

Abstract
The spinal cord is composed of distinct neuronal groups with well-defined anatomic connections. In some transgenic models of Alzheimer’s disease (AD) amyloid plaques develop in this structure, although the underlying cellular mechanism remains elusive. We attempted to explore the origin, evolution and modulation of spinal β-amyloid (Aβ) deposition using transgenic mice harboring five familiar AD-related mutations (5XFAD) as an experiential model. Dystrophic neuritic elements with enhanced β-secretase-1 (BACE1) immunoreactivity (IR) appeared as early as 2 months of age, and increased with age up to 12 months examined in this study, mostly over the ventral horn (VH). Extracellular Aβ IR emerged and developed during this same period, site-specifically co-existing with BACE1-labeled neurites often in the vicinity of large VH neurons that expressed the mutant human APP. The BACE1-labled neurites almost invariably colocalized with β-amyloid precursor protein (APP) and synaptophysin, and frequently with the vesicular glutamate transporter-1 (VGLUT). Reduced IR for the neuronal specific nuclear antigen (NeuN) occurred in the VH by 12 months of age. In 8 month-old animals surviving 6 months after an unilateral sciatic nerve transection, there were significant increases of Aβ, BACE1 and VGLUT IR in the VN of the ipsilateral relative to contralateral lumbar spinal segments. These results suggest that extracellular Aβ deposition in 5XFAD mouse spinal cord relates to a progressive and amyloidogenic synaptic pathology largely involving presynaptic axon terminals from projection neurons in the brain. Spinal neuritic plaque formation is enhanced after peripheral axotomy, suggesting a retrograde transneuronal modulation on pathogenesis.
Keywords: Alzheimer’s disease, amyloidogenesis, BACE1, neuritic dystrophy, synaptoplasticity
Introduction
β-Amyloid (Aβ) peptides are produced following β-secretase-1 (BACE1 or BACE) and γ-secretase mediated β- and γ-site cleavages of the amyloid precursor protein (APP) (Vassar et al., 2009; Chow et al., 2010), and are catabolized likely via enzymatic degradation, glial phagocytosis and vascular clearance (Miners et al., 2011; Thal, 2012). Because of their amphiphilic nature, Aβ peptides tend to aggregate into large insoluble forms leading to the formation of extracellular plaques, a hallmark neuropathology in Alzheimer’s disease (AD) (Braak et al., 2011; Jucker and Walker, 2011). There exist different hypotheses explaining why and how plaques develop in the brain. The classic amyloid hypothesis posits a rise of free-floating interstitial Aβ as the initial step for plaque development, which in familial AD is caused by increased production and in sporadic AD by reduced clearance, of the peptides (http://www.alzforum.org/res/adh/cur/knowntheamyloidcascade.asp). Alterations in Aβ composition (e.g., Aβ42/Aβ40 ration) or fibrillary property are also considered as factors driving plaque pathogenesis (Chiang et al., 2008; Okereke et al., 2009; Kuperstein et al., 2010; Hata et al., 2011). The sink hypothesis suggests reduced brain-to-blood efflux, or increased periphery-to-central transportation, of Aβ as the cause of cerebral amyloidosis (Zhang and Lee, 2011). The prion hypothesis suggests that Aβ peptides may undergo a self-propagating conformational change resulting in amyloidosis in a transmissible manner (Frost and Diamond, 2010; Polymenidou and Cleveland, 2012). Pharmaceutical strategies aiming at these different amyloidogenic events are being developed for AD treatment and prevention. As many anti-Aβ therapies fail to reverse or slow down neurological and cognitive declines in clinical trials, earlier drug administration is considered to be essential, although some call for a better understanding of the amyloid pathogenic process and its role in AD etiology (Pimplikar, 2009; Struble et al., 2010; D’Alton and George, 2011; Panza et al., 2011; No authors listed, 2011).
As the initiating and rate-limiting enzyme for Aβ genesis, BACE1 may serve an ideal marker to elucidate plaque pathogenesis. BACE1, especially its upregulation if occurs, would provide biochemical and anatomic clues for in situ Aβ overproduction and potentially plaque formation. Accordingly, we studied plaque pathogenesis by correlating BACE1 to Aβ and neuronal/synaptic changes in transgenic AD mouse and primate brains (Zhang et al., 2009, 2010; Cai et al., 2010, 2012a, 2012b). BACE1 elevation appears to be anatomically linked to synaptic/axonal terminal pathogenesis as well as local Aβ deposition in the brain. As neuronal organization and connectivity are rather complex in a given brain region, it is somewhat difficult to identify particular cellular/pathway-based factors that might modulate cerebral plaque development.
The spinal cord is a part of the central nervous system with its neuronal organization and connectivity well-established anatomically. Amyloid plaques can be found in the spinal cord in AD patients (Bugiani et al., 1989; Ogomori et al., 1989). Recent studies also report the presence of amyloid plaques in the spinal cord of transgenic (Tg) mice expressing familial AD (FAD) related APP and presenilins, including the Tg2576, 2XFAD and 5XFAD strains (Wirths et al., 2006; Seo et al., 2010; Jawhar et al. 2012). In the present study we explored plaque pathogenesis in the spinal cord of 5XFAD mice. Age-related spinal amyloidogenesis occurred in association with a presynaptic axon terminal pathology involving largely the descending glutamatergic projection systems, which could be enhanced after a traumatic injury to motoneurons axons.
Materials and Methods
Animals and surgery
Animal use in the present study was in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Animal Care and Use Committee of Southern Illinois University at Carbondale and the Ethics Committee for Animal Use at Central South University. A small colony of 5XFAD [B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas/J] mice was established using breeders from the Jackson Laboratory (Bar Harbor, ME) (Oakley et al., 2006). For analysis of age-related plaque development, male transgenic animals were examined at 1–2, 4, 8 and12 months of age (n=4–6 per age point). Male C57BL/6J mice at matched age points (n=4 per age group) were used as controls (for the lack of AD-like pathology) (Zhang et al., 2009). Twelve 2 month-old male transgenics were used for study of the effect of unilateral peripheral nerve injury. For surgery the animals were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). The left hind limb was shaved and swabbed with an antiseptic solution (Bethadine). A longitudinal skin incision was made around the back of the upper thigh, and the hamstring and gluteal muscles were bluntly separated to expose the sciatic nerve. The nerve was transected, with approximately 2 mm nerve segment removed. The muscles and skin were sutured. Animals were allowed to survive until 4 (n=2), 6 (n=4) and 8 (n=4) months of age (2 other operated animals died accidently).
Tissue preparation
Mice were deeply anesthesized with sodium pentobarbital (100 mg/kg, i.p.) and perfused with 4% paraformaldehyde in 0.1 M phosphate buffer. The spinal cord together with the left dorsal root ganglias (for orientation purpose) was dissected out, post-fixed in the perfusion fixative overnight, and cryoprotected in 30% sucrose at 4 °C until tissue sank. Cross-sections of the lumber spinal segments (L1–5) were processed and analyzed in the present study. Twelve sets of 30 μm thick sections were collected in order in 0.01 M phosphate-buffered saline (PBS, pH7.4) in cell culture plates. Additional 6 sets of 12 μm and 6 sets of 6 μm thick sections were collected by thaw-mounting on positively charged microslides. Accordingly, each set consisted of equally-spaced sections at ~500 μm intervals.
Immunohistochemistry
Sets of the 30 μm sections from each animal were used for Nissl stain, immunohistochemistry (including BACE1, Aβ antibodies 6E10, 12F4, Ter42, NeuN, see Table-1) with the avidin-biotin complex (ABC) method and histochemistry for nicotinamide adenine dinucleotide phosphate diaphorase (NADPH-d). For BACE1 labeling sections were first treated in 50% formamide and 50% 2XSSC (300 mM sodium chloride, 30 mM sodium citrate) at 65 °C for 60 (30 μm sections) and 20 minutes (6 μm sections) (Zhang et al., 2009). For Aβ antibody immunohistochemistry, sections were pretreated with 50% formic acid at room temperature followed by several washes with 0.05 M Tris-HCl buffer (pH=8.5). After antigen retrieval, sections were preincubated in 1% H2O2 in PBS for 30 minutes and in 5% horse serum in PBS with 0.3% Triton X-100 for 1 hour. Sections were then reacted with a primary antibody at pre-optimized concentration overnight at 4 °C. Sections were further reacted with a biotinylated universal secondary antibody (horse anti-goat, rabbit and mouse IgGs) at 1:400 for 2 hours, and subsequently with the ABC reagents (1:400) (Vector Laboratories, Burlingame, CA) for another hour, at room temperature. Immunoreaction product was visualized in 0.003% H2O2, 0.05% 3,3-diaminobenzidine (DAB). Three 10-minute washes with PBS were used between incubations. After staining, the sections were allowed to air-dry, cleared and coverslippered.
Table 1.
Primary antibodies used in the present study
| Antibody | Source | Product # | Dilution |
|---|---|---|---|
| mouse anti-Aβ1–5, 3D6 | Elan | Lot#1132 | 1:3000 |
| mouse anti-Aβ1–42, 12F4 | Signet | 39240 | 1:4000 |
| mouse anti-Aβ1–16, 6E10 | Signet | 39320 | 1:4000 |
| mouse anti-amyloid precursor protein, 22C11 | Millipore | MAB348 | 1:1000 |
| rabbit anti-BACE1 (a.a. 46–163) | H. Cai | 1:2000 | |
| mouse anti-calbindin (CB) | Sigma | C9848 | 1:4000 |
| goat anti-choline acetyltransferase (ChAT) | Millipore | AB1447 | 1:1000 |
| mouse anti-glutamate decarboxylase 67 (GAD67) | Millipore | MAB5406 | 1:4000 |
| mouse anti-growth- associated protein-43 (GAP43) | Sigma | G9264 | 1:4000 |
| rabbit anti-glial fibrillary acidic protein (GFAP) | Sigma | G9269 | 1:4000 |
| mouse anti-microtubule associated protein-2 (MAP2) | Sigma | M9942 | 1:1000 |
| mouse anti-parvalbumin (PV) | Sigma | P3088 | 1:4000 |
| mouse anti-synaptophysin (SYN) | Millipore | MAB329 | 1:4000 |
| mouse anti-vesicular glutamate transporter-1 (VGLUT1) | Millipore | MAB5502 | 1:2000 |
Double immunofluorescence was carried out by a pretreatment of thaw-mount sections in PBS containing 5% donkey serum to lower non-specific binding. Then the sections were incubated with a pair of primary antibodies raised in different species (rabbit, mouse or goat) overnight at room temperature. This was followed by a 2 hour reaction with Alexa Fluor® 488 and Alexa Fluor® 594 conjugated donkey anti-mouse, rabbit or goat IgGs (1:400, Invitrogen, Carlsbad, CA). Sections were then counter-stained with Bisbenzimide (Hoechst 33342, 1:50000), washed, and mounted with anti-fading medium before microscopic examination.
The BACE1 antibody used in the present study was raised in rabbit against a synthetic peptide corresponding to amino-acid residues 46–163 of human BACE1. It detects a 70 kDa band in brain homogenates by western blot, which is absent in BACE1 knockout tissue (Laird et al., 2005; Xiong et al., 2007). This antibody does not visualize any specific reactivity in BACE1 knockout mouse brain and spinal cord (Zhang et al., 2009). Other primary antibodies were commesurized, with their specificity initially tested by preabsorption of the primary antibody with a neutralizing antigen peptide and omission of the primary antibody in the immunolabeling protocol. Neither condition yielded specific immunolabeling in brain sections. In each batch of peroxidase or immunofluorescent stain, several spine cord sections were processed along with the experimental sections, excluding the exposure to the primary antibody. These latter sections were used to define the levels of background reactivity in densitometric analysis.
NADPH-diaphorase histochemistry
Sections were incubated in 0.05 M Tris-HCl buffered saline (pH 8.0, TBS) containing 0.3% Triton X-100, 1 mM nicotinamide adenine dinucleotide phosphate diaphorase (β-NADPH-d, N7505, Sigma-Aldrich), 0.8 mM nitroblue tetrazolium (NBT, N6639, Sigma-Aldrich) and 5% dimethyl sulfoxide at 37 °C for 1 hour. Selected sections were further immunostained for BACE1 using the DAB-peroxidase method to assess colocalization.
Imaging, quantitative analysis and figure preparation
All sections were examined on Olympus (BX60 and BX53) fluorescent microscopes equipped with image analysis systems (Optronics, Goleta, CA; CellSens Standard, Olympus, Japan). Images were captured with 2X to 40X objective lens. Optic density of IR over the spinal grey matter was obtained using the OptiQuant analysis software (Parkard Instruments, Meriden, CT). Specific labeling densities (expressed as digital light units per squire mm, DLU/mm2) in area of interest in transgenic spinal cord was calculated by subtracting either the background (obtained from sections processed with the exclusion of the primary antibody) or cutoff (obtained from age-matched non-transgenic tissue) signal from total density values. In other cases, percent area occupied by immunolabeled elements (Aβ, BACE1, NeuN) was measured using a threshold selection approach (detailed later). For comparison between different labelings, specific densities were normalized to the mean of controls correspondingly. Student’s t-test (two-tailed paired analysis, Excel 2007) or one-way ANOVA with Bonferroni post-hoc comparison (Prism GraphPad 4.1, San Diego, CA) were used for statistical evaluation of mean differences, with p<0.05 considered statistically significant.
Results
Overall BACE1 expression pattern in non-transgenic and 5XFAD mouse spinal cord
In non-Tg spinal cord, BACE1 IR was present over the grey matter, with little labeling in the white matter (Fig. 1A), consistent with the pattern shown in a previous report using a different antibody (Lee et al., 2005). The labeling was generally diffuse or neuropil-like across the ventral horn (VH), intermediate zone (IZ) and dorsal horn (DH). Lamina I and II of the DH exhibited a noticeably higher density than the remaining grey matter areas (Fig. 1A, C). Faintly labeled perikaryal profiles were identifiable at high magnification, especially in the VH. These profiles were relatively large in size, therefore would likely represent the ventral motoneurons (Fig. 1B). No apparent cellular profiles were seen in the IZ and DH (Fig. 1C). It should be noted that fairly distinct perikaryal BACE IR existed in the dorsal root ganglia (DRG) (Fig. 1D–I). These labeled somata were considerably large and resembled the primary sensory neurons.
Fig. 1.
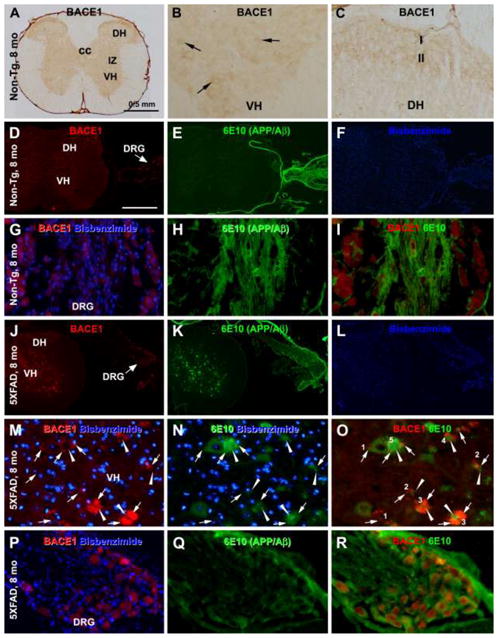
Characterization of overall BACE1 alteration and transgenic human APP expression in 5XFAD relative to non-transgenic spinal cord and dorsal root ganglia (DRG) with the peroxidase-DAB method (A-C) and immunofluorescent. Bisbenzimide (blue) nuclear counterstain is shown in same imagines for histological orientation (D–R). BACE1 immunoreactivity (IR) in non-transgenic (non-Tg) spinal cord shows largely a neuropil-like pattern restricted to the grey matter, with lamina I and II of the dorsal horn (DH) exhibited a heavier IR (A–D). Faintly stained perikarya are seen in the ventral horn (VH, arrows, B). BACE1 labeled somata are present in the DRG (D, G). 6E10 IR is absent in the spinal cord, although putative non-specific IR occurs in the DRG in fiber bundles and likely satellite glia (E, H, I). In transgenic spinal cord, distinct profiles are labeled by BACE1 and 6E10 antibodies (J–L). At high magnification, BACE1 labeled elements are swollen and sprouting neurites and clusters arranged in rosette-like configuration (M). 6E10 IR colocalizes in some of these neurites (M–O, arrows), but also occurs in neuronal perikarya, and appears as extracellular fibrillary elements (arrowheads) with a spreading configuration (N, O). There appears to be a trend of emergence and buildup of the extracellular deposits from small to large-sized dystrophic neuritic clusters (follow the profiles marked with 1 to 5 in O). The amyloid deposits occur commonly around the central zone of the neuritic cluster but may spread to the periphery. cc: central cannel. 6E10 IR exists in DRG neuronal somata colocalizing with BACE1 in transgenic materials (P–R). Scale bar = 0.5 mm in (A) applying to (D–F, J–L); equivalent to 50 μm for remaining panels.
In 5XFAD mouse spinal cord, heavily labeled BACE1 immunoreactive profiles occurred in the grey matter (Fig. 1J). These profiles existed as isolated spheroids or swollen processes, as well as rosette-like clusters of neurites in varying size. They showed age-related changes and were identified as dystrophic axonal neurites site-specifically associated with plaque development (Fig. 1M–O), which will be detailed in following sections. As with the non-Tg mice, BACE1-labeled somata were found in the DRG in 5XFAD mice. However, there were no apparent age and genotype-related alteration regarding this perikaryal BACE1 IR (Fig. J, P–R). Also similar to non-transgenics, faint BACE1 IR was seen in some VH neurons in 5XFAD mice (e.g., Figs. 1M; 2A), which exhibited little age-related changes by microscopic examination.
Fig. 2.
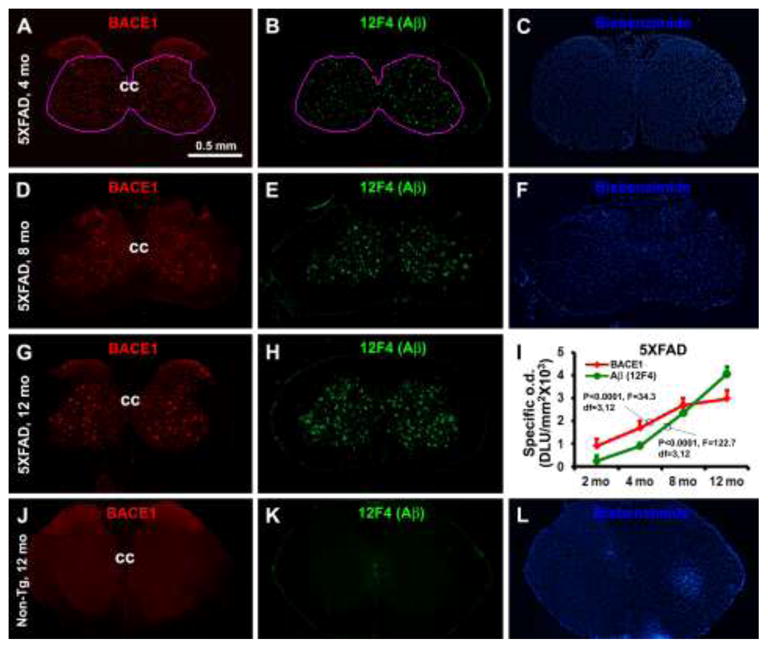
Representative images and densitometry showing an age-dependent increase of BACE1 expression and extracellular Aβ deposition in 5XFAD mouse spinal cord. Panels A–H show the pattern and abundance of BACE1 and 12F4 labeled extracellular Aβ in lumber spinal cord cross sections, with increased amount of labeled profiles noticeable from 4, to 8 and to 12 months of age for both markers. Panel I illustrates the specific densities measured over the area defined in (A and B), as a function of age. There exists an age-dependent increased of BACE1 and 12F4 reactivities, as indicated by ANOVA analysis. Note that the mean density of amyloid deposit exceeds that of BACE1 after but not before 8 months of age. Panels J–L show the levels of BACE1 and 12F4 reactivities in a batch-processed section from a non-transgenic spinal cord, as an example for defining the cutoff threshold for calculating the “specific densities” in transgenic tissue. Bisbenzimide nuclear stain is included for histological orientation (C, F, L). Abbreviations are as defined in Fig. 1. Scale bar = 0.5 mm in (A) applying to all image panels.
Expression of transgenic APP in 5XFAD mouse spinal cord
The monoclonal Aβ antibody 6E10 was used to assess the cellular expression of putative mutant human APP in 5XFAD spinal cord, using non-Tg tissue as assay control (Winton et al., 2011). In non-Tg spinal cord at any age, no 6E10 IR was found in the grey or white matter of the spinal cord (Fig. 1E, F). However, there was 6E10 IR in the DRG that appeared in dorsal root fiber bundles and around BACE1-labeled somata (likely non-specific reactivity). The BACE1-labeled DRG somata per se did not exhibit apparent 6E10 IR (Fig. 1H, I). In 5XFAD spinal cord, 6E10 IR occurred in neuronal somata over the grey matter in pre-plaque young animals (as early as 1 month-old, not shown) as well as older ones with spinal amyloid pathology. Shown as an example (Fig. 1J–O), 6E10 IR was present in neuronal somata and proximal dendrites, in addition to extracellular Aβ deposits, in the spinal cord from 8-month-old transgenics. The labeled neurons varied in size and were distributed largely in the VN (Fig. 1K, N). It should be noted that 6E10 IR also colocalized with BACE1 IR in dystrophic neurites (Fig. 1M–O, arrows). Moving from small/isolated BACE1/6E10 labeled neuritic spheroids to large neuritic clusters (arrows), there was a trend of appearance and buildup of local extracellular Aβ deposits (arrowheads) seen in double labeling preparation (Fig. 1M–O; follow the profiles marked with 1→2→3→4→5 in O). Different from the pattern seen in non-Tg samples, 6E10 IR occurred in the somata of DRG neurons, colocalizing with BACE1 IR (Fig. 1P–R).
Age-related increase of BACE1 and extracellular Aβ IR in 5XFAD mouse spinal cord
A small amount of discretely distributed BACE immunoreactive neuritic elements were identifiable in the VH in 5XFAD mice as early as 2 months of age (data not shown). These neurites were clearly present in the VH, IZ and deep DH in 4 month-old transgenics (Fig. 2A–C). A small amount of extracellular Aβ deposits also occurred at this age regionally corresponded with the BACE1 labeled neurites (Fig. 2B), as visualized with the 12F4 antibody that labels extracellular Aβ but rarely neuronal somata (Cai et al., 2010). The amount of BACE1-labled neurites and Aβ deposits were increased at 8 months, and further at 12 months, of age in the VH, IZ and deep DH (Fig. 2D–H). BACE1 labeled neurites and Aβ deposits were absent in lamina I and II at all ages examined (Fig. 2. Neither BACE1 labeled neuritic profile nor extracellular Aβ deposits were detectable in the spinal cord in double immunofluorescent preparation in non-Tg mice at all age time points examined (Figs. 1A–C, 2J–L).
A correlated densitometry was carried out in the transgenics to determine the trend of changes in BACE1 relative to extracellular Aβ deposits with age. Four equally-spaced (500 μm) lumber spinal sections in each animal were used for quantification (n=4 per age point). The densities of BACE1 and 12F4 IR were measured correlatively over the VH, IZ and deep DH with a threshold selection approach, with the area of interest defined by selecting the region with plaques as the measuring template for both BACE1 and 12F4 IR (Fig. 2A, B). The optic densities over the same area in batch-processed non-transgenic spinal cord were used as cutoff levels to calculate “specific” or elevated BACE1 and 12F4 IR in the transgenics. A progressive increase in specific BACE1 (P<0.0001, F=34.3, DF=3, 12) and 12F4 (P<0.0001, F=122.7, DF=3, 12) IR occurred from 4, to 8, and to 12 months of age in the transgenics (Fig. 1I). Different from that seen in 4 and 8 month-old animals, the relative density of 12F4 IR became higher than that of BACE1 IR at 12 months of age (Fig. 1I).
Cellular localization of BACE1/Aβ antibody IR in 5XFAD mouse spinal cord
Previously we showed a differential Aβ antibody labeling at axon terminals expressing increased BACE1 in the forebrain in several transgenic AD models (Zhang et al., 2009; Cai et al., 2012a, b). This phenomenon was also found in the spinal cord of 5XFAD mice. Thus, in addition to 6E10 (Fig. 1M–O), BACE1 IR co-localized with 4G8 (Fig. 3A–C) or 3D6 (Fig. 3D–F) IR in swollen/sprouting neurites. Of note, this intra-axonal Aβ antibody labeling was generally weaker relative to extracellular Aβ fibrils. Also, small/isolated BACE1 immunoreactive spheroids were often not associated with local extracellular Aβ IR, whereas large spheroids, especially rosette-like neuritic clusters, were generally surrounded by Aβ deposits (Figs. 1O; 3C, F). In the latter case, the Aβ deposits concentrated at the center, but could spread across and beyond the periphery, of a given neuritic cluster (Figs. 1M–O; 3A–F). The neuritic spheroids and clusters were also distinctly labeled by the 22C11 antibody (Fig. 3G–I).
Fig. 3.
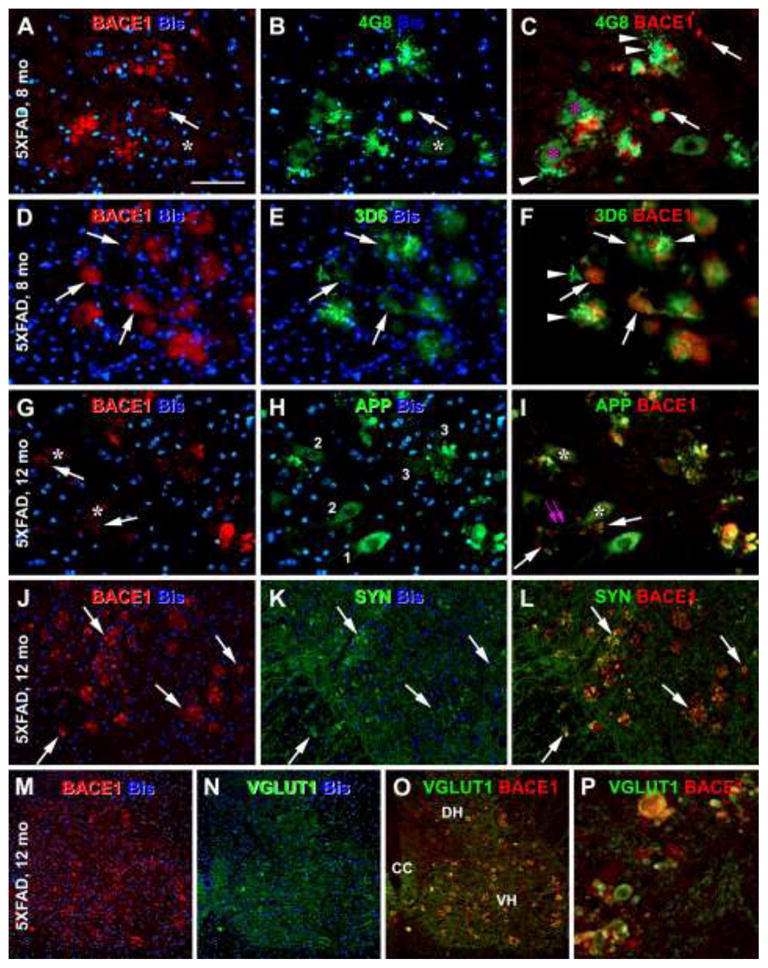
Microanatomic characterization of BACE1 and Aβ antibody labeling in the ventral horn (VH) of the spinal cord in 5XFAD mice at representative ages as indicated. Panels A–C show the presence of 4G8 labeled extracellular Aβ deposits around BACE1 labeled neuritic clusters. Some neurites coexpress BACE1 and 4G8 reactivities (arrows). Neuronal somata and primary dendrites are labeled by 4G8, whereas they show little BACE1 IR (asterisks). Panels D–F show a colocalization of BACE1 with 3D6 IR in swollen/sprouting neurites (arrows), with 3D6 also clearly visualizing local extracellular Aβ deposits around neuritic cluster (arrowheads). Unlike 6E10 (see Fig. 1N, O) and 4G8 (B, C), 3D6 does not label perikaryal profiles. Panels G–I show colocalization of BACE1 and β-amyloid precursor protein (APP) detected by 22C11 in dystrophic neurites. The neuritic clusters or neuritic plaques (arrows) are often juxtaposed to the somata (asterisks, A–C, G–I) or dendrites (I, paired purple arrows) of large ventral horn neurons. Panels J–L and M–P show frequent colocalization of BACE1 with synaptophysin (J–L) and vesicular glutamate transporter-1 (VGLUT1) (M–P) in swollen/sprouting axon terminals. Abbreviations are the same as defined in Fig. 1. Scale bar = 50 μm in (A) applying to all panels.
A close spatial relationship between neuritic plaques and large VH neurons was noticeable in the spinal cord. Thus, in BACE1/6E10 or BACE1/4G8 double labeled sections (note: 3D6 IR is absent in neuronal somata), the neuritic plaques (dystrophic neurites and extracellular Aβ) were commonly present in proximity of the somata or primary dendrites of large multipolar neurons in the VH (Figs. 1O; 3C, F). Similarly, neuritic clusters coexpressing BACE1/APP occurred in the vicinity of large VH neurons (Fig. 3I). Notably, APP expression in neuronal somata juxtaposed to neuritic plaques appeared to be reduced relative to those distributed apart from neuritic plaques (Fig. 3G–I, comparing somata labeled with 1 to 3).
BACE1-labeled swollen/sprouting neurites in 5XFAD mouse spinal cord colocalized with synaptophysin (Fig. 3J–L). Among specific neuronal phenotype markers, BACE1 immunoreactive neurites mostly co-expressed VGLUT1 (Fig. 3M–P). There was infrequent colocalization of BACE with choline acetyltransferase (ChAT) (Fig. 4A–C), GAD67 (Fig. 4D–F), calbindin (Fig. 4G–I), NADPH-d (Fig. 4J–L) and PV (not shown). No colocalization was found between BACE1 and MAP2 (not shown) or BACE and GFAP (Fig. 4M–O).
Fig. 4.
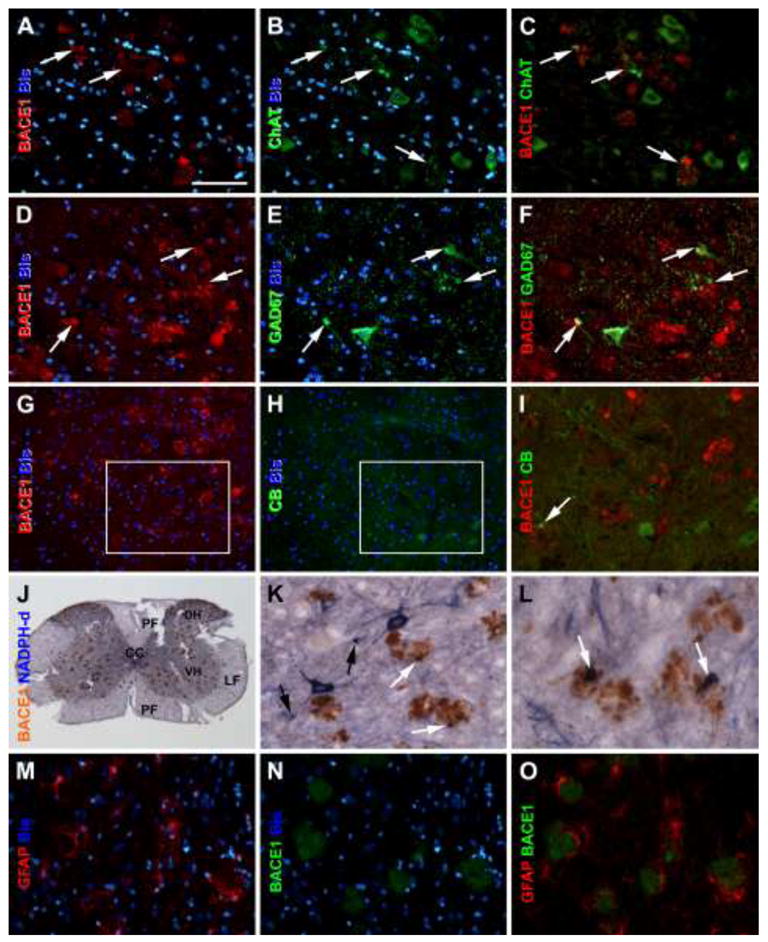
Representative images from an 8 month-old 5XFAD mouse showing infrequent colocalization (pointed by arrows) of BACE1 with cholinergic and some interneuron markers in the ventral horn. Choline acetyltransferase (ChAT) labeling occurs in motoneurons and colocalize in a small subset of BACE1 labeled neuritic profiles (A–I). Similarly, a small amount of BACE1 labeled neurites coexpress glutamate decarboxylase 67 (GAD 67) (D–F), calbindin (CB) (G–I) and nicotinamide adenine dinucleotide phosphate diaphorase (NADPH-d) (J–L). Some isolated swollen NADPH-d processes are also present in the ventral horn (black arrows in J, L). No colocalization exists between BACE and glial fibrillary acidic protein (GFAP) (M–O). Scale bar = 50μm in (A) applying to other high magnification panels, equivalent to 0.5 mm for (J).
Age-related neuronal loss in 5XFAD mouse spinal cord
Neuronal degeneration has been reported in the brain in 5XFAD mice (Oakley et al., 2006; Ohno et al., 2007; Jawhar et al., 2012). We assessed the timing and extent of neuronal loss in 5XFAD mouse spinal cord, in relative to neuritic and amyloid pathology, using a systematic approach. The analyses were conducted in 4 equally-spaced (500 μm apart) lumber spinal sections double-labeled for BACE1 with NeuN and BACE1 with 12F4 (bisbenzimide counterstained). C57BL/6J mice were used as control for neuronal count. NeuN immunoreactive perikarya containing clearly present nuclei were countered manually in the ventral left or right quadrant (largely corresponding to the VN, including a small portion of IZ) of the spinal grey matter in reference to the central cannel, in transgenic and non-transgenic mice at 2, 4, 8 and 12 months of age. “Percent area occupied by labeled elements” was also used for comparative analysis of NeuN, BACE1 and Aβ IR in 5XFAD mice at 8 and 12 months age points (Fig. 5A–L).
Fig. 5.
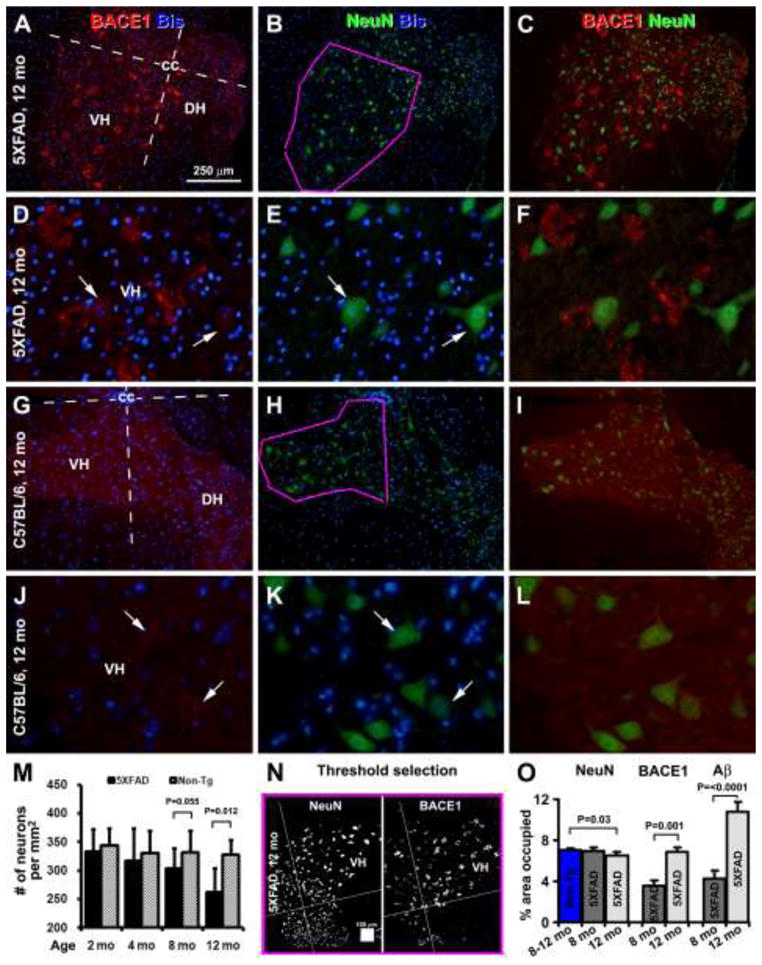
Images, quantification methodology and histograms demonstrating age-related loss of neurons in the ventral horn of 5XFAD mice relative non-transgenic counterparts. Measurements were carried out in a ventral quadrant of the grey matter in reference to the central cannel (cc). The ventrolateral border of measuring area corresponds to that of the ventral horn (as outlined in purple in B and H). Cell count was carried out in this area in transgenics and controls at 2, 4, 8 and 12 months of age (n=3), with perikarya exhibiting immunoreactivity for neuron-specific nuclear antigen (NeuN) but also containing bisbenzimide positive nuclei recorded (M). Correlative analysis of area occupied by NeuN, BACE and 12F4 Aβ (images not shown) immunoreactivity was conducted in 8 and 12 month-old transgenics using a threshold-selection approach; the cutoff level was obtained from batch-processed sections not exposed to primary antibodies (N, O). A reduced density of NeuN positive profiles is noticeable in images from a 12 month-old transgenics (B, E) relative to age-matched control (H, K). Numerical density of NeuN positive neurons is reduced in 5XFAD mice at 8 and 12 months of age relative to controls (M). Area occupied by all NeuN positive elements is reduced in 12 month-old transgenics relative to control (O). Area occupied by BACE1 and 12F4 positive profiles is increased significantly in the transgenics from 8 to 12 months of age (O). DH: dorsal horn. Scale bar = 250 μm in (A) applying to (B, C, G–I), equal to 50 μm for (D–F, J–L).
Compared to non-transgenic counterparts, numeric density of neurons in VH was comparable between 5XFAD and non-transgenic mice at 2 (333.5±38.9 vs 344.5±29.2) and 4 (317.4±55.7 vs 330.7±38.9) months of age (Fig. 5M), but tended to reduce in the former afterwards. Thus, the density reduced in 5XFAD relative to controls with a marginal statistical significance at 8 months (302.9±35.7 vs 331.7±37.3, P=0.055), but significantly at 12 months (262.4±41.6 vs 327.4±25.5, P=0.012), of age (Fig. 5M). The percent area occupied by NeuN immunoreactive elements (including perikarya that passing or not passing the cell nuclei) reduced in 5XFAD relative to controls at 12 months of age (6.54±0.32% vs 7.05±0.14%, P=0.030), but not at 8 months (6.97± 0.35% vs 7.05±0.14%, P=0.356; Fig. 5N, O). The percent area occupied by BACE1-labeled neurites increased at 12 (6.83±0.48%) relative to 8 (3.58±0.52%) months of age in the transgenics (P<0.001; Fig. 5O). The percent area occupied by extracellular Aβ IR increased at 12 (10.83±0.89%) relative to 8 (4.28±0.73%) months in the transgenics (P<0.0001; Fig. 5O).
Increased plaque formation after peripheral nerve injury in 5XFAD mouse spinal cord
Experimental effects were examined in 4–8 month-old transgenics after unilateral sciatic nerve transection at 2 month, considering the intense neuropathology at 12 months of age (see Fig. 2) that might cause a ceiling effect. In two animals examined after surviving 2 months post axotomy, there was no microscopically impressive difference in immunolabelings between the ipsilateral and contralateral grey matter (therefore not included in analysis). Quantification was thus carried out to determine axotomy-induced changes at 4 and 6 months post axotomy (Fig. 6). Specific density of labeling in area of interest was calculated by subtracting background from total optic density readings, with the former obtained from batch-processed sections excluding exposure to primary antibody. For statistical comparison, data were normalized to the mean density (defined as 100%) obtained from the contralateral VH in the 6 month-old group, correspondingly for each type of immunolabeling.
Fig. 6.
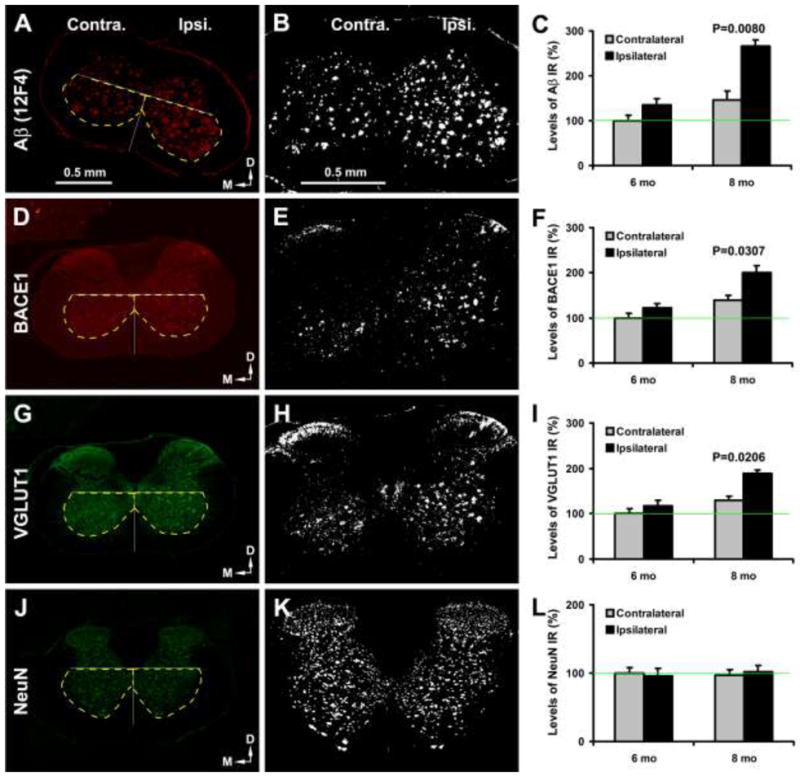
Increased amyloid and neuritic pathology in 5XFAD mouse spinal cord following sciatic nerve transection. The left panels show images from an 8 month-old transgenics surviving 6 months after unilateral sciatic nerve cut. The middle panels show threshold-based selection of labeled profiles. Specific optic density was calculated with a cutoff set at the level of non-specific reactivity measured in sections processed in the absence of primary antibody. Area of interest includes the ventral quadrants relative to central cannel (cc), as marked by yellow broken lines in the left panels. Histograms show relative mean specific densities normalized to the mean (defined as 100%, green lines in histograms) from the contralateral ventral horn in 6 month-old transgenics. The densities of Aβ (A–C), BACE1 (D–F) and VGLUT1 (G–I) IR are significantly higher in the ipsilateral (ipsi.) than contralateral (contra.) ventral horn in 8 month-old transgenics (with difference of Aβ density approaching significance in the 6 month-old group). No difference exists for NeuN density between the two sides in 6 and 8 month-old animals (J–L). Scale bars (0.5 mm) in (A) and (B) apply to the same vertical panels of images, respectively.
Specific density of extracellular Aβ deposits (12F4 IR) was higher in the ipsilateral (135.9 ± 13.5%, mean±S.D., same format below) than contralateral (100±11.5%) VH in the 6 month-old group (P=0.076, paired Student-t test, same test below), more prominent in the 8 month-old group (265.6.9 ±14.9% vs 146.3±19.6%; P=0.008) (Fig. 6A–C). Specific density of BACE1-labeled dystrophic neurites increased in the ipsilateral (121.8±9.7%) relative to contralateral (100±10.3%) VH at 6 months (P=0.095), and 8 months (201.2±14.8% vs 138.7±11.1%; P=0.030) (Fig. 6D–F). A trend of increase in VGLUT specific density in ipsilateral (117.7 ± 11.2%) relative to contralateral (100±9.0%) sides was detected at 6 months (P=0.153), and was significant at 8 months of age (188.6±8.5% vs 129.5±9.7%; P=0.020) (Fig. 6G–I). No difference was detected for the specific NeuN IR density between the ipsilateral and contralateral sides at 6 (96.0±9.3% vs 100±8.4%; P=0.730) or 8 (102.2±13.7% vs 97.4±7.4%; P=0.643) months of age (Fig. 6J–L).
Discussion
Spinal Aβ deposition largely correlates with glutamatergic axonopathy in 5XFAD mice
The major origin of plaque-associated Aβ remains a subject of ongoing research, while a variety of sources have been proposed (Fiala, 2007). For instance, it is suggested that peripheral Aβ may contribute to central plaque formation. This would require active transportation of plasma Aβ across brain-blood barrier against a steep concentration gradient (from pg/ml levels in blood to ng/ml levels in brain or CSF) (Deane et al., 2009; Silverberg et al., 2010; Seppälä et al., 2011; Figurski et al., 2012). Notably, Aβ plaques are rarely reported in the periphery, whereas many other β-sheet proteins can aggregate to form amyloidosis in bodily tissues/organs (Merlini et al., 2011). Glial cells are suggested to produce Aβ and perhaps contribute to plaque formation (Zhao et al., 2011). However, these cells are also considered as the principal machinery for Aβ clearance (Thal et al., 2012). Intracellular Aβ released from neuronal somata and dendrites could lead to extracellular Aβ accumulation and plaque formation. However, studies indicate that some prominent Aβ antibody labeling in neuronal somata has a poor value in predicting regional cerebral plaque formation over time in human or transgenic mouse brain (Wegiel et al., 2007; Aho et al., 2010; Winton et al., 2011; Cai et al., 2012a).
In the present study, we observe increased BACE1 IR in the spinal cord of 5XFAD mice as early as 2 months of age, distributed mostly over the ventral horn. The density or occupying area of BACE1-labeled profiles increase with age until at least 1 year-old. These profiles are individually surrounded by, but not precisely overlap with, extracellular Aβ deposits. Correlative densitometry shows a parallel increase in the amount of BACE1-labeled elements and Aβ deposits with age. The density of extracellular Aβ IR tends to exceed that of BACE1 IR after 8 months of age, likely representing a time-dependent accumulative effect of the insoluble Aβ deposits. The BACE1-labeled elements also exhibit 22C11 and 6E10 IR, suggesting an overexpression of the transgenic human APP or perhaps murine APP as well. These profiles are further identified to be swollen/sprouting, or dystrophic, axon terminals that are predominantly glutamatergic and often occur around the somata of VH neurons. A small fraction of the BACE1-labeled terminals colocalize with GABAergic and cholinergic markers (e.g., GAD, CB, NADPH-d, ChAT), therefore may derive from spinal interneurons and motoneurons (i.e., their axonal collaterals) (Xie et al., 2003; Brownstone and Bui, 2010).
In contrast to dystrophic axonal neurites, neuronal somata and dendrites in transgenic spinal cord exhibit minimal BACE1 IR, nor change in labeling intensity, relative to non-transgenic counterparts. Spinal cord white matter exhibits little BACE1 IR in non-transgenic and 5XFAD mice. Interestingly, the somata and likely central terminals (i.e., in lamina I and II) of DRG neurons exhibit fairly strong constitutive BACE1 expression in both genotypes. Yet, no or minimal extracellular Aβ IR occurs in the DRG or lamina I and II in the transgenics, despite an expression of the putative transgenic APP (6E10 IR) in the DRG neurons. Taken these findings together, it appears that plaque-forming Aβ products in 5XFAD mouse spinal cord may largely derive from dystrophic presynaptic axon terminals originated from glutamatergic neurons in the brain, which may include projection neurons in the motor cortex and perhaps some subcortical structures that innervate the spinal cord neurons.
As with a report in Tg2576 nice (Seo et al., 2010), an age-related loss of neuronal somata is detectable in the VH in 5XFAD mice relative to non-transgenic controls. Thus, the numerical density or occupying area of NeuN immunoreactive somata tends to decline at 8 months of age, and is significantly reduced at 12 months. In addition, a reduction in APP expression is noticeable in neuronal perikarya apposing to, relative to those apart from, neuritic plaques. Thus, established amyloid plaques may affect the viability of nearby neurons but likely cause neuronal death at a certain stage. Because neuritic plaques begin to develop long before neuronal loss, it appears unlikely that spinal plaque formation is initially seeded by Aβproducts released from dying or dead neurons (Fiala, 2007).
Motoneuron axotomy promotes spinal neuritic plaque pathogenesis in 5XFAD mice
The perisomatic distribution of neuritic plaques and the predominant glutamatergic identity of dystrophic neurites appear to suggest a certain synaptic mechanism involved in plaque development in the spinal cord. Previous studies show that peripheral nerve injury causes synaptic reorganization on the somata and dendrites of motoneurons in the spinal cord as well as neurons in higher brain centers (Sanes et al., 1990; Brumovsky et al., 2007; Navarro et al., 2007; Zelano et al., 2009; Alvarez et al., 2011). We therefore explored whether sciatic nerve axotomy may influence neuritic and amyloid plaque pathogenesis in the VH of the lumber spinal cord.
The amounts of extracellular Aβ deposits, BACE1 and VGLUT1 labeled dystrophic neurites tend to increase in the ipsilateral relative to contralateral lumbar VH in 6 month-old transgenics 4 months after unilateral sciatic nerve transection. These changes become statistically significant by 6 months after axotomy. In contrast, no differences are found between the ipsilateral and contralateral VH in the amount of NeuN IR in 6 and 8 month-old transgenics following axotomy. The enhanced neuritic (BACE1 and VGLUT1 labeled) and amyloid pathogenesis might occur as a reactive response in the presynaptic axon terminals on the somata and dendrites of the motoneurons subjected to axotomy (Brännström et al., 1992; Rose and Odlozinski, 1998). It is possible that the transgene expression (e.g., 6E10 IR) in pre- and/or post-synaptic components might somehow promote a dystrophic and amyloidogenic response in some central excitatory axonal terminals innervating spinal motoneurons after these neurons are traumatized. The lack of neuron loss in the VH is consistent with previous reports showing well survival of spinal motoneurons after axotomy in adult rodents (Vanden Noven et al., 2003; Xu et al., 2010).
It should be noted that a previous study shows APP and Aβ antibody labeled neurites in the VH of aged rat spinal cord, which is increased after periphery axotomy, in the absence of motoneuron degeneration (Xie et al., 2003). In the present study we observe labeling of dystrophic neurites by 6E10, 3D6 and 4G8 in the spinal cord. Early-onset intra-axonal Aβ antibody labeling also occurs in the cerebral cortex and hippocampus in several transgenic AD models, which is associated with or followed by local extracellular amyloidosis (Zhang et al., 2009; Cai et al., 2012a). However, in the olfactory bulb glomeruli, axonal terminal Aβ antibody labeling persists in the absence of local extracellular amyloidosis (Cai et al., 2012b). We have hypothesized that this type intra-axonal/synaptic labeling might relate to a cross-reactivity of some Aβ antibodies to the APP β-site cleavage fragment, β-CTF (in addition to APP), although we do not rule out a possibility that the labeling represents true intraneuronal Aβ (Cai et al., 2012a, b). In comparison, the lack of extracellular Aβ deposition in aged rat spinal cord could be because normal rodents do not produce a sufficient amount of the most fibrillary Aβ42 species (Xie et al., 2003). Alternatively, BACE1 upregulation might be not prominent in spinal neurites in aged rats following axotomy, thus rendering a low vulnerability of amyloidosis in wild-type rodent.
In summary, this study identifies early and progressive presynaptic terminal pathology involving predominantly the glutamatergic phenotype in the spinal cord of 5XFAD mice, which is associated with BACE1 and potentially APP β-CTF elevations as well as local extracellular Aβ deposition. Sciatic nerve axotomy promotes the axon terminal pathology but also plaque pathogenesis in lumber spinal cord. Together with previous findings in the brain (Zhang et al., 2009, 2010; Cai et al., 2010, 2012a, b; Yan et al., 2012), the data point to a common mechanism for neuritic plaque development in the central nervous system, which is compatible with an increasing notion that AD is a synaptic disease tightly associated with amyloidogenesis within neuronal elements (Palop et al., 2007; Goto et al., 2008; Marcello et al., 2012; Spires-Jones and Knafo, 2012). If the elevated BACE1, it’s immediate (i.e., β-CTF) or end (Aβ) products play a harmful role to synapses (Saganich et al., 2006; Tamayev et al., 2012), pharmacological inhibition of excessive BACE1 would be an ideal early therapeutic option to prevent synaptic pathogenesis but block amyloid plaque formation in AD (Vassar et al., 2009).
Acknowledgments
This work was supported by the Illinois Department of Public Health (X.X.Y.), the National Natural Science Foundation of China (#81171091 to X.X.Y. and #81171160 to X.G.L), Hunan Natural Science Foundation (11jj5072 to J.M.L) and the intramural research program of National Institute on Aging, NIH (Z01-IAAG000944-04 to H.C.). We thank Elan Pharmaceuticals for providing the 3D6 antibody.
Contributor Information
Jian-Ming Li, Neuroscience Research Center, Changsha Medical University, Changsha, Hunan 410219, China. Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China.
Zhi-Qin Xue, Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China. Department of Anatomy, Xinjiang Medical University, Urumqi, Xinjiang 830011, China.
Si-Hao Deng, Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China.
Xue-Gang Luo, Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China.
Peter R. Patrylo, Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University School of Medicine, Carbondale, IL 62901, USA
Gregory W. Rose, Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University School of Medicine, Carbondale, IL 62901, USA
Huaibin Cai, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD 20892, USA.
Yan Cai, Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China.
Xiao-Xin Yan, Email: yanxiaoxin@csu.edu.cn, Department of Anatomy and Neurobiology, Central South University Xiangya School of Medicine, Changsha, Hunan 410013, China. Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University School of Medicine, Carbondale, IL 62901, USA.
References
- Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I. Immunohistochemical visualization of amyloid-beta protein precursor and amyloid-beta in extra- and intracellular compartments in the human brain. J Alzheimers Dis. 2010;20:1015–1028. doi: 10.3233/JAD-2010-091681. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Titus-Mitchell HE, Bullinger KL, Kraszpulski M, Nardelli P, Cope TC. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J Neurophysiol. 2011;106:2450–2470. doi: 10.1152/jn.01095.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brännström T, Havton L, Kellerth JO. Changes in size and dendritic arborization patterns of adult cat spinal alpha-motoneurons following permanent axotomy. J Comp Neurol. 1992;318:439–451. doi: 10.1002/cne.903180408. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Brownstone RM, Bui TV. Spinal interneurons providing input to the final common path during locomotion. Prog Brain Res 2010. 2010;187:81–95. doi: 10.1016/B978-0-444-53613-6.00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumovsky P, Watanabe M, Hökfelt T. Expression of the vesicular glutamate transporters-1 and -2 in adult mouse dorsal root ganglia and spinal cord and their regulation by nerve injury. Neuroscience. 2007;147:469–490. doi: 10.1016/j.neuroscience.2007.02.068. [DOI] [PubMed] [Google Scholar]
- Bugiani O, Giaccone G, Frangione B, Ghetti B, Tagliavini F. Alzheimer patients: preamyloid deposits are more widely distributed than senile plaques throughout the central nervous system. Neurosci Lett. 1989;103:263–268. doi: 10.1016/0304-3940(89)90110-9. [DOI] [PubMed] [Google Scholar]
- Cai Y, Xiong K, Zhang XM, Cai H, Luo XG, Feng JC, Clough RW, Struble RG, Patrylo PR, Chu Y, Kordower JH, Yan XX. β-Secretase-1 elevation in aged monkey and Alzheimer’s disease human cerebral cortex occurs around the vasculature in partnership with multisystem axon terminal pathogenesis and β-amyloid accumulation. Eur J Neurosci. 2010;32:1223–1238. doi: 10.1111/j.1460-9568.2010.07376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Zhang XM, Macklin LN, Cai H, Luo XG, Oddo S, Laferla FM, Struble RG, Rose GM, Patrylo PR, Yan XX. BACE1 elevation is involved in amyloid plaque development in the triple transgenic model of Alzheimer’s disease: Differential Aβ antibody labeling of early-onset axon terminal pathology. Neurotox Res. 2012a;21:160–174. doi: 10.1007/s12640-011-9256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Xue ZQ, Zhang XM, Li MB, Wang H, Luo XG, Cai H, Yan XX. An age-related axon terminal pathology around the first olfactory relay that involves amyloidogenic protein overexpression without plaque formation. Neuroscience. 2012b;215:160–173. doi: 10.1016/j.neuroscience.2012.04.043. [DOI] [PubMed] [Google Scholar]
- Chiang PK, Lam MA, Luo Y. The many faces of amyloid beta in Alzheimer’s disease. Curr Mol Med. 2008;8:580–584. doi: 10.2174/156652408785747951. [DOI] [PubMed] [Google Scholar]
- Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12:1–12. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alton S, George DR. Changing perspectives on Alzheimer’s disease: thinking outside the amyloid box. J Alzheimers Dis. 2011;25:571–581. doi: 10.3233/JAD-2011-110089. [DOI] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol. 2007;114:551–571. doi: 10.1007/s00401-007-0284-8. [DOI] [PubMed] [Google Scholar]
- Figurski MJ, Waligórska T, Toledo J, Vanderstichele H, Korecka M, Lee VM, Trojanowski JQ, Shaw LM Alzheimer’s Disease Neuroimaging Initiative . Improved protocol for measurement of plasma β-amyloid in longitudinal evaluation of Alzheimer’s Disease Neuroimaging Initiative study patients. Alzheimers Dement. 2012;8:250–260. doi: 10.1016/j.jalz.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata S, Fujishige S, Araki Y, Taniguchi M, Urakami K, Peskind E, Akatsu H, Araseki M, Yamamoto K, Martins RN, Maeda M, Nishimura M, Levey A, Chung KA, Montine T, Leverenz J, Fagan A, Goate A, Bateman R, Holtzman DM, Yamamoto T, Nakaya T, Gandy S, Suzuki T. Alternative processing of γ-secretase substrates in common forms of mild cognitive impairment and Alzheimer’s disease: evidence for γ-secretase dysfunction. Ann Neurol. 2011;69:1026–1031. doi: 10.1002/ana.22343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Niidome T, Hongo H, Akaike A, Kihara T, Sugimoto H. Impaired muscarinic regulation of excitatory synaptic transmission in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Eur J Pharmacol. 2008;583:84–91. doi: 10.1016/j.ejphar.2008.01.030. [DOI] [PubMed] [Google Scholar]
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2012;33:196. e29–40. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, Epis R, Saraceno C, Di Luca M. Synaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol. 2012;970:573–601. doi: 10.1007/978-3-7091-0932-8_25. [DOI] [PubMed] [Google Scholar]
- Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–1933. doi: 10.1200/JCO.2010.32.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Barua N, Kehoe PG, Gill S, Love S. Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70:944–959. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- Navarro X, Vivó M, Valero-Cabré A. Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol. 2007;82:163–201. doi: 10.1016/j.pneurobio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- The amyloid cascade hypothesis has misled the pharmaceutical industry. Biochem Soc Trans. 2011;39:920–923. doi: 10.1042/BST0390920. [No authors listed] [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogomori K, Kitamoto T, Tateishi J, Sato Y, Suetsugu M, Abe M. Beta-protein amyloid is widely distributed in the central nervous system of patients with Alzheimer’s disease. Am J Pathol. 1989;134:243–251. [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okereke OI, Xia W, Selkoe DJ, Grodstein F. Ten-year change in plasma amyloid beta levels and late-life cognitive decline. Arch Neurol. 2009;66:1247–1253. doi: 10.1001/archneurol.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, Frisardi V, Imbimbo BP, Seripa D, Solfrizzi V, Pilotto A. Monoclonal antibodies against β-amyloid (Aβ) for the treatment of Alzheimer’s disease: the Aβ target at a crossroads. Expert Opin Biol Ther. 2011;11:679–686. doi: 10.1517/14712598.2011.579099. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol. 2009;41:1261–1268. doi: 10.1016/j.biocel.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209:889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PK, Odlozinski M. Expansion of the dendritic tree of motoneurons innervating neck muscles of the adult cat after permanent axotomy. J Comp Neurol. 1998;390:392–411. doi: 10.1002/(sici)1096-9861(19980119)390:3<392::aid-cne7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13236. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JN, Suner S, Donoghue JP. Dynamic organization of primary motor cortex output to target muscles in adult rats. I. Long-term patterns of reorganization following motor or mixed peripheral nerve lesions. Exp Brain Res. 1990;79:479–491. doi: 10.1007/BF00229318. [DOI] [PubMed] [Google Scholar]
- Seo JS, Leem YH, Lee KW, Kim SW, Lee JK, Han PL. Severe motor neuron degeneration in the spinal cord of the Tg2576 mouse model of Alzheimer disease. J Alzheimers Dis. 2010;21:263–276. doi: 10.3233/JAD-2010-091528. [DOI] [PubMed] [Google Scholar]
- Seppälä TT, Koivisto AM, Hartikainen P, Helisalmi S, Soininen H, Herukka SK. Longitudinal changes of CSF biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2011;25:583–594. doi: 10.3233/JAD-2011-101911. [DOI] [PubMed] [Google Scholar]
- Silverberg GD, Miller MC, Messier AA, Majmudar S, Machan JT, Donahue JE, Stopa EG, Johanson CE. Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol. 2011;69:98–108. doi: 10.1097/NEN.0b013e3181c8ad2f. [DOI] [PubMed] [Google Scholar]
- Spires-Jones T, Knafo S. Spines, plasticity, and cognition in Alzheimer’s model mice. Neural Plast. 2012;2012:319836. doi: 10.1155/2012/319836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struble RG, Ala T, Patrylo PR, Brewer GJ, Yan XX. Is brain amyloid production a cause or a result of dementia of the Alzheimer’s type? J Alzheimers Dis. 2010;22:393–399. doi: 10.3233/JAD-2010-100846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, D’Adamio L. β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med. 2012;4:171–179. doi: 10.1002/emmm.201100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp Neurol. 2012;236:1–5. doi: 10.1016/j.expneurol.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Vanden Noven S, Wallace N, Muccio D, Turtz A, Pinter MJ. Adult spinal motoneurons remain viable despite prolonged absence of functional synaptic contact with muscle. Exp Neurol. 1993;123:147–156. doi: 10.1006/exnr.1993.1147. [DOI] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Wegiel J, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimäki T, Kivimäki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. 2007;113:389–402. doi: 10.1007/s00401-006-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wirths O, Weis J, Kayed R, Saido TC, Bayer TA. Age-dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging. 2007;28:1689–1699. doi: 10.1016/j.neurobiolaging.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yao Z, Chai H, Wong WM, Wu W. Potential roles of Alzheimer precursor protein A4 and beta-amyloid in survival and function of aged spinal motor neurons after axonal injury. J Neurosci Res. 2003;73:557–564. doi: 10.1002/jnr.10667. [DOI] [PubMed] [Google Scholar]
- Xiong K, Cai H, Luo XG, Struble RG, Clough RW, Yan XX. Mitochondrial respiratory inhibition and oxidative stress elevate beta-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp Brain Res. 2007;181:435–446. doi: 10.1007/s00221-007-0943-y. [DOI] [PubMed] [Google Scholar]
- Xu QG, Forden J, Walsh SK, Gordon T, Midha R. Motoneuron survival after chronic and sequential peripheral nerve injuries in the rat. J Neurosurg. 2010;112:890–899. doi: 10.3171/2009.8.JNS09812. [DOI] [PubMed] [Google Scholar]
- Yan XX, Cai Y, Zhang XM, Luo XG, Cai H, Rose GM, Patrylo PR. BACE1 elevation is associated with aberrant limbic axonal sprouting in epileptic CD1 mice. Exp Neurol. 2012;235:228–237. doi: 10.1016/j.expneurol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelano J, Berg A, Thams S, Hailer NP, Cullheim S. SynCAM1 expression correlates with restoration of central synapses on spinal motoneurons after two different models of peripheral nerve injury. J Comp Neurol. 2009;517:670–682. doi: 10.1002/cne.22186. [DOI] [PubMed] [Google Scholar]
- Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lee DH. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist. 2011:17163–173. doi: 10.1177/1073858410381532. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Cai Y, Xiong K, Cai H, Luo XG, Feng JC, Clough RW, Struble RG, Patrylo PR, Yan XX. Beta-secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci. 2009;30:2271–2283. doi: 10.1111/j.1460-9568.2009.07017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Xiong K, Cai Y, Cai H, Luo XG, Feng JC, Clough RW, Patrylo PR, Struble RG, Yan XX. Functional deprivation promotes amyloid plaque pathogenesis in Tg2576 mouse olfactory bulb and piriform cortex. Eur J Neurosci. 2010;31:710–721. doi: 10.1111/j.1460-9568.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]