

Abstract
Differences in chromatin organization are key to the multiplicity of cell states that arise from a single genetic background, yet the landscapes of in vivo tissues remain largely uncharted. Here we mapped chromatin genome-wide in a large and diverse collection of human tissues and stem cells. The maps yield unprecedented annotations of functional genomic elements and their regulation across developmental stages, lineages, and cellular environments. They also reveal global features of the epigenome, related to nuclear architecture, that also vary across cellular phenotypes. Specifically, developmental specification is accompanied by progressive chromatin restriction as the default state transitions from dynamic remodeling to generalized compaction. Exposure to serum in vitro triggers a distinct transition that involves de novo establishment of domains with features of constitutive heterochromatin. We describe how these global chromatin state transitions relate to chromosome and nuclear architecture, and discuss their implications for lineage fidelity, cellular senescence and reprogramming.
INTRODUCTION
Since the initial sequencing of the human genome a decade ago, our understanding of the primary DNA sequence has advanced profoundly (Lander, 2011). Sequence signals and multi-species conservation have enabled precise annotation of protein coding genes and the identification of increasing numbers of non-coding RNAs, regulatory elements and motifs. Systematic genotyping studies have identified common variants associated with complex diseases and recurrent mutations that confer growth advantage in cancer.
However, entirely sequence-directed investigations cannot address the fundamental question of how one genome can give rise to a large and phenotypically-diverse collection of cells and tissues during embryonic development. Nor can they explain how environmental conditions further shape these phenotypes and affect disease risks (Feinberg, 2007). An understanding of the regulatory networks and epigenetic mechanisms that underlie context-specific gene expression programs and cellular phenotypes remains a critical scientific goal with broad implications for human health.
Genomic DNA is organized into chromatin, which adopts characteristic configurations when DNA interacts with transcription factors (TFs), RNA polymerase or other regulators (Margueron and Reinberg, 2010). Charting these configurations with genome-wide maps of histone modifications (`chromatin state maps') thus represents an effective means for identifying functional DNA elements and assessing their activities in a given cell population (Zhou et al., 2010). Signature patterns of `active' chromatin marks demarcate poised or active promoters, transcribed regions and candidate enhancers. Other modifications reveal distinct modes of chromatin repression, such as those mediated by Polycomb regulators or heterochromatin proteins.
Recent studies have applied chromatin profiling to characterize enhancer dynamics and epigenetic regulatory mechanisms in differentiation, cellular reprogramming and disease processes (Ernst et al., 2011; Hawkins et al., 2010b; The ENCODE Project Consortium, 2012). However, the overwhelming focus of such studies on in vitro cells has constrained our ability to detect and characterize regulatory elements in the human genome, and to understand how global features of the epigenome impact cellular phenotypes across different lineages, developmental stages and environmental conditions.
Here we present a resource of over 300 chromatin state maps for a phenotypically-diverse collection of human tissues, blood lineages and stem cells, produced by the NIH Roadmap Epigenomics Mapping Consortium (Bernstein et al., 2010). The maps depict the distributions of major histone modifications and provide a systematic view of the dynamic chromatin landscapes of in vivo tissues. We use the maps to identify and characterize ~400,000 cell type-specific distal regulatory elements, many of which can be tied to upstream TFs or signaling pathways, and whose activity patterns provide a precise fingerprint of cell phenotype. We also describe global chromatin state transitions that distinguish groups of cells representative of different developmental stages or environmental conditions, and investigate their implications for lineage fidelity, nuclear architecture, cellular senescence and reprogramming. This extensive catalog of in vivo chromatin states thus presents a unique resource of genomic annotations for biomedical research, along with novel epigenetic features that vary markedly across cellular states.
RESULTS
Charting chromatin landscapes of human tissues, blood lineages and stem cells
We acquired chromatin state maps for 29 tissues and cell types spanning a wide range of developmental stages, lineages and derivations (Figure 1A). We used chromatin immunoprecipitation and high-throughput sequencing (ChIP-seq) to map histone modifications associated with diverse regulatory and epigenetic functions, including H3K4me1 (H3 lysine 4 mono-methylation), H3K4me3, H3K9me3, H3K27me3, H3K36me3, H3K9ac (lysine 9 acetylation) and H3K27ac. Procedures were optimized for different tissue preparations and to accommodate for limiting samples (Experimental Procedures). We also incorporated datasets for in vitro cultured cells into our analysis (Ernst et al., 2011). The resource contains over 300 chromatin state maps that significantly expand coverage of the human epigenome (Table S1). All datasets were publically released upon verification at www.roadmapepigenomics.org and are also available at http://www.broadinstitute.org/pubs/epigenomicsresource.
Figure 1. Chromatin state maps for in vivo tissues, stem cells and primary culture models.
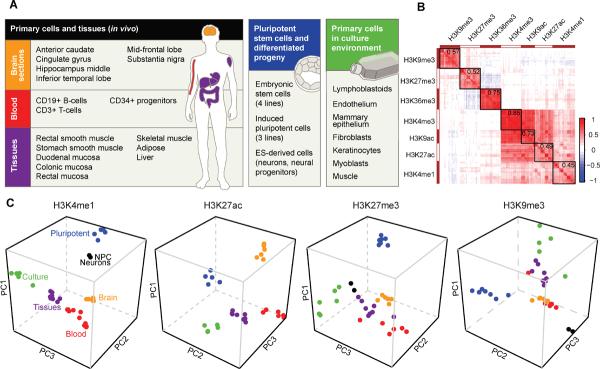
(A) Over 300 chromatin state maps were generated for human tissues, stem cells and cultured primary cells. In the schematic, tissues and cells with related phenotypes are grouped and color-coded. (B) Cross-correlation map generated by clustering ~200 histone modification profiles based on pair-wise correlations. Heat indicates degree of positive (red) or negative (blue) correlation between datasets. Mean correlation values over all datasets for each modification are indicated at the upper right of the corresponding block. (C) Projection plots show PCA coordinates for each tissue and cell type (colored as in `A'). The data indicate that coherent variations in the chromatin landscape distinguish cells from different developmental stages, lineages and growth environments. See also Figure S1 and Table S1
We applied automated methods to characterize the chromatin landscapes and relate them to underlying cellular phenotypes. First, we clustered the profiles based on pair-wise correlations (Figure 1B). The modifications organize into separate clusters, reflecting their associations to distinct genomic features. Modifications associated with promoter (H3K4me3, H3K9ac), transcript (H3K36me3) and distal element (H3K4me1, H3K27ac) activity correlate positively with one another, but show varying degrees of exclusivity with repressive marks (H3K27me3, H3K9me3).
Next, we used principal component analysis (PCA) to measure and visualize differences between cell types. Treating each histone modification separately, we computed weighted combinations of enrichment levels in genomic windows (PC1, PC2, and PC3) that capture a large proportion of the variation between cell types (Figure S1; Experimental Procedures). The PCA shows a striking capacity to segregate cells and tissues based on fundamental characteristics, as seen in three-dimensional projections of PC coordinates (Figure 1C). This is particularly evident for modifications associated with regulatory activity (H3K4me1, H3K27ac), which distinguish five groups of phenotypically-related tissue and cell types (see `Patterns and determinants…', below). PC projections for several modifications are notable for stark translations of certain cellular groups along PC1, indicative of major differences in chromatin state. In particular, the marked separation of pluripotent stem cells from other cell and tissue types evident in the H3K27me3 and H3K4me1 projections portends a global re-organization that accompanies developmental specification (see `Developmental…'). Furthermore, a clear separation of cultured cells in the H3K9me3 projection signifies a distinct re-organization induced by in vitro culture (see `Culture environments…'). The PCA also provides a general tool for comparing newly characterized cell types against representative chromatin state maps in this resource.
Patterns and determinants of regulatory activity and potential in the human genome
Enhancers and other distal regulatory elements are critical for context-specific gene regulation, but have yet to be systematically charted in primary human cells and tissues. Such elements are associated with characteristic chromatin marks, including H3K4me1 and H3K27ac, which facilitate their identification (Bulger and Groudine, 2011; Hawkins et al., 2010b).
We annotated candidate regulatory elements by calling H3K4me1 peaks in 30 cell types. After filtering out peaks that overlap a transcription start site (TSS), we identified an average of ~94,000 distal sites per cell type. Integrating all sites marked by H3K4me1 in at least two cell types reveals ~377,000 putative distal regulatory elements, with a median size of 1.2 kb. The elements are highly tissue-specific, with 56% marked in 3 or fewer cell types. Clustering on H3K4me1 patterns revealed 23 major clusters of elements with related cell type-specificities (Figure 2A). The biological relevance of individual clusters is supported by the identities of proximal genes, which are expressed at higher levels in the corresponding cell types and enriched for related functional annotations (Table S2; Experimental Procedures). Roughly half of all H3K4me1 sites also carry H3K27ac in at least a subset of cell types, and thus represent candidate enhancers (Figure 2B). Notably, nearly half of the candidate enhancers are specific to in vivo tissues, blood lineages or brain sections.
Figure 2. Genome-wide annotation of tissue-specific distal regulatory elements.
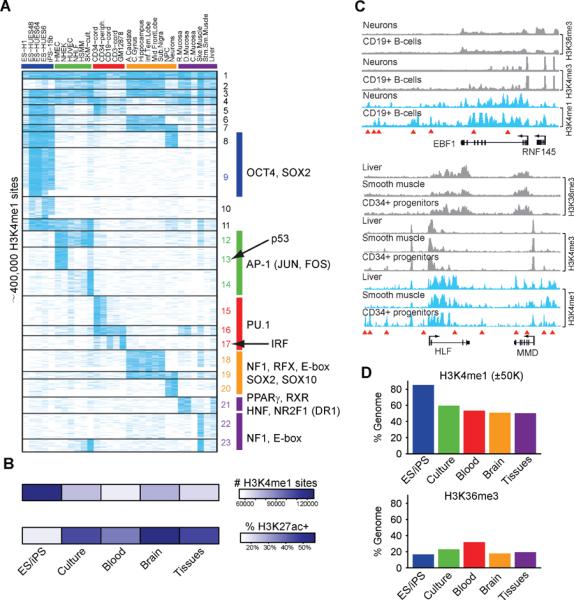
(A) Heatmap depicts H3K4me1 signals (blue) for ~400,000 distal elements (rows) across cell types (columns) arranged by phenotypic groups as in Figure 1A. Clusters of distal elements with similar cell type-specificities (horizontal lines) are enriched for the indicated TF motifs (right). The data emphasize the importance of tissue diversity for understanding distal regulatory elements, a large fraction of which is specific to in vivo tissues. (B) Heatmap indicates the total number of H3K4me1 sites (top) and fraction of H3K4me1 sites that are also enriched for H3K27ac (bottom). Values represent averages for each cell group. (C) H3K36me3, H3K4me3 and H3K4me1 signal tracks are shown for the EBF1 and HLF/MMD loci in the indicated cell types. Red triangles indicate cell type-specific H3K4me1 sites. Although EBF1 is expressed in neurons and CD19+ B-cells, distal H3K4me1 patterns vary markedly between these cell types. Similarly, HLF and MMD are expressed in liver, muscle and CD34+ progenitors, despite stark differences in distal element patterning. (D) Bar graphs show the proportions of genome within 50 kb of an H3K4me1 site (top) or within an H3K36me3 interval (bottom). Values represent averages for each cell group. The prevalence and distribution of H3K4me1 sites suggest that pluripotent cells have more accessible chromatin, but H3K36me3 patterns suggest that total gene activity is similar to other cells. See also Figure S2 and Table S2–S3.
To identify underlying sequence determinants, we scanned the candidate enhancers for TF consensus motifs. We identified significantly enriched motifs for each of the 23 clusters of commonly regulated elements (Figure 2A, Table S3; Experimental Procedures). The corresponding motif instances tend to be highly conserved and to coincide with dips in the chromatin profiles indicative of TF interactions (He et al., 2010). A sampling of predicted TF-motif interactions were also verified using TF binding data (Figure S2, (The ENCODE Project Consortium, 2012)). The data implicate known and potentially novel roles for specific TFs as regulators of cell type-specific distal elements and gene expression programs. To highlight one example, brain and neural stem cell-specific elements are enriched for NF1, RFX, SOX2, SOX10 and E-box motifs (Figure 2A; clusters 18–20). In several cases, a given TF motif is enriched in multiple un-related clusters, and thus implicated under distinct cellular contexts. A case in point is SOX2, a multifunctional TF with roles in pluripotent stem cells and neural lineages. The SOX2 motif is enriched in pluripotent- (cluster 9) and neural-specific (cluster 20) distal elements, suggesting that specificity among these clusters may involve proximal sequence signals. Indeed, 25% of SOX2 motifs in `pluripotent' elements coincide with OCT4 motifs, while a majority of SOX2 motifs in `neural' elements instead coincide with PAX2 motifs. Further complexity is evident at the level of enhancer usage as many loci contain multiple elements whose activity patterns vary even between cell types in which nearby genes are active (Figure 2C). These and other examples suggest a prominent role for combinatorial TF activities and complex distal element patterning in directing gene expression programs in human cells (Bulger and Groudine, 2011).
In addition to lineage-specific TFs, the motif enrichments implicate signaling and environmental response pathways activated in specific contexts. Clusters of distal elements broadly associated with primary cells cultured in serum are enriched for motifs recognized by AP-1 (clusters 12–14), a classical integrator of extrinsic growth stimuli and environmental stress (Angel and Karin, 1991). A related cluster is also enriched for the p53 motif (cluster 13), consistent with documented activation of p53 senescence programs in these primary cell models (Rheinwald et al., 2002).
The large number and diverse activity patterns of distal elements prompted us to examine their global distributions. We calculated the proportion of the genome that lies within 50 kb of an H3K4me1+ element in each cell type. This proportion remains relatively constant at ~50% across the differentiated tissues and cell types. However, H3K4me1 sites are more prevalent and dispersed in pluripotent cells, such that a full 85% of the genome is within 50 kb of a site (Figure 2D). This pattern does not appear to reflect increased gene activity as the proportion of genome marked by H3K36me3 in pluripotent cells (~17%) is similar to the average for differentiated cells (~23%). Moreover, the proportion of elements with concomitant H3K27ac is notably lower for H3K4me1 sites in pluripotent stem cells (Figure 2B), indicating that many may represent `poised' enhancers or other sites of accessible chromatin. Thus, much of the genome in pluripotent cells appears to be coincident or proximal to accessible chromatin.
Pluripotent cell chromatin is refractory to Polycomb-repressed state
The dramatically reduced prevalence of accessible chromatin in differentiated cells prompted us to test for concomitant changes in repressive chromatin. We focused on marks associated with Polycomb-repression (H3K27me3) and constitutive heterochromatin (H3K9me3). Both modifications contribute to stable gene repression through interactions with protein complexes involved in chromatin compaction (Margueron and Reinberg, 2010; Simon and Kingston, 2009).
The PCA statistics (Figure 1C) indicate that, like H3K4me1, global H3K27me3 patterns are distinct in pluripotent cells. Indeed, we find that the changes in H3K4me1 patterns are complemented by a profound reorganization of the H3K27me3 landscape. In embryonic (ES) and induced pluripotent stem (iPS) cells, this surrogate of Polycomb activity is confined to peaks at `bivalent' GC-rich promoters that also carry H3K4me3. This punctate pattern contrasts with a much broader distribution of H3K27me3 in differentiated cells and tissues (Figure 3A). We used a customized approach to quantify coverage in each cell type (Figure 3B and S3; Experimental Procedures). This confirmed that H3K27me3 affects a considerably larger proportion of genome in differentiated cells (~40%) than pluripotent cells (~8%). This dramatic shift is supported by western blots showing that acid extracted histones from differentiated cells have higher H3K27me3 levels than similar extracts from ES cells (Figure S3).
Figure 3. Global chromatin restriction during developmental specification.
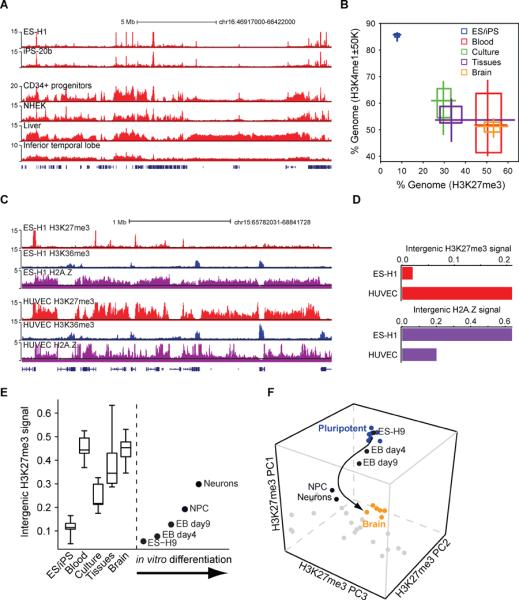
(A) H3K27me3 signal tracks for a 20 Mb region of chromosome 16 are shown for pluripotent (top) and differentiated (bottom) cell types. (B) Two-dimensional box plot compares the fraction of genome within 50 kb of an H3K4me1 site (y-axis) and the proportion of genome enriched for H3K27me3 (x-axis). For each phenotypic cell group, shaded squares and midpoint indicate 25th, 50th and 75th percentiles, while the crosses designate minimum and maximum values. Specification is accompanied by marked restriction of accessible chromatin and increased prevalence of the Polycomb-repressed state. (C) H3K27me3, H3K36me3 and H2A.Z signal tracks for a 7 Mb region of chromosome 15 are shown for ES cells and endothelial cells. (D) Bar plot contrasts normalized intergenic H2A.Z and H3K27me3 signal distributions in ES and differentiated cells (see Figure S3). These normalized signal distributions are a sensitive indicator of chromatin state transitions. H2A.Z is distributed broadly in pluripotent cells, indicative of genome-wide remodeling, but is confined to sites of regulatory activity in differentiated cells. (E) Box plots indicate normalized intergenic H3K27me3 signal distributions for cells in each phenotypic group. Boxes indicate 25th, 50th and 75th percentiles, while whiskers indicate minimum and maximum values. Corresponding values for ES cell derivatives are indicated at right. (F) H3K27me3 profiles for ES cell derivatives are projected onto PC space as in Figure 1C. The arrow emphasizes the developmental progression. See also Figure S3.
The focal distribution of H3K27me3 in pluripotent cells could reflect reduced Polycomb activity. However, EZH2 and other Polycomb factors are highly expressed in ES cells. This led us to consider an alternate model in which the highly dynamic chromatin in pluripotent cells (Meshorer et al., 2006) is refractory to the compaction associated with Polycomb repression. To explore this, we mapped H2A.Z, a histone variant associated with nucleosome exchange and remodeling (Talbert and Henikoff, 2010), in representative cell types (Figure 3C–D and S3). Consistent with prior studies (Hardy et al., 2009), H2A.Z is depleted within elongating transcripts in all cell types examined. However, the global H2A.Z distribution diverges markedly between pluripotent and differentiated cells. In ES and iPS cells, the variant marks promoters and distal elements, but is also distributed throughout intergenic regions. In differentiated cells, H2A.Z is instead confined to promoters and distal elements. The broad H2A.Z distribution suggests that chromatin exchange is prevalent throughout the genome in embryonic cells. Although H2A.Z may be compatible with punctate Polycomb sites in ES cells (Creyghton et al., 2008), dynamic chromatin is likely incompatible with the stable interactions required for Polycomb spreading and compaction (Simon and Kingston, 2009). Thus, pervasive exchange may underlie the constrained H3K27me3 distribution and the uniquely accessible chromatin landscape in pluripotent cells.
Developmental specification is associated with progressive chromatin restriction
To gain insight into the timing of the developmental transition, we profiled H3K27me3 in a series of cell populations representing successive stages of specification. These include (i) embryoid bodies (EBs) isolated from differentiating ES cells at day 4; (ii) EBs isolated at day 9; (iii) neural progenitors derived from ES cells through a three-week differentiation procedure; and (iv) neurons differentiated from these progenitors. We found that ES cell differentiation is accompanied by progressive enrichment of H3K27me3 across the genome, with subtle but significant changes in EBs, and profound alterations in neural progenitors and neurons (Figure 3E and 3F). The latter populations exhibit a diffuse H3K27me3 distribution akin to other differentiated cell types. This suggests that re-organization of the chromatin landscape begins early in development and is recapitulated by in vitro differentiation of ES cells.
We next examined the locations and characteristics of genomic loci that gain H3K27me3 in the differentiated populations. We focused on a set of ~3000 loci (100 kb size) with variable activity across the phenotypic groups. We scored each locus for (i) distal element H3K4me1 levels, (ii) promoter H3K4me3 levels, (iii) transcript H3K36me3 levels, and (iv) overall H3K27me3 levels within each phenotypic group, and clustered them accordingly (Figure 4A; Experimental Procedures). The resulting cluster diagram conveys the variable patterning of these loci. Promoter, transcript and distal element activities are highly concordant within a given locus, but correlate negatively with the extent of H3K27me3 coverage. These patterns provide a systematic view of how Polycomb-repressed chromatin is engaged in specific lineages to maintain silencing of gene loci with functions in alternate lineages.
Figure 4. Epigenetic states relate to context-specific genome regulatory programs.
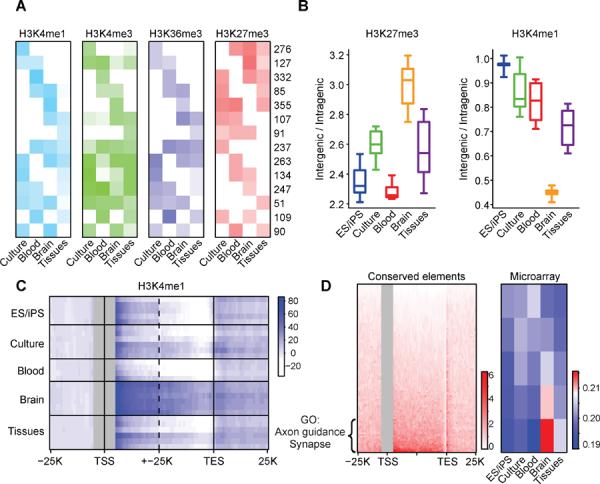
(A) Chromatin states are depicted for a set of 100 kb loci with variable activity patterns. Rows correspond to clusters of loci with similar cell type-specificities. Heatmap depicts relative levels of H3K4me1 over distal elements (light blue), H3K4me3 over promoters (green), H3K36me3 over gene bodies (dark blue), and overall H3K27me3 (red) for each cluster in the indicated phenotypic group. Numerical values indicate the number of loci in each cluster. Promoter, gene and distal element activities are largely concordant within a locus, but are exclusive with H3K27me3. (B) Boxplots show H3K27me3 coverage of intergenic regions relative to gene bodies (left), and H3K4me1 peak density in intergenic regions relative to gene bodies (right). Brain sections are notable for a high prevalence of repressive chromatin throughout intergenic regions and a relative confinement of H3K4me1 sites within genes. (C) Heatmap shows composite H3K4me1 profiles over genes and flanking regions (TSS ± 15 kb and TES ± 15 kb; all genes > 15 kb) for each cell type (rows). Brain sections display higher H3K4me1 signals in gene bodies, even when TSS proximal regions (± 5 kb) are masked (gray). (D) Heatmap (left panel) shows the distribution of highly-conserved non-coding sequence elements over gene bodies and flanking regions (TSS ± 25 kb and TES ± 25 kb) for all genes >15 kb (rows). The genes are ordered according to the density of conserved elements within their introns. The top quintile of genes (n=1760) is strongly enriched for functional annotations related to brain physiology, including axon guidance (p < 10−13) and synapse (p < 10−9), and exhibit higher RNA expression in brain (heatmap at right; red indicates high expression; blue indicates low expression). These data suggest that a highly restrictive chromatin structure in specialized brain cells favors access to intronic regions and may influence the function of sequence elements that mediate corresponding regulatory programs. See also Figure S3.
Chromatin restriction can also proceed beyond this initial developmental re-organization, under certain contexts. In particular, the brain sections exhibit uniquely high H3K27me3 coverage over intergenic regions, relative to annotated genes (Figure 4B). Expansion of the Polycomb-repressed state is accompanied by a dramatic restriction of accessible chromatin, such that ~70% of H3K4me1 sites in brain reside within transcriptional units (Figure 4B and 4C). To test the generality of this finding, we examined H3K4me1 profiles for assorted mouse tissues (Shen et al., 2012). Out of 19 cell and tissue type examined, we found that cerebellum and cortex have the highest proportions of H3K4me1 sites within genic regions (Figure S3). The brain sections are unique among tissues in the resource in that they comprise specialized, terminally differentiated cell types – primarily neurons and glial cells. We reasoned that the restrictive chromatin environment in these cells might obstruct access to intergenic sequences, and thus favor recognition of functional elements within introns. In support of this possibility, we find that the genes with a high density of conserved non-coding sequences in their introns are expressed at higher levels in brain and enriched for functional annotations related to neuronal physiology (Figure 4D; Experimental Procedures).
In summary, our data suggest that developmental specification is accompanied by a striking transition from a permissive chromatin state with widespread remodeling to a restrictive state with pervasive Polycomb repression. Chromatin restriction may also proceed significantly further in certain specialized cells, with brain sections in particular exhibiting severe sequestration of intergenic sequences and evidence for preferential utilization of regulatory elements within introns.
Relating macro-scale chromatin features to nuclear architecture
The coarse partitioning of the genome between loci of coordinated gene regulatory activity and large Polycomb-repressed regions prompted us to investigate macro-scale patterns of histone modification. Focusing on ~3000 1 megabase intervals, we quantified relative coverage by each modification, and clustered the intervals accordingly. We found that most intervals are dominated by a single coherent chromatin state, allowing us to segregate them into four groups: (1) `active' loci with high H3K36me3 and H3K4me1 coverage; (2) `Polycomb-repressed' loci with high H3K27me3; (3) Heterochromatic loci with high H3K9me3; and (4) `Null' loci devoid of histone modification (Figure 5).
Figure 5. Macro-scale chromatin features and nuclear architecture.
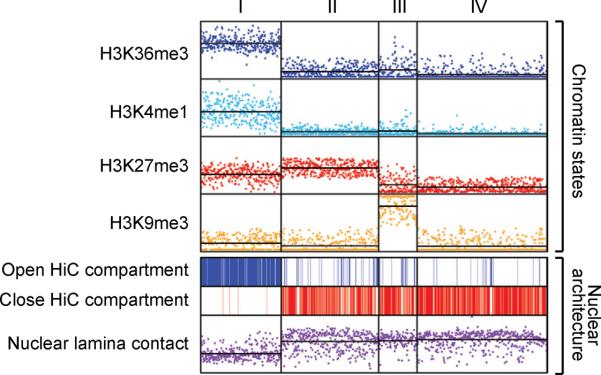
Non-overlapping 1 Mb genomic windows (n=2725) were clustered by their coverage by four histone modifications. Roughly half of the windows clustered into four main states: (I) `Active' loci with high H3K36me3 and H3K4me1; (II) `Polycomb-repressed' loci with high H3K27me3; (III) Heterochromatic loci with high H3K9me3; and (IV) `Null' loci devoid of histone modification. The top panel shows coverage for each interval (data points) and average coverage for each cluster (horizontal lines) by the indicated modification. The bottom panel indicates intervals that occupy active (top) or inactive (middle) nuclear compartments, and shows enrichment for nuclear lamina contacts (bottom). The data relate macro-scale modification patterns to genome compartmentalization and nuclear architecture.
Several lines of evidence suggest that the macro-scale chromatin patterns reflect chromosomal and nuclear architecture. The chromatin states align with chromosome banding patterns, with active (1) and inactive states (2–4) respectively enriched in light and dark bands. All three inactive states are also enriched for contacts with the nuclear lamina (Guelen et al., 2008). We also examined the relationship between the macro-scale chromatin domains and chromosomal interactions (Lieberman-Aiden et al., 2009). The interaction maps indicate that the genome can be partitioned into two compartments such that contacts within each compartment are enriched and contacts between compartments are depleted. We found that the chromatin state-based classification scheme mirrors the HiC compartmentalization, with active states coinciding with one compartment and repressive states with the other (Figure 5). In fact, the chromatin states enable prediction of HiC compartments with an overall accuracy of 83%. These correspondences suggest that macro-scale chromatin patterns are reflective of chromosomal and nuclear architecture and that the compendium of chromatin maps may thereby provide insight into architectural changes between cellular states.
Culture environments trigger macro-scale chromatin state changes
Although the macro-scale chromatin patterns are each recovered in all of the differentiated cell types, one configuration – the pure H3K9me3 state – varies markedly in its prevalence. It is rare in brain sections and other in vivo models, but ~50-fold more prevalent in cultured primary cells (Figure S4). Visual inspection of the profiles confirms expansive regions of modest but contiguous H3K9me3 enrichment that are particularly pronounced in cultured cells (Figure 6A). We called H3K9me3-enriched intervals in each cell type and merged overlapping intervals to collate a set of 296 domains (median size 1.4 Mb). We then calculated normalized H3K9me3 signals for each domain in each cell type, and clustered the domains accordingly (Experimental Procedures).
Figure 6. Macro-scale chromatin aberrations in cultured cells.
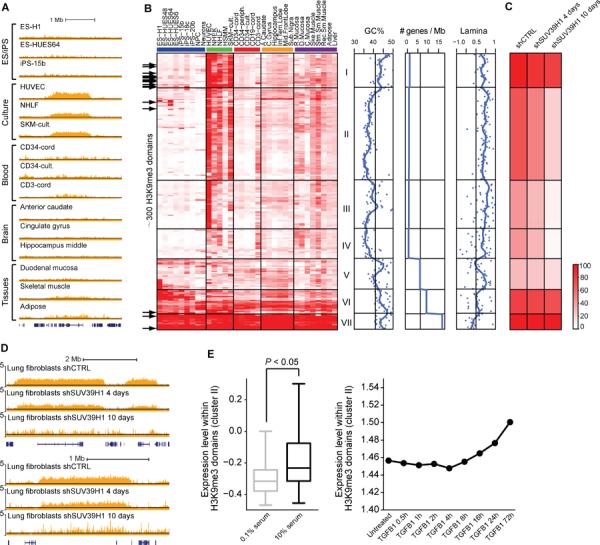
(A) H3K9me3 signal tracks for representative cell types are shown for a 3.5 Mb region of chromosome 16 that contains a culture-specific H3K9me3 domain. (B) Heatmap shows normalized H3K9me3 signals for 296 H3K9me3 domains (rows) in the indicated cell types (columns). The domains are clustered into seven groups based on their cell type-specificities. GC content, gene density and nuclear lamina contact enrichment are plotted for each cluster (right). Black arrows (left) indicate domains that coincide with loci found to have aberrant DNA methylation patterns in iPS cells (Lister et al., 2011). (C) For each cluster in (B), heatmap shows H3K9me3 signals in lung fibroblasts after 4 or 10 days of Suv39h1 knock-down. (D) H3K9me3 signal tracks for two culture-specific domains are shown for lung fibroblasts after 4 or 10 days of Suv39h1 knock-down. (E) Boxplot (left) shows expression levels of genes within culture-specific H3K9me3 domains (cluster II) in fibroblasts cultured in high or low serum. P-value of Wilcoxon rank-sum test is shown. Median expression levels for these genes are also shown for lung adenocarcinoma cells undergoing TGF-β-mediated EMT. See also Figure S4 and Table S4.
Two clusters comprise constitutive H3K9me3 domains (Figure 6B, clusters VI, VII; Table S4). The corresponding loci include olfactory receptor, zinc finger and protocadherin gene clusters and several imprinted loci, which have previously been associated with H3K9me3 (Magklara et al., 2011; O'Geen et al., 2007). The remaining clusters comprise domains with variable enrichments across the cell types. These domains tend to be near telomeres (cluster I), AT-rich (cluster II–IV), gene poor (clusters I–IV) and in contact with the nuclear lamina. Their H3K9me3 signals are most pronounced in in vitro cultured cells, such as endothelial cells (HUVEC), keratinocytes (NHEK) and fibroblasts (NHLF) (Figure 6B; clusters I–IV). Preferential association of variable H3K9me3 domains with the culture environment is further supported by a direct comparison of surgically resected skeletal muscle against cultured skeletal muscle cells (Figure S4).
Although they are representative of different lineages, the cell types with pronounced H3K9me3 domains are all grown as adherent cultures in the presence of serum or other potent growth stimuli. By contrast, hematopoietic cells grown in suspension and stem cells grown in defined media without serum lack the variable H3K9me3 domains. Growth stimuli have previously been linked to global chromatin changes. TGF-β-mediated epithelial-to-mesenchymal transition (EMT) leads to a global increase in euchromatin marks and a reduction in the lamina-associated modification H3K9me2 (Guelen et al., 2008; McDonald et al., 2011). Although we find that the genome-wide patterns of H3K9me3 and H3K9me2 are distinct (Figure S4), we nonetheless considered whether the culture-induced H3K9me3 domains might relate to EMT-like nuclear architecture changes. In support of this possibility, we found that inhibition of TGF-β signaling in WI-38 fibroblasts leads to a reduction in H3K9me3 domain signals (Figure S4). Furthermore, EMT and serum-exposure both lead to subtle increases in the median expression of genes underlying H3K9me3 domains (Figure 6E). Finally, we note that the H3K9me3 domains occupy the same inactive compartment as the nuclear lamina in the chromatin interaction data (Figure 5; Figure 6B). These findings are suggestive of a model in which growth stimuli in culture media promote alterations to nuclear architecture and lamina contacts that render formerly inert loci susceptible to H3K9me3 modification.
In addition to having related growth environments, the affected models are all non-transformed, primary cells that will undergo senescence-related growth arrest (Figure S4). Cellular senescence is also associated with global architecture changes – specifically, the formation of `senescence-associated heterochromatin foci (SAHFs)', which are DAPI-dense nuclear structures that stain for H3K9me3 and related chromatin markers (Adams, 2007). Moreover, oncogene-induced senescence is dependent on the H3K9 methyltransferase Suv39h1 (Braig et al., 2005). We tested whether Suv39h1 mediates the culture-specific domains by profiling H3K9me3 in fibroblasts after shRNA knock-down. We observed markedly reduced H3K9me3 in the culture-specific domains, indicating that Suv39h1 is required for their maintenance (Figure 6C, D). We suggest that context-specific induction of H3K9me3 domains by Suv39h1 in cells subjected to growth stimuli in non-physiologic environments may underlie their vulnerability to senescence-associated chromatin changes (see Discussion).
In contrast to the primary cell models, ES and iPS cells lack the variable H3K9me3 domains (Figure 6B). Since iPS cells are typically derived from cultured fibroblasts, this implies that chromatin in these regions is repaired during cellular reprogramming. However, evidence suggests that the domains may be repaired inefficiently and thus present an impediment to reprogramming. Specifically, recent studies have highlighted more than 20 `hotspots' of aberrant epigenomic reprogramming that frequently exhibit aberrant DNA methylation patterns in iPS cells, relative to ES cells (Lister et al., 2011; Ruiz et al., 2012). Remarkably, we find that essentially all of these hotspots overlap regions with H3K9me3 domains in differentiated cells (Figure 6B). Recent work has also shown that suppression of Suv39h1 enhances reprogramming (Onder et al., 2012). Thus, although H3K9me3 patterns are largely reset in iPS cells, the culture-specific domains may present a barrier to reprogramming and potentially retain a memory of architectural aberrations in the pre-programmed donor cell.
DISCUSSION
Discerning the principles by which a single genome can give rise to a multiplicity of cellular states remains a critical goal. Model organism studies have presented paradigms by which interactions among TFs, epigenetic regulators and genome sequence elements mediate lineage-specific gene expression and dynamic responses to environmental stimuli. Yet our understanding of these components and their functions in humans has lagged, with limited existing knowledge derived largely from in vitro models.
This study aimed to characterize systematically the sequence elements, regulators and epigenetic states that modulate human genome function in native contexts. We mapped chromatin states across a diverse collection of in vivo populations, including blood lineages, brain sections, gastrointestinal tissues, adipose, liver and muscle, and compared them to pluripotent and differentiating stem cells and other in vitro counterparts. We used the maps to locate and classify regulatory sequences, and to characterize large-scale epigenetic states and their dynamics across a breadth of cellular phenotypes.
The distal element annotations extend prior studies of in vitro cells with a broad survey of in vivo tissues. Integration of genome-wide profiles for H3K4me1 and H3K27ac across 29 cell and tissue types yielded nearly 400,000 putative distal elements, roughly half of which have enhancer-like chromatin patterns. The elements show exquisite tissue specificity, with most showing activity in just a few cell types. By analyzing their tissue-specificities and the underlying DNA sequences, we predict upstream factors that drive genome regulatory programs in specific cellular contexts. These include master regulator TFs that dictate specific lineages, as well as regulators such as AP1 whose activity patterns appear to reflect increased mitogenic signaling triggered by the culture environment. The large proportion of candidate enhancers specific to in vivo tissues should be valuable for human genetics given their potential to facilitate the identification and interpretation of causal sequence variants from genome-wide association studies (Ernst et al., 2011; Gaulton et al., 2010; The ENCODE Project Consortium, 2012).
Our study also helps resolve controversy regarding how chromatin is reorganized during developmental specification. Prior studies have been equivocal with regard to whether specification involves expansion of repressive chromatin domains or, alternatively, is dominated by localized state changes (Hawkins et al., 2010a; Lienert et al., 2011; Wen et al., 2009). We critically addressed this issue by comparing the distributions of multiple histone modifications and a marker of chromatin exchange across tissues and cells at various stages of commitment. We implemented a statistical model to quantify differences in the chromatin landscapes and verified inferences by measuring global modification levels with western blots.
We conclude that specification is accompanied by a stark transition in the epigenetic landscape from a uniquely accessible state to increasingly restrictive configurations (Figure 7). In embryonic cells, active and inactive loci both appear subject to dynamic chromatin remodeling that is likely incompatible with repressive chromatin compaction (Simon and Kingston, 2009). Accordingly, the Polycomb mark is largely confined to poised promoters in ES and iPS cells. Differentiated cells present an inverse pattern with chromatin exchange confined to loci under active regulation and much of the remaining landscape affected by Polycomb repression. Our findings build upon prior reports of expanded H3K27me3 domains in differentiated cells (Hawkins et al., 2010a; Pauler et al., 2009), and suggest a prominent role for hyper-dynamic chromatin in pluripotent cells as a hindrance to stable silencing (Meshorer et al., 2006). However, our analyses do not support prior claims that expanded H3K9me3 domains arise upon specification, as we find that such features are instead triggered by the culture environment (see below). Further study is needed to clarify how developmental and environmental cues alter H3K9me2 patterns, which appear distinct from the other repressive states.
Figure 7. Model for large-scale chromatin state transitions.
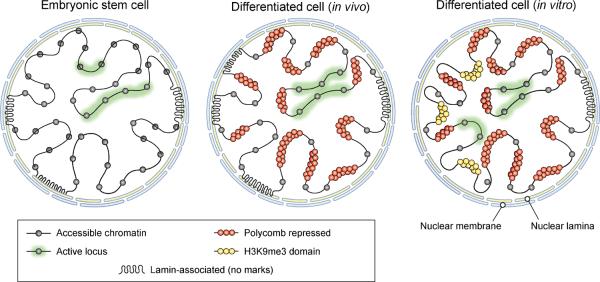
Illustration depicts large-scale chromatin patterns and their relative prevalence in cells from different developmental stages or environments. Although the amount of genome sequence engaged in gene regulatory activity is relatively consistent, the chromatin configuration of inactive regions varies considerably. In ES cells (left), inactive regions are diffusely enriched for markers of chromatin exchange and accessibility. In differentiated cells acquired in vivo (middle), inactive loci instead tend to adopt a Polycomb-repressed chromatin state. In differentiated cells cultured in vitro (right), large domains enriched for the heterochromatin marker H3K9me3 arise in regions associated with the nuclear lamina.
In certain contexts, chromatin restriction can proceed well beyond the primary transition that accompanies lineage commitment. Case in point is the brain sections wherein intergenic regions are almost entirely covered by Polycomb-repressed chromatin and, conversely, a large majority of accessible chromatin sites occur within genes. The brain samples are composed of neurons, glia and other highly specialized cell types that may be permissive to the accumulation of repressive chromatin. A corollary of the chromatin patterns is that intronic regulatory elements may be more accessible and more readily engaged in such cells. Indeed, we find that introns of genes with neuronal functions have a relatively higher density of conserved non-coding sequence elements, raising the provocative concept that epigenomic landscapes shape the evolution of genome sequence.
Chromatin architecture changes have also been associated with cellular responses to environmental cues, yet their influences on specific genomic loci have remained vague. Here we describe a set of megabase-sized H3K9me3 domains that arise in primary cultured cells, but which are rare or absent in tissues, blood lineages and stem cells (Figure 7). The domains likely reflect nuclear architecture changes as they correspond to gene-poor regions that contact the lamina and occupy the same inactive compartment. Prior indications that TGF-β-mediated EMT and cellular senescence perturb nuclear architecture prompted us to examine whether the culture-specific domains might relate to such processes (Adams, 2007; McDonald et al., 2011). We found that EMT modestly increases the expression of genes within the domains and, moreover, that TGF-β inhibition lowers their H3K9me3 signals. The domains are dependent on Suv39h1, a histone methyltransferase that mediates oncogene-induced senescence (Braig et al., 2005). Cellular senescence is associated with the formation of nuclear foci (SAHFs) that stain for H3K9me3 and are thought to arise by rearrangement of pre-existing regions of histone modification (Chandra et al., 2012). We speculate that H3K9me3 domains represent an initial response to growth stimuli and non-physiologic environments that primes cells for senescence-associated events. Regardless, the direct identification of genomic regions as candidate targets of EMT and pre-senescence changes should facilitate the study of processes that are fundamental to human health, aging and cancer.
The fate of culture-specific H3K9me3 domains during cellular reprogramming presents an interesting question. Although they are prominent in fibroblasts, the domains appear to be erased during reprogramming, as they are absent in iPS cells. Yet the domains contain within them essentially all of differentially-methylated regions found to distinguish iPS cells from ES cells, which have not undergone reprogramming (Lister et al., 2011; Ruiz et al., 2012). Somatic cell reprogramming is enhanced by inhibition of TGF-β signaling and suppression of the H3K9 methyltransferase Suv39h1 (Maherali and Hochedlinger, 2009; Onder et al., 2012). Moreover, H3K9me3 heterochromatin domains have recently been found to impede the initial binding of pluripotency TFs during this process (Soufi et al., 2012). These correspondences raise the intriguing possibility that H3K9me3 domains present a hindrance to reprogramming and possibly retain an epigenetic memory of pre-senescence changes in the donor cell. Although further study is clearly needed to appreciate their significance, the identification and initial characterization of these macro-scale chromatin aberrations provides a starting point for such investigations.
EXPERIMENTAL PROCEDURES
Data access
All datasets were publically released upon verification at www.roadmapepigenomics.org and are available at NCBI's GEO database (GSE17312, GSE19465 and GSE25249) and a dedicated website http://www.broadinstitute.org/pubs/epigenomicsresource.
Sample acquisition and production mapping
ES and iPS cell lines were cultured in serum-replacement media (Bock et al., 2011; Boulting et al., 2011). EBs were generated by in vitro differentiation of H9 ES cells in low attachment plates in EB differentiation media. Neural Progenitors and neurons were derived from H9 ES cells by in vitro differentiation for 3 and 5 weeks, respectively (Dhara and Stice, 2008). Blood lineages were isolated from cord or peripheral bloods. Liver, adipose, skeletal muscle and gastrointestinal tissues were harvested at surgical resection. Brain sections were obtained post-mortem within hours of death.
Tissue and cell preparations were subjected to ChIP-seq, as described (Adli et al., 2010; Ku et al., 2008). Aligned reads were used to derive 25-bp resolution density maps. Non-centered PCA was carried out on each modification separately based on the number of reads in all non-overlapping 1 kb windows. Detailed descriptions are presented in Supplemental Information.
Distal regulatory element and repressive chromatin states
We used a scanning window approach to call H3K4me1 and H3K27ac peaks (Guttman et al., 2010). After excluding sites within 2.5 kb of a TSS, we designated H3K4me1 sites as candidate distal elements and clustered (K-means) them by their cell type-specificities. We quantified their predictive power for gene activity by correlating the state of each element and the expression of the nearest TSS. We scanned the distal element clusters for over-represented TF motifs, and validated a sampling of predicted TF-enhancer interactions using published TF binding profiles (The ENCODE Project Consortium, 2012).
We quantified H3K27me3 and H3K36me3 distributions in each cell type by modeling foreground and background signals, and then used a scanning procedure to call intervals of contiguous enrichment. We clustered 2,976 100 kb intervals with variably expressed genesor bivalent chromatin in ES cells by their chromatin states to elucidate context-specific chromatin regulation and repression. We also clustered all 1 Mb windows based on their relative coverage by each modification to characterize macro-scale chromatin features. To collate H3K9me3 domains, we masked repeat elements and used a 100 kb window-scanning procedure to call intervals in each cell type. Detailed descriptions are presented in the Supplemental Information.
Suv39h1 knockdown and TGF-β inhibitor experiments
WI-38 fibroblasts were subjected to Suv39h1 knockdown with two shRNAs, as described (Onder et al., 2012). For TGF-β inhibition, WI-38 cells were treated with 2 μM or 4 μM TGF-β RI Kinase Inhibitor II (Calbiochem #616452) for 6 days. H3K9me3 was profiled by ChIP-seq, as described above.
Supplementary Material
A resource of chromatin state maps for phenotypically-diverse human tissues
Annotation of regulatory elements across development stages and environments
Developmental specification is accompanied by progressive chromatin restriction
Chromatin architecture changes in cultured cells have implications for reprogramming
ACKNOWLEDGEMENTS
We acknowledge members of the Broad Institute's Epigenomics Program and Genome Sequencing and Analysis Program, and the NIH Epigenomics Mapping Consortium for constructive comments. We thank Kevin Eggan for ES and iPS lines, Allen Powe and Steve Stice for neural cells, Greg Lauwers and the MGH Tissue Repository for tissue procurement, and David Flowers, Irwin Bernstein, John Stamatoyannopoulos and Shelly Heimfeld for blood samples. We also thank Leslie Gaffney and Lauren Solomon for assistance with figures. This research was supported by the NIH Common Fund (U01 ES017155), the National Human Genome Research Institute (U54 HG004570), the National Heart, Lung and Blood Institute (U01 HL100395), the National Institute on Aging (P30 AG10161), the Howard Hughes Medical Institute, the Starr Cancer Consortium and the Burroughs Wellcome Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene. 2007;397:84–93. doi: 10.1016/j.gene.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M, Zhu J, Bernstein BE. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nature methods. 2010;7:615–618. doi: 10.1038/nmeth.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008;135:649–661. doi: 10.1016/j.cell.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhara SK, Stice SL. Neural differentiation of human embryonic stem cells. J Cell Biochem. 2008;105:633–640. doi: 10.1002/jcb.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, et al. A map of open chromatin in human pancreatic islets. Nature genetics. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature biotechnology. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Jacques PE, Gevry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, Robert F. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009;5:e1000687. doi: 10.1371/journal.pgen.1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell stem cell. 2010a;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010b;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS genetics. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienert F, Mohn F, Tiwari VK, Baubec T, Roloff TC, Gaidatzis D, Stadler MB, Schubeler D. Genomic Prevalence of Heterochromatic H3K9me2 and Transcription Do Not Discriminate Pluripotent from Terminally Differentiated Cells. PLoS Genet. 2011;7:e1002090. doi: 10.1371/journal.pgen.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magklara A, Yen A, Colquitt BM, Clowney EJ, Allen W, Markenscoff-Papadimitriou E, Evans ZA, Kheradpour P, Mountoufaris G, Carey C, et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011;145:555–570. doi: 10.1016/j.cell.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nature reviews Genetics. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18:867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, Green R, Farnham PJ. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS genetics. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, Sommer A, Aszodi A, Jenuwein T, Barlow DP. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome research. 2009;19:221–233. doi: 10.1101/gr.080861.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, De Luca M, Catricala C, O'Toole KM. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol. 2002;22:5157–5172. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Diep D, Gore A, Panopoulos AD, Montserrat N, Plongthongkum N, Kumar S, Fung HL, Giorgetti A, Bilic J, et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:16196–16201. doi: 10.1073/pnas.1202352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature reviews Molecular cell biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell. 2012;151:1–11. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nature reviews Molecular cell biology. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nature genetics. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2010;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.