1.2 were purchased from Biolegend, to CD24 (HSA) from BD Biosciences, and to CD25, CD103, CD39, Klrg1 from eBiosciences. All results were analyzed by using a BD LSR2 and Diva software.
pSTAT5 activity
Intracellular FACS staining for pSTAT5 was performed as previously described (17). In brief, IL-2-stimulated cells were harvested and fixed with 1.5% PFA at 37°C for 10 minutes and further incubated with 0.5 ml of methanol on ice for 30 minutes. After washing twice with PBS containing 0.02% NaN3 and 0.5% BSA, cells were stained with antibodies to surface and intracellular markers for 1 hour at room temperature in the dark. To assess pSTAT5 in vitro, thymocytes were cultured with IL-2 (10 ng/ml) for 15 min prior to paraformaldehyde fixation. To assess pSTAT5 activity in vivo, cell suspensions were immediately prepared in ice cold RPMI 1640 containing 5% FCS and a 100 μl aliquot (~2% of the suspension) was transferred into a test tube and the cells were fixed by addition of 900 μl of 1.5% paraformaldehyde.
Bone morrow chimeras
Bone marrow chimeras were generated as previously described (3). Briefly, Thy1.1 B6 mice (900 rads), TCRα-/- (600 rads), and Rag2-/- γc-/- (600 rads) received a single dose of total body irradiation. 24 hours later, these mice received a mixture of the indicated T cell-depleted bone marrow (5 × 106 total cells) by i.v. injection into the tail vein. The mice were maintained with gentamycin (1mg/dl) containing drinking water.
Statistical analysis
Data were analyzed by using Prism 5.0 by either a one-way ANOVA with Tukey's multiple comparison test when more than 2 groups were compared or by an unpaired t-test when 2 groups were compared. Significant difference is designated as * P< 0.05, ** P< 0.01, *** P< 0.001, **** P< 0.0001.
Results
IL-2 influences the growth of developing Tregs
We have previously shown that 2Rβ-/- mice do not develop a normal compartment of Treg cells (11). However, polyclonal T cell activation and autoimmunity rapidly ensue in these mice, raising the possibility that Treg development may be indirectly affected by this inflammatory environment. To test this possibility, we compared mainstream T cell development by 3-8 week old untreated autoimmune 2Rβ-/- and autoimmune-free “cured” 2Rβ-/- mice. The latter mice received Tregs by the transfer of unfractionated syngeneic CD45.1-congenic CD4+ T cells shortly after birth. Past studies have established that Tregs are the key cell population that prevents this autoimmunity (11).
When these mice were compared to control WT C57BL/6 mice, the proportion and distribution of double negative (DN), double positive (DP), CD4 single positive (SP), CD8 SP thymocytes were within the normal range (Fig. 1A). However, thymic cellularity was somewhat, but equivalently, reduced in 2Rβ-/- and “cured” 2Rβ-/- mice (Fig. 1B). This effect might reflect some inefficiency related to lack of expression of IL-2Rβ on lymphoid precursor cells. As expected, Treg development was impaired with a 2-3 fold reduction in the proportion of CD4+ Foxp3+ T cells from 2Rβ-/- and “cured” 2Rβ-/- mice (Fig. 1C, 1D). However, a substantial fraction of the CD4+ Foxp3+ cells in the thymus of the “cured” IL-2Rβ-/- mice were CD45.1+ donor cells. When considering only the recipient derived IL-2Rβ-/- thymocytes, “cured” 2Rβ-/- mice contained a significantly (p<0.0001) lower fraction (Fig. 1D) of CD4+ Foxp3+ T cells than found in untreated 2Rβ-/- mice. As 2Rβ-/- and “cured” 2Rβ-/- mice contain similar numbers of CD4 SP (Fig. 1A, B), this difference also represents a proportional decrease in CD4+ Foxp3+ T cell numbers. This finding suggests that factors associated with the accompanying autoimmunity in untreated 2Rβ-/- mice may increase the number of developing CD4+ Foxp3+ cells.
FIGURE 1. Thymic Treg compartment in the absence of IL-2Rβ signaling.

(A) Distribution of DN, DP, SP CD4, SP CD8 thymocytes (% positive cells are shown in each quadrant) and (B) total thymic cellularity for the indicated mice. (C) Gating strategy and (D) the frequency of Foxp3+ cells in SP CD4 T cells in the thymus of the indicated mice. Data (B, D) are means ± SD from 5-8 mice/group.
IL-2-induced signaling promotes T cell proliferation (27) and survival (28). Thus, the lower numbers of CD4+ Foxp3+ thymocytes in 2Rβ-/- and “cured” 2Rβ-/- mice, might reflect impaired proliferation or survival. By evaluating the proliferation marker Ki67 and anti-apoptotic marker Bcl-2, we found that the expression of Bcl-2 by 2Rβ-/- and host-derived “cured” 2Rβ-/- CD4+ Foxp3+ cells was high and comparable to WT Tregs (Fig. 2A Top, 2B Left). In contrast, compared to Foxp3+ thymocytes from WT B6 mice, only host-derived IL-2Rβ-/- CD4+ Foxp3+ cells from “cured” 2Rβ-/- mice exhibited a lower fraction of Ki67+ cells (Fig. 2A Bottom, 2B Right). This finding implicates IL-2 for expansion of developing Tregs in the thymus. The similar percentage of Ki67+ CD4+ Foxp3+ cells from WT and untreated 2Rβ-/- mice is consistent with the notion that the inflammatory environment associated with autoimmunity in IL-2Rβ-/- mice somewhat increased the overall numbers of CD4+ Foxp3+ thymocytes in an IL-2-independent manner. The donor-derived WT Tregs in the thymus of “cured” IL-2Rβ-/- mice showed a slightly different pattern, primarily in detection of a readily measured fraction of Bcl2lo cells (Fig. 2A) and the MFI of Bcl-2 on average was lower, but this difference was not statistically significant (Fig. 2B).
FIGURE 2. IL-2 in the regulation of Treg proliferation and Bcl-2 expression.

(A) Representative histograms and (B) expression of Bcl-2 and Ki67 in thymic Tregs from the WT, 2Rβ-/- mice and the host and donor compartment of “cured” 2Rβ-/- mice. The MFI for Bcl-2 is based on the total Tregs in the respective histogram. Data are means ± SD from 5-8 mice/group.
Phenotypic distinct CD4+ Foxp3+ thymocytes in mice lacking IL-2Rβ signaling
FACS analysis of CD4+ Foxp3+ thymocytes indicates that the level of Foxp3 and CD25 was lower in the absence of IL-2Rβ-dependent signaling when compared to WT Tregs (Fig. 3A). Quantitative analysis showed a 2-fold reduction of Foxp3 expression by CD4+ Foxp3+ cells from 2Rβ-/- and host-derived “cured” 2Rβ-/-mice (Fig. 3A). In addition, very few CD4+ Foxp3lo thymocytes expressed CD25 in the absence of IL-2Rβ signaling (Fig. 3A). This finding is consistent with the role of IL-2 to up-regulate expression of CD25 (29). More recently, genome wide expression profiling showed that CD103, which marks activated Tregs, depends on IL-2R signaling (17). Expression of CD103 by CD4+ Foxp3+ thymocytes was also highly reduced in the absence of IL-2R signaling (Fig. 3B). In each of these cases mentioned above, an identical requirement for IL-2Rβ signaling was noted regardless of whether the Foxp3+ cells were obtained from untreated autoimmune or “cured” 2Rβ-/- mice, suggesting that these activities are non-redundantly dependent upon IL-2. Since high Foxp3 expression is a determinant check-point for Treg lineage commitment (22, 24, 25), these findings also suggest that the developmental progression of Treg cells is impaired in the absence of IL-2Rβ signaling. It is also noteworthy that the donor-derived WT Tregs in the thymus of IL-2Rβ-/- mice expressed a phenotype that is markedly distinct from the host-derived IL-2Rβ-/- Foxp3+ cells (Fig. 3A, B), as these donor cells expressed the highest levels of Foxp3, CD25, and CD103, consistent with their peripheral origin and responsiveness to IL-2, which readily occurs in IL-2Rβ-deficient mice (30).
FIGURE 3. IL-2 regulates expression of Foxp3, CD25 and CD103 in thymic Tregs.

(A) Representative dot plots and MFI of Foxp3 and percent of CD25 expression by the indicated Treg populations. (B) Representative histograms and percent of CD103 expression from the indicated thymic Tregs. Data are means ± SD from 3-8 mice/group.
Functional programming of developing Tregs depends on IL-2Rβ signaling
A numbers of molecules have been associated with mediating Treg suppressive function (31). Three of these, CTLA4, CD39, and CD73, are readily measured by FACS analysis. In comparison to the WT Tregs, 2Rβ-/- and “cured” 2Rβ-/- CD4+ Foxp3lo T cells showed a marked and an equivalent reduction in expression of each of these suppressive mediators (Fig. 4). Thus, IL-2/IL-2R signaling is essential for the functional programming of thymic Treg cells. This result also indicates that the uncontrolled autoimmunity associated with untreated 2Rβ-/- mice is likely due to limited production of thymic CD4+ Foxp3lo T cells that do not express normal levels of suppressive molecules.
FIGURE 4. IL-2 regulates the thymic Treg functional program.
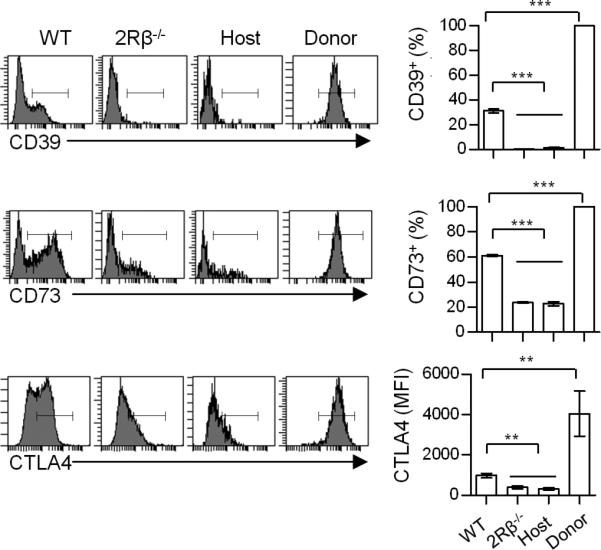
Representative histograms and MFI or percentages of CD39, CD73 and CTLA4 in thymic Tregs from WT, 2Rβ-/-, and the host and donor compartment of “cured” 2Rβ-/- mice. Data are means ± SD from 3-8 mice/group.
WT thymic Foxp3lo CD25- Tregs express properties similar to CD4+ Foxp3+ IL-2Rβ-/- thymocytes
The preceding experiments indicate that the thymus from 2Rβ-/- and “cured” 2Rβ-/- mice harbor mostly CD4+ Foxp3lo CD25- T cells with normal expression of Bcl-2, but impaired expression of CD103, CTLA4, CD39, and CD73. When one examines Tregs from the thymus of WT mice, a substantial fraction (20-30%) is also Foxp3lo and CD25neg/lo. Therefore, we tested whether WT Foxp3lo and CD25neg/lo Tregs exhibited properties related to CD4+ Foxp3lo IL-2Rβ-/- thymocytes. When WT Tregs were gated on CD25- Foxp3+ cells (Fig. 5A, left), most were Foxp3lo (Fig. 5A, right). Reciprocally, when WT Tregs were gated on Foxp3lo cells (Fig. 5B, left), the large majority were CD25- (Fig. 5B, right). Therefore, there is a readily measurable fraction of WT CD4+ Foxp3lo CD25- thymocytes. WT CD25- (Fig. 5A) or WT Foxp3lo (Fig. 5B) Tregs each showed reduced expression of CTLA4, CD103, CD39, and CD73, but similar expression of Bcl-2 and Ki67, when compared to CD25+ or Foxp3hi Tregs. This pattern of expression is highly analogous to that of CD4+ Foxp3lo IL-2Rβ-/- thymocytes and indicates these CD25- or Foxp3lo Tregs are not an aberrant cell population only associated with 2Rβ-/- mice. Collectively, these data demonstrate that CD4+ Foxp3lo CD25- thymocytes represent a normal thymocyte population that arises as a consequence of Treg development and suggest that these Tregs in WT mice arise because they have not received a productive IL-2 signal.
FIGURE 5. Properties of CD25- vs. CD25+ and Foxp3lo vs. Foxp3hi thymic Tregs in normal mice.

Representative dots plots (left) for CD25- vs. CD25+ (A) or Foxp3lo vs. Foxp3hi (B) by thymic Tregs. Expression (right) of the indicated markers for CD25- and CD25+ (A) or Foxp3lo and Foxp3hi (B) Tregs. Data are means ± SD from 5-6 mice/group.
The relationship between Foxp3lo CD25- and Foxp3hi CD25hi thymocytes
CD4+ CD25+ Foxp3- thymocytes contain precursors to Tregs (18). This population was readily found in the thymus of IL-2Rβ-/- mice (Fig. 6, P1 gate). As thymocytes mature, their expression of CD24 (HSA) is down-regulated. Correspondingly, CD4+ CD25+ Foxp3- cells from WT and IL-2Rβ-/- mice expressed predominately high levels of CD24. The CD4+ CD25+ Foxp3hi cells that dominate the P2 gate were mostly CD24- (Fig. 6), consistent with more mature T cells, although more cells with an immature CD24 phenotype were seen in the thymus of IL-2Rβ-/- mice. The CD4+ Foxp3lo CD25- cells in the P3 gate from WT and IL-2Rβ-/- mice identically down-regulated CD24, which indicates that most cells have undergone thymic maturation processes. However, optimal expression of CTLA4 was found only for CD4+ CD25+ Foxp3+ cells from WT mice. Based on CD24 expression, CD4+ Foxp3+ CD25- vs. CD25+ thymocytes have undergone a similar level of maturation, but based on CTLA4 expression only the latter developed into functionally mature Tregs.
FIGURE 6. Thymic maturation of Treg subpopulations in the presence and absence of IL-2R signaling.
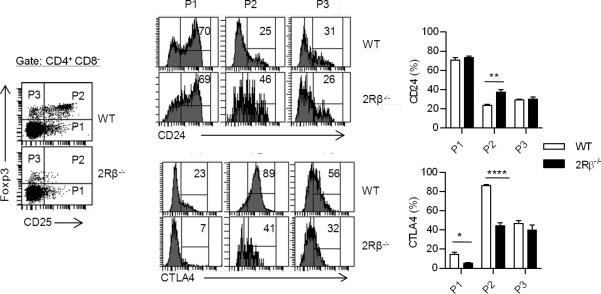
Representative dot plots (left) for the indicated thymic populations from WT and 2Rβ-/- mice and histograms (middle) for CD24 and CTLA4 expression by the cells within the indicated gated regions of the dot plots. The percent of cells within the P1-3 gates for CD24 and CTLA4 are shown to the right. Data are means ± SD from 4-5 mice/group.
The efficient down-regulation of CD24 by CD4+ CD25- Foxp3+ cells in the P3 gate raises the possibility that these cells are not a developmental precursor to fully mature CD4+ CD25hi Foxp3+ CTLA4hi cells. To address this issue, the appearance of these CD25- vs. CD25+ Foxp3+ cells was examined early during thymic development in WT mice (Fig. 7). Past studies using mice that expressed a Foxp3/GFP reporter showed that Tregs first appear in the thymus on day 2 after birth (32). We confirmed this result using WT mice expressing a Foxp3/RFP reporter (Fig. 7A). The RFP reporter is expressed through an IRES knocked into the 3’ untranslated region of Foxp3 (33), avoiding potential complications associated with aberrant activity of the Foxp3/GFP fusion molecule that has recently been shown to alter Foxp3 function (34, 35). Importantly, when Foxp3+ Treg cells first appear, most cells were CD25hi (Fig. 7B). In 2-3 day old neonatal mice, we noticed that the MFI for RFP staining was approximately 25% lower than found in adult mice (not shown). This lower level is likely related to the RFP reporter because expression of Foxp3 protein was at equivalent levels for Tregs from 3 and ≥30 day old mice (not shown). Moreover, the distribution of CD25hi Tregs was identical for 3 and >30 day old mice (Fig. 7C). Collectively, these data indicate that CD25hi Tregs dominant the thymic Treg pool as they initially develop and suggest that CD25- Treg cells are not a mandatory precursor cell to the CD25hi population. If this latter possibility occurred, CD4+ CD25- Foxp3+ cells are expected to first dominant the neonatal pool of thymic Treg cells. In addition, the detection of a normal fraction of CD4+ CD25- Foxp3+ T cells early in ontogeny indicates that these cells are thymic-derived and not the result of T cells that have circulated from the periphery into the thymus.
FIGURE 7. Treg developmental progression and maturation in the neonatal thymus of WT mice.

In each panel, thymocytes were always first gated on CD4+ CD8- thymocytes. The number (A) and expression of CD25 (B) by Foxp3+ cells from Foxp3/RFP reporter mice of the indicated age. Data are means ± SD from 4-6 mice/group, except day 1 (n=2). (C) Comparison of CD25 expression by Foxp3+ T cells from mice of the indicated age after intracellular staining for Foxp3 protein. (D) Representation of the CD4+ CD25- Foxp3- population from Foxp3/RFP reporter mice of the indicated age. (E-F) Expression of CD39 and CD103 by Foxp3+ cells from Foxp3/RFP reporter mice of the indicated age. Data (C-F) are means ± SD from 3-6 mice/group.
We also made several other observations in this ontogeny analysis of Treg development. First, the CD4+ CD25+ Foxp3- Treg precursor pool comprises a substantially greater relative proportion of CD4 SP thymocytes (Fig.7D) in 3 day old neonatal mice when compared to ≥30 day old mice. This result may reflect the need for rapid production of Tregs in the neonatal period to catch up with the more rapid development of conventional T cells. Second, expression of Treg functional molecules, as represented by CD39, is delayed relative to the detection of Foxp3+ T cells (Fig. 7E), indicating that functional maturation of Tregs takes more time than induction of Foxp3 expression. Lastly, the proportion of CD103+ Tregs was markedly diminished in the neonatal thymus (Fig. 7F). This finding suggests that these cells take more time to develop and may represent a unique thymic-derived Treg subpopulation. However, we cannot rule out that these cells may also represent peripheral Tregs that have migrated back into the thymus, particular when considering that most donor-derived WT Tregs found in the thymus in “cured” IL-2Rβ-/- were CD103+ (Fig. 3B).
The effect of sub-optimal IL-2Rβ signaling on Treg development
We recently developed transgenic mice (Y3) on the IL-2Rβ-/- background whose T cells express Y→F mutations in 3 tyrosine residues (Y341, Y395, Y498) of IL-2Rβ that are required for normal recruitment of the adaptor Shc and STAT5 as a consequence of the IL-2/IL-2R interaction. Even though this mutation leads to weak transient IL-2-induced pSTAT5, Y3 mice have no outward problems in Treg thymic development or peripheral homeostasis as reflected in Treg number, frequency or Foxp3 levels (17). To further define the efficiency of this weak IL-2R signaling during thymic Treg development, CD4+ Foxp3hi vs. Foxp3lo thymocytes from WT, Y3, and 2Rβ-/- mice (Fig. 8A) were examined for expression of several molecules dependent on IL-2R signaling (Fig. 8B). When compared to the numerous Foxp3lo and the few Foxp3hi T cells in the thymus of 2Rβ-/- mice, weak IL-2R signaling associated with Y3 IL-2Rβ readily supported increased expression of CD25, CD103, and CTLA4. This effect was most striking for Foxp3hi Tregs and was generally comparable to expression of these molecules by WT Tregs, although CD25 by Y3 Foxp3hi Tregs was slightly but not significantly lower. Y3 Tregs showed reduced proliferation when compared to WT and untreated 2Rβ-/- CD4+ Foxp3+ cells. As Ki67 expression was also somewhat reduced by the Foxp3+ cells from “cured” 2Rβ-/- Tregs (Fig. 2), this result suggests that weak IL-2R signaling does not fully support proliferation of developing Tregs. In contrast, Bcl-2 levels were comparable for WT, Y3, and 2Rβ-/- Foxp3hi vs. Foxp3lo thymocytes. These data further link IL-2R signaling with Foxp3hi Tregs and indicate the functional programming and CD103 activated Tregs are readily supported by Y3 IL-2Rβ.
FIGURE 8. Characterization of thymic Tregs with sub-optimal IL-2Rβ signaling.

(A) Representative dot plots and (B) expression of the indicated molecules by Foxp3lo vs. Foxp3hi thymic Tregs from the indicated mice. Data are means ± SD from 3-5 mice/group. Statistically significant differences are shown for comparisons between WT and Y3.
pSTAT5 activation by Tregs in vivo
pSTAT5 activation in vivo was assessed for WT, Y3 and 2Rβ-/- CD4+ Foxp3+ thymocytes by immediately evaluating pSTAT5 expression without the addition of IL-2 (Fig. 9A, B). In comparison to CD4+ Foxp3+ 2Rβ-/- T cells, Y3 Tregs showed substantial pSTAT5 that was nearly comparable to that associated with WT Tregs. The lack of pSTAT5 by Foxp3+ cells from 2Rβ-/- mice indicates that this in vivo pSTAT5 associated with WT and Y3 Tregs is primarily the result of IL-2R signaling. As previously noted (17), in vitro stimulation of Foxp3+ thymocytes with IL-2 resulted in most WT and Y3 Tregs to activate pSTAT5, although the percentage and level of pSTAT5+ cells was somewhat reduced for Y3 Tregs (Fig. 9A,B). Collectively, the nearly normal thymic development, as assessed by CTLA4, CD103, and CD25 expression, associated with Y3 Tregs is accounted for relatively normal pSTAT5 signaling.
FIGURE 9. pSTAT5 activity of thymic Treg cells.

(A). Representative histograms and (B) percent and MFI of pSTAT5 expression by the indicated mice. Data are means ± range from 2 mice/group. (C) Percent and (D) relative MFI (thymus:spleen) of pSTAT5 expression directly ex vivo by WT B6 and Y3 thymic and splenic Tregs. Data are means ± SD from 5 mice.
Past work indicated that an equivalent level of IL-2 in vitro activated greater pSTAT5 in thymic Tregs when compared to peripheral Tregs from normal mice (17), suggesting that during thymic development Tregs potentially generate greater IL-2R signaling and are highly sensitive to an environment that is limiting in IL-2. This trend was also readily apparent when evaluating pSTAT5 activation in vivo. Tregs directly isolated from the thymus of WT mice showed a greater percentage (Fig. 9C) of pSTAT5+ cells when compared to Tregs from the spleens whereas Tregs from Y3 thymus showed a similar percentage of pSTAT5+ cells in the thymus and spleen (Fig. 9C). Importantly, when compared to splenic Tregs, the level of pSTAT5 was nearly 2-fold greater for Tregs from the thymus of WT and Y3 mice (Fig. 9D). Thus, in the steady state in vivo, relatively normal pSTAT5 activation is observed by thymic Y3 Tregs, including a higher activation of this IL-2R-associated signaling molecule.
Competitive thymic development and weak IL-2R signaling
Although several properties associated with Y3 Tregs appeared outwardly normal, Y3 Tregs showed somewhat lower proliferation as assessed by Ki67 expression. To more broadly evaluate the efficiency of IL-2R signaling associated with Y3 IL-2Rβ, mixed bone marrow chimeras were prepared to compare the development of Y3 vs. WT Tregs in the same environment (Fig. 10A). Development by WT CD45.1+ B6 Tregs was compared to CD45.2+ Tregs expressing transgenic WT (Y0) or Y3 IL-2Rβ (on the IL-2Rβ-/- genetic background) after a 1:1 mixture of the respective T cell-depleted bone marrow was transferred into lethally irradiated Thy-1.1+ or sublethally irradiated Rag 2-/- γc-/- recipients. Y3 mice express levels of transgenic IL-2Rβ that is similar to WT B6 mice whereas those of Y0 are approximately 2-fold higher (17). In other experiments, development by CD45.1+ WT B6 Tregs was compared to CD45.2+ WT B6 Tregs or Y3 Tregs after a 1:1 or 1:4 mixture of the respective T cell-depleted bone marrow was transferred into sublethally irradiated TCRα-/- recipients. The gating strategy to follow donor thymic- and spleen-derived SP CD4+ Foxp3- conventional T cells and CD4+ Foxp3+ Tregs is shown in Fig. 10B.
FIGURE 10. Effectiveness of low IL-2R signaling for Treg thymic development and peripheral homeostasis in competitive chimeric mice.

(A) Experimental design for the mixed bone marrow chimeras. (B) Representative histograms to follow donor-derived SP CD4+ Foxp3- T cells and CD4+ Foxp3+ Tregs in the thymus and spleen. (C) The relative development of Tregs in the thymus and spleen of chimeric mice. The x-axis shows the indicated CD45.2 donor bone marrow, the ratio (CD45.2:CD45.1) after mixture with CD45-1+ WT B6 bone marrow, and the irradiated recipient. The y axis represents Efficiency of Treg reconstitution of the indicated CD45.2 Tregs and was calculated by the formula: Efficiency= (% TregCD45.2/% TregCD45.1)/(% SP CD4+ T conventionalCD45.2/% SP CD4+ T conventionalCD45.1). Values >1 or <1 for Efficiency represent over or under-representation of the CD45.2+ donor-derived Treg population, respectively. (D) The expression of CD25 by CD45.2 Tregs from the indicated chimeras. The expression of CD25 by the CD45.2+ Tregs was normalized to the CD45.1+ Tregs within the same mouse, i.e. Relative CD25 MFI= MFICD45.2/MFICD45.1. Data (C,D) are means ± SD from 3-5 mice/group.
Thymic reconstitution was largely comparable (60-100 × 106 cells) when recipients were examined 8 weeks post-bone marrow transfer. The relative representation of CD45.2+ Y0, Y3 and B6 CD4 SP T cells and Tregs to control CD45.1+ CD4+ SP T cells and Tregs from the thymus and spleen was evaluated for each recipient. We expected that the ratios of experimental CD45.2+ to control CD45.1+ T cells to be 1 in the thymus when equal numbers of bone marrow were transferred, if the both bone marrow donors contained an equivalent number of precursor cells. However, these ratios were often lower than 1 when Y0 or Y3 bone marrow was mixed with CD45.1+ WT bone marrow, suggesting that the numbers of progenitors were lower in mice with the IL-2Rβ-/- genetic background. Overall this lower ratio was consistent when examining major thymic subpopulations, i.e. DN, DP, CD4 and CD8 SP cells (not shown).
To correct for this effect and assess the relative efficiency that Y3 Tregs compete with WT CD45.1 Tregs in the thymus and spleen, we considered the relative representation of Y3 Tregs to Y3 conventional CD4+ SP T cells from these tissues (Fig. 10C, left). As a control, CD45.2+ WT Tregs competed almost equivalently with CD45.1+ WT Treg at 1:1 or 4:1 ratio of donor cells in the thymus based on the relative development of SP CD4+ thymocytes. In contrast, there was a consistent trend in all experiments for a lower relative representation of Y3 Tregs when they developed in the thymus in competition with WT CD45.1+ Tregs. To assess the extent constitutive expression of transgenic IL-2Rβ might influence this type of competitive experiment, the development of Tregs in the thymus was compared from mixed chimeras derived from Y0 (expressing WT transgenic IL-2Rβ) and CD45.1+ WT B6 bone marrow. These experiments showed a preference for Tregs expressing the WT Y0 IL-2Rβ transgene, consistent with higher levels of this transgenic IL-2Rβ. Collectively, these data indicate that impaired IL-2R signaling by Y3 thymocytes support Treg development in a competitive setting with WT Tregs, but WT Tregs are somewhat favored. This finding is in striking contrast to the spleen where nearly the entire Treg pool is derived from WT Tregs (Fig. 10C, right). Thus, impaired IL-2R signaling is much more efficient in maintaining thymic Treg development in comparison to peripheral homeostasis.
The expression of CD25, which depends on IL-2R signaling, by Y3 thymic and peripheral Tregs is comparable to normal WT Tregs in a non-competitive setting (17). Therefore, we assessed CD25 expression by the various Treg populations as an indication of the level of IL-2R signaling in these mixed chimeras (Fig. 10D). CD25 levels by CD45.2+ donor-derived Tregs was normalized to control CD45.1+ WT Tregs. In all cases the levels of CD25 by Y3 Tregs were decreased. In the thymus, CD25 levels were directly proportional to the overall representation of Y3 Tregs with greater CD25 levels by Y3 Tregs correlating with greater representation of Y3 Tregs. In the spleen, although very few Y3 Tregs were found, the levels of CD25 was similar to that in the thymus. This latter finding is consistent with these splenic and thymic Tregs receiving a similar IL-2R signal. This finding further supports the notion that thymic Treg development is generally effective with low IL-2R signaling.
Discussion
Current work indicates that IL-2 provides essential signals during thymic Treg development through activation of STAT5 (15). IL-2 has generally been thought to function as a growth/survival signal as Tregs develop in the thymus. This conclusion is primarily based on the finding that the thymus of IL-2- or IL-2R-deficient mice harbor somewhat lower numbers of CD4+ Foxp3+ Treg cells. The detection of CD4+ Foxp3+ Tregs in the periphery, although at a markedly reduced proportion, of IL-2 and IL-2R-deficient mice has led to the common view that autoimmunity associated with these animals primarily represents a defect in Treg peripheral homeostasis (4). However, recent work showed that when peripheral Treg numbers and proportions were restored to normal levels in IL-2-/- mice by abrogating Bim expression, severe lethal autoimmunity still occurred (21). This effect was associated with impaired Treg function, although another contributing factor may also be increased Th17 cells that were not antagonized by the lack of IL-2 (30).
In this report we directly show that the thymic development of CD4+ Foxp3+ Tregs is highly dysregulated in IL-2Rβ-/- mice. Although IL-2Rβ is also involved in IL-15 signaling, we have interpreted this impairment to primarily reflect a role for IL-2 because IL-15 knockout mice exhibit normal Treg production and do not exhibit autoimmunity (36). Our data are consistent with a thymic Treg developmental block in IL-2Rβ-/- mice that gives rise predominantly to functionally immature CD4+ Foxp3lo CD25- Tregs that expressed markedly reduced levels of key molecules, i.e. CTLA, CD39, and CD73, that mediated suppressive function. Low levels of these molecules were also detected in IL-2-/- Bim-/- mice (21). These findings coupled with our results indicate that the impaired function of normal levels of CD4+ Foxp3lo CD25- T cells in the periphery of these double knockout mice is the result of impaired functional programming of Tregs during thymic development. Thus, the main reason for autoimmunity in the absence of IL-2R signaling is because the Tregs produced in the thymus are developmentally blocked and largely non-functional.
The detection of primarily CD4+ Foxp3lo CD25- Tregs in the thymus of IL-2Rβ-/- mice does not represent some aberrant cell populations due to high cytokine production as a consequence of autoimmunity. In this regard, most of the properties associated with these Tregs were also observed when we evaluated thymic host-derived CD4+ Foxp3+ T cells from IL-2Rβ-/- mice that were rendered autoimmune-free by providing them WT Tregs at birth. In addition, the thymus of WT mice also harbors a substantial proportion of CD4+ Foxp3lo CD25lo cells with low expression of CTLA4, CD39, and CD73. Thus, the detection of these Tregs in the thymus of IL-2Rβ-/- mice likely reflects some block during Treg development.
A simple explanation of the impaired expression of Treg functional molecules in developing Tregs is that their lower levels of Foxp3 is not sufficient to properly induced and reinforce the Treg suppressive program (25). It is important to note that we also detected a small fraction of Foxp3hi Tregs in the thymus of IL-2Rβ-/- mice. These cells, however, still expressed a low level of CTLA4 even though CTLA4 is a direct target of Foxp3 regulation (37) and an increased proportion of these cells were CD24hi, indicative of a more immature thymocyte population. This finding indicates that normal levels of Foxp3 are not sufficient to induce the Treg suppressive program without IL-2R signaling and that these cells are not equivalent to CD4+ Foxp3hi CD25hi Tregs in WT mice. This observation also raises the possibility that IL-2 exerts several important effects during thymic development to yield a mature functional Treg that goes beyond its known role in contributing to activation of Foxp3 through STAT5 (19).
Our findings also support a role for IL-2 in the expansion of developing Tregs. This conclusion is based on the lower levels of the Ki67 proliferative marker by the developmentally impaired Tregs found in the thymus of “cured” 2Rβ-/- mice. These Tregs from untreated 2Rβ-/- mice, however, showed similar Ki67 expression to total WT Tregs (Fig. 2) or to WT Foxp3lo and Foxp3hi thymocytes, suggesting that some factors present due to the concomitant autoimmunity associated with IL-2Rβ-deficiency may somewhat compensate for impaired IL-2R signaling by increasing the numbers of immature Tregs. We cannot rule out that the selective reduced impaired host Treg compartment of “cured” 2Rβ thymus might actually reflect competition for resources or niche due to the presence of donor WT Tregs used to prevent autoimmunity that also populate the thymus. However, we also noted that Foxp3lo and Foxp3hi Y3 thymic Tregs expressed lower Ki67, which is consistent with a role for IL-2 in supporting proliferation of developing Tregs. Although Bcl-2 is an important anti-apoptotic gene and its expression is regulated by IL-2 (38), we found normal Bcl-2 levels in developing Tregs from untreated 2Rβ-/-, “cured” 2Rβ-/- and Y3 mice, indicating that IL-2 is not required for expression of Bcl-2 by thymic Tregs and suggesting that IL-2R signaling may not importantly shape survival of developing Tregs.
An important feature of developing Tregs is that these cells are exquisitely sensitive to IL-2. This was shown in two ways. First, WT thymic Tregs showed increased pSTAT5 activation when compared to peripheral Tregs. Second, in competitive bone marrow chimeras, thymic Treg development was supported to a much greater level than peripheral Treg homeostasis by Tregs expressing the Y3 mutant IL-2Rβ. Thus, this increased IL-2R sensitivity to pSTAT5 activation is very effective in driving Treg development in the thymus. The molecular basis of this phenomenon will be of interest to determine.
Past work indicates that CD4+ CD25+ Foxp3- thymocytes contain precursors for the Treg lineage (18, 19). Our findings provide some information concerning the developmental origin of CD4+ Foxp3lo Tregs present in the thymus of WT and 2Rβ-/- mice. It is unlikely that CD4+ Foxp3lo CD25- cells represent a mandatory developmental intermediate between CD4+ CD25+ Foxp3-thymocytes and mature CD4+ Foxp3hi CD25+ Tregs. First, ontogeny studies indicate that the appearance of CD4+ Foxp3lo CD25- cells does not precede, but rather is concurrent with CD4+ Foxp3hi CD25hi Tregs. Second, the pattern of expression of CD24 by CD4+ Foxp3lo CD25- cells from the thymus of WT and 2Rβ-/- mice were identical to that by CD4+ Foxp3hi CD25hi Tregs from WT mice, where mature CD24- cells predominated. This latter result suggests that the CD4+ Foxp3lo CD25- cells undergo similar levels of some Treg maturation processes, but fail to optimally express Treg functional molecules. Given the high relationship between CD4+ Foxp3lo CD25- cells from WT and 2Rβ-/- mice, the failure in full maturation of these cells is primarily accounted by a lack of IL-2R signaling. The production of CD4+ Foxp3lo CD25- T cells likely depends on IL-7R signaling in the context of TCR and co-stimulatory signals, as no Foxp3+ cells are found when both IL-2 and IL-7 are absent (12). Therefore, we favor an alternative scenario where these CD4+ Foxp3lo CD25- Tregs represent aborted products of the Treg developmental scheme. In this model, IL-2 would directly drive maturation of CD4+ Foxp3- CD25+ thymocytes into mature CD4+ Foxp3hi CD25+ Tregs, where Foxp3 and CD25 are direct targets of IL-2R-dependent STAT5 activation (39). However, some thymocytes through the selection process are also destined to become Tregs, but without an IL-2 signal their full development is prevented and they are arrested at a CD4+ Foxp3lo CD25- stage. Due to their high Bcl-2 levels these cells survive and seed the periphery but cannot suppress peripheral autoreactive T cells due to impaired expression of Treg functional molecules. In WT mice, the detection of these cells in the thymus may simply represent developing Tregs that have never received an IL-2R signal. However, we cannot rule out that some of these CD4+ Foxp3lo CD25- T cells are derived from the dedifferentiation of mature CD4+ Foxp3hi CD25+ Tregs. We think that this is not a major source of CD4+ Foxp3lo CD25- thymocytes because these cells are readily found in 2Rβ-/- mice that contain very few mature CD4+ Foxp3hi CD25+ Tregs and because recent studies indicate that thymic-derived Foxp3+ Tregs are a stable lineage (40).
Abbreviations used in this article
- DN
double negative
- DP
double positive
- Tregs
T regulatory cells
- MFI
mean fluorescent intensity
- RFP
red fluorescent protein
- SP
single positive
Footnotes
This work was supported by grants from the NIH (R01CA45957 and R01AI055815).
References
- 1.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 2.Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal period. J. Exp. Med. 2005;201:769–777. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayer AL, Yu A, Malek TR. Function of the IL-2R for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J. Immunol. 2007;178:4062–4071. doi: 10.4049/jimmunol.178.7.4062. [DOI] [PubMed] [Google Scholar]
- 4.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 6.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 9.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 10.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 11.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 12.Bayer AL, Lee JY, de la Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 2008;181:225–234. doi: 10.4049/jimmunol.181.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Allmen CE, Schmitz N, Bauer M, Hinton HJ, Kurrer MO, Buser RB, Gwerder M, Muntwiler S, Sparwasser T, Beerli RR, Bachmann MF. Secretory phospholipase A2-IID is an effector molecule of CD4+CD25+ regulatory T cells. PNAS. 2009;106:11673–11678. doi: 10.1073/pnas.0812569106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 16.Yu A, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 2006;177:5115–5121. doi: 10.4049/jimmunol.177.8.5115. [DOI] [PubMed] [Google Scholar]
- 17.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lio CWJQ, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, Farrar MA. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, Plas DR, Hildeman DA. A major role for Bim in regulatory T cell homeostasis. J. Immunol. 2011;186:156–163. doi: 10.4049/jimmunol.1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O'Gorman WE, Abbas AK. Cutting Edge: Mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J. Immunol. 2010;185:6426–6430. doi: 10.4049/jimmunol.0903940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 23.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 25.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 26.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian EL, Perlmutter RM, Taniguchi T. 3 Distinct lL-2 signaling pathways mediated by Bcl-2, c-Myc, and Lck cooperate in hematopoietic-cell proliferation. Cell. 1995;81:223–231. doi: 10.1016/0092-8674(95)90332-1. [DOI] [PubMed] [Google Scholar]
- 28.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 29.Malek TR, Ashwell JD. Interleukin 2 upregulates expression of its receptor on a T cell clone. J. Exp. Med. 1985;161:1575–1580. doi: 10.1084/jem.161.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea J J. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med. 2005;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. PNAS. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–741. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, Bell BD, Ziegler SF, Huehn J, Pardoll DM, Vignali DA. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–730. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol. Lett. 2007;114:1–8. doi: 10.1016/j.imlet.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD. Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J. Biol. Chem. 2006;281:36828–36834. doi: 10.1074/jbc.M608848200. [DOI] [PubMed] [Google Scholar]
- 38.Mandal M, Kumar R. Bcl-2 modulates telomerase activity. J. Biol. Chem. 1997;272:14183–14187. doi: 10.1074/jbc.272.22.14183. [DOI] [PubMed] [Google Scholar]
- 39.Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol. Rev. 2011;241:63–76. doi: 10.1111/j.1600-065X.2011.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]