

Abstract
Biased codon usage in protein-coding genes is pervasive, whereby amino acids are largely encoded by a specific subset of possible codons. Within individual genes, codon bias is stronger at evolutionarily conserved residues, favoring codons recognized by abundant tRNAs. Although this observation suggests an overall pattern of selection for translation speed and/or accuracy, other work indicates that transcript structure or binding motifs drive codon usage. However, our understanding of codon bias evolution is constrained by limited experimental data on the fitness effects of altering codons in functional genes. To bridge this gap, we generated synonymous variants of a key enzyme-coding gene in Methylobacterium extorquens. We found that mutant gene expression, enzyme production, enzyme activity, and fitness were all significantly lower than wild-type. Surprisingly, encoding the gene using only rare codons decreased fitness by 40%, whereas an allele coded entirely by frequent codons decreased fitness by more than 90%. Increasing gene expression restored mutant fitness to varying degrees, demonstrating that the fitness disadvantage of synonymous mutants arose from a lack of beneficial protein rather than costs of protein production. Protein production was negatively correlated with the frequency of motifs with high affinity for the anti-Shine-Dalgarno sequence, suggesting ribosome pausing as the dominant cause of low mutant fitness. Together, our data support the idea that, although a particular set of codons are favored on average across a genome, in an individual gene selection can either act for or against codons depending on their local context.
Keywords: Methylobacterium extorquens, codon usage bias, fitness, codon usage evolution, ribosome sequestration
Introduction
As a widespread phenomenon observed from bacteria to mammals, within- and between-genome variation in codon usage has been studied extensively during the past few decades (Ikemura 1985; Bulmer 1991; Akashi 1994; Sharp 2005; Kimchi-Sarfaty et al. 2007). However, it remains unclear why some codons are used frequently while others are rare in protein-coding genes. Synonymous mutations (which change codons but not amino acids) are traditionally thought to be under weak selection and have thus served as a neutral baseline for tests of selection. However, it is increasingly clear that changing codon composition and order of occurrence in the transcript (which we collectively refer to as “codon usage”) can have large effects on protein expression and function (Parmley and Hurst 2007; Hershberg and Petrov 2008; Plotkin and Kudla 2010). A recent experiment emphasized this by showing striking similarities between the distributions of fitness effects of synonymous versus nonsynonymous substitutions in ribosomal proteins of Salmonella enterica Serovar Typhimurium (Lind, Berg, et al. 2010). If comparable results hold for other proteins across multiple species, we need to change our assumptions about the fitness effects of synonymous mutations and their long-term influence on codon usage and genome evolution.
Population genetic models propose that biased codon usage evolves largely due to mutational biases favoring AT- or GC-rich codons, countered by weak-to-moderate selection acting on specific codons (Bulmer 1991; Yang and Nielsen 2008). The importance of selection on codon composition is demonstrated by within- and across-genome comparisons showing that codon bias is strongest in genomic regions that experience strong purifying selection. For instance, codon bias is typically stronger in highly expressed protein-coding genes (Ikemura 1985). Even within genes, there appears to be variation in the strength of selection on codon usage: evolutionarily conserved amino acid residues show stronger bias than evolutionarily variable residues (Akashi 1995; Drummond and Wilke 2008). However, the relative influence of physiological processes leading to the evolution and maintenance of biased codon usage remain unclear.
Various hypotheses posit that selection can act upon specific codons at different stages of protein production (supplementary table S1, Supplementary Material online). First, protein production may be limited by translation initiation, causing selection on 5′ mRNA folding and stability rather than direct selection on codons per se (Gold 1988). Second, rare codons serviced by low-abundance tRNAs can slow down translation because ribosomes must wait longer for the appropriate charged tRNA to elongate the nascent polypeptide (Curran and Yarrus 1989; Sørensen et al. 1989). On the other hand, glutamic acid codons recognized by the same tRNA are translated at different rates (Sørensen and Pedersen 1991), and two recent studies claim that the translation time for synonymous codons in yeast and bacteria is invariable (Li et al. 2012; Qian et al. 2012). Even if ribosomes do pause during translation, such pauses may actually be critical to allow the protein to fold correctly (Komar 2009). However, in most cases ribosomal pausing is thought to be detrimental: it can hasten mRNA degradation, sequester ribosomes on slow transcripts, compromise protein function by increasing the probability of mistranslation or altering cotranslational folding kinetics (Cortazzo et al. 2002), and lead to mistranslation-induced protein misfolding (Drummond and Wilke 2008). Thus, selection for translational speed and accuracy (“efficiency”) is expected to generally increase concordance between the cellular tRNA pool and codon usage (“translational selection”). Finally, the balance between translation initiation and elongation may be tuned via a “ramp” of codons serviced by rare tRNAs at the 5′-end of transcripts, which may minimize ribosome collisions or sequestration during elongation (Tuller et al. 2010).
Despite myriad hypotheses about mechanisms underlying selection on codon usage, the paucity of experimental manipulations of codons in native, functional genes that have coevolved with their genome leaves many key questions unaddressed. What magnitude of selective difference is possible between synonymous alleles with varying codon usage? Is the wild-type (WT) codon usage optimal with respect to fitness? Are codons that are serviced by more abundant tRNAs universally preferred over those serviced by rare tRNAs? How much does codon position (and not just composition) matter? Which level(s) of the central dogma (mRNA or protein levels, enzyme activity) are affected by synonymous variants? Are fitness differences driven by insufficient beneficial protein, or excess cost of production? To move toward an integrated picture of the mechanistic bases of selection upon codon usage, it is important to address these questions for variety of native genes in multiple species.
As a step in this direction, we tested the fitness effects of altering codon usage in a highly expressed enzyme-coding gene (∼2% of total cell protein, Vorholt et al. 2000) in the α-proteobacterium Methylobacterium extorquens AM1. For the majority of amino acids, the relative occurrence of the most frequently used codon in M. extorquens genes is >70% (fig. 1A and B), indicating a substantially skewed codon usage (supplementary fig. S1, Supplementary Material online). M. extorquens has evolved to utilize single-carbon (C1) compounds such as methanol via a series of specific metabolic pathways (Chistoserdova et al. 2003). We targeted one of the genes in the formaldehyde oxidation pathway, fae, which encodes the 18 kDa formaldehyde activating enzyme FAE. FAE is essential for growth on methanol but is dispensable during growth on multicarbon substrates (Vorholt et al. 2000). fae has highly biased codon usage (supplementary fig. S1, Supplementary Material online), as expected from the commonly observed pattern of strong codon bias in highly expressed genes (Drummond and Wilke 2008). fae was thus ideal for our experiments because its codon usage represents highly expressed M. extorquens genes, and its high expression level and substrate-specific essentiality render it tractable for protein and fitness measurements.
Fig. 1.

Codon usage bias metrics. The relationship between commonly used indices of codon bias and the index of relative codon usage in Methylobacterium extorquens AM1 used in this study (odds ratio of finding a specific codon at conserved rather than variable residues in protein-coding genes across the genome). (A) Relative frequency of codon usage for each amino acid, calculated for a list of 39 highly expressed ribosomal protein coding genes (from the codon usage bias database, http://cub-db.cs.umt.edu/index.shtml, last accessed December 16, 2012). (B) Relative frequency of codon usage for each amino acid, calculated for 1,022 coding regions (GenBank) and (C) relative synonymous codon usage (RSCU), the frequency of each codon normalized by the total number of synonymous codons for that amino acid. (D) The frequency of optimal codons (Fop) for each of the synonymous fae alleles used in this study; bars are colored by strain. In (A–C), red points indicate the most commonly used codon in the respective set of genes. Data in (B) and (C) are based on the entire M. extorquens genome.
To sample across a wide range of possible codon usage in fae within a minimal set of alleles, we designed and synthesized six synonymous fae alleles (table 1). To ensure that codon differences between alleles were evolutionarily relevant, we characterized “rare” codons as those significantly depleted at conserved amino acid residues, and “frequent” codons as those significantly enriched at conserved residues (see Results). The alleles varied from 0% to 100% rare codons, including a trio of variants with 50% rare codons with identical codon composition but different placement of rare versus frequent codons. We replaced the chromosomal copy of WT fae with these mutant alleles or a WT control (retaining the native promoter), and quantified selection on codon usage using each mutant’s growth rate on methanol (Supplementary methods, Supplementary Material online). Our results uncovered extreme fitness differences between synonymous gene variants, including a nearly complete lack of growth with an allele encoded only by frequent codons. These synonymous alleles exhibited different mRNA levels, but varied even more in production of FAE protein and enzyme activity. In all mutants, growth limitation was a result of insufficient enzyme activity rather than excessive costs of protein production. These results indicate a complicated mapping of codon usage to gene expression and fitness, and suggest that different synonymous alleles may be suboptimal in distinct ways.
Table 1.
Experimental Strains with Synonymous fae Alleles.
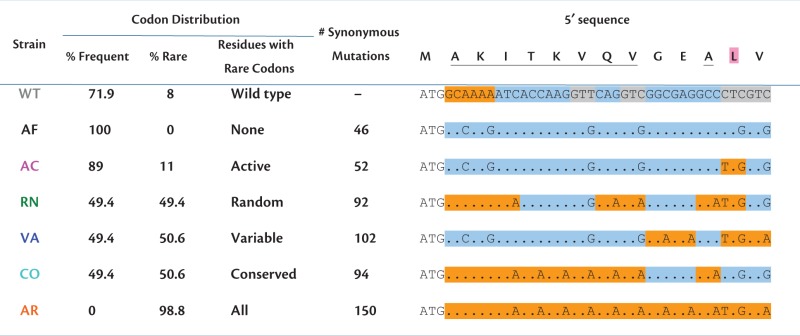 |
Note.—To demonstrate our codon alterations, the first 14 residues at the 5′-end of the gene are shown, with conserved residues underlined and enzyme active sites highlighted in pink. Sequence highlights: orange, rare codons; blue, frequent codons; gray, intermediate codons. In strains RN and AR, two intermediate codons were introduced to retain restriction sites for cloning. For further details of allele design, see Supplementary methods and information, Supplementary Material online.
Results
Design and Synthesis of Synonymous fae Alleles with Altered Codon Usage
To guide our manipulation of codon usage, for each amino acid, we first identified codons that are most enriched (“frequent codons”) or depleted (“rare codons”) at conserved amino acid residues relative to variable residues across all protein-coding genes in M. extorquens (Supplementary methods and supplementary table S2, Supplementary Material online). Codon bias calculated in this way is well correlated with most other commonly used metrics of codon bias, as well as codon usage of highly expressed genes of M. extorquens (fig. 1). Each M. extorquens gene can thus be characterized in terms of the proportion of amino acids that are encoded by frequent, rare, and “intermediate” codons (for amino acids with >2-fold degeneracy, those codons that are neither most frequent nor most rare).
Given our goal to explore the fitness consequences across the widest possible range of codon usage, we generated synonymous variants of fae with varied proportion and position of frequent versus rare codons. WT fae is largely composed of frequent codons (72%), with a few rare codons (8%), and some intermediate codons (20%). We synthesized six alleles carrying different codon combinations: AF (All Frequent), AR (All Rare), CO (“Conserved sites rare”, 50% rare), VA (“Variable sites rare”, 50% rare), RN (“Randomly picked”, 50% rare), and AC (“Active sites rare”). Our synthesized fae alleles (table 1) thus test both the effects of extreme codon usage (0–100% rare codons), as well as positional effects (rare codons located at enzyme active sites, or conserved vs. variable residues). Note that all three 50% rare codon versions have an identical codon composition (except three codons that are different in RN) because the location of frequent or rare codons was independently manipulated for each amino acid residue. We incorporated a C-terminal FLAG tag in each allele for FAE protein quantification (supplementary table S3, Supplementary Material online) and replaced the native fae allele with each synonymous variant (retaining the native promoter) to create chromosomal mutant strains using an fae knockout strain (“Del”). We confirmed that the FLAG tag does not affect fitness, by comparing the strain carrying the WT fae construct and FLAG-tag (“WT”) with the parent WT strain without any FLAG-tags (“WT*”; see Results).
Fitness Effects of fae Alleles: Both Frequent and Rare Codons Can Be Detrimental
As FAE is necessary for growth on methanol, we quantified the fitness effects of our codon manipulations using the growth rate of mutants on methanol, which is strongly correlated with competitive fitness (Pearson’s r = 0.99, P < 0.001 supplementary fig. S2, Supplementary Material online). We found that none of the variants were neutral with respect to growth rate. The strain with the WT construct was significantly faster than all other variants (fig. 2A; analysis of variance [ANOVA] for effect of strain on growth rate: P = 2.2 × 10−16; all Tukey HSD corrected pairwise differences in growth rate between strains are significant with P < 0.001, except WT*–WT with P = 0.07, and CO–AR, VA–RN, and comparisons between AC, Del, and AF with P > 0.3).
Fig. 2.

Fitness effects of synonymous fae alleles. Points show mean ± SE (standard error) (n = 4) for each strain, labeled and colored as in table 1. The open triangle represents the WT strain without a FLAG tag (WT*) and the dashed line shows data for a Δfae (knockout) strain (Del). (B–E) Data are shown relative to WT. (A) Growth rate (per hour) as a function of the proportion of rare codons in fae. (B) Growth rate as a function of the amount of mRNA, as measured from quantitative real time PCR. (C) Growth rate as a function of the amount of FLAG-tagged FAE protein as quantified from Western blots. (D) Average amount of FAE protein produced per mRNA in each strain. (E) Enzyme activity in protein extracts from different strains (rate of conversion of substrate to product per milligram total protein; n = 3). We did not measure enzyme activity for strains AC and AF because they did not produce detectable FAE protein; however, we measured activity in the fae knockout strain (Del) to serve as a control for the spontaneous reaction that occurs in cell extracts but that is insufficient to permit growth on methanol (Vorholt et al. 2000).
Overall, the proportion of rare codons was not significantly correlated with growth rate (Pearson’s r = 0.59, P = 0.58). This was driven by the fact that the two alleles with the greatest number of frequent codons—and the highest sequence similarity to WT (AF and AC, table 1)—barely grew at all in methanol (fig. 2A). The remaining strains, with 50% and 100% rare codons, grew at a rate one-third to one-half that of WT.
Considering the specific role of codon position, we compared the growth rate of strains CO, VA, and RN, the three versions with nearly identical codon composition (each with 50% rare codons). We found that strain CO had significantly higher growth rate than both VA and RN (Tukey-adjusted pairwise comparisons; P < 0.01 in both cases). In fact, CO and VA had identical codon composition; but the combination of rare codons at variable residues and frequent codons at conserved residues (VA) was more detrimental than the inverse codon distribution (CO). This observation is contrary to the expectation from genome-wide analyses that associate frequent codons with conserved residues (Akashi 1994; Stoletzki and Eyre-Walker 2007; Drummond and Wilke 2008). Together, these results indicate that substituting WT codons with either more frequent or rarer codons imposed a fitness disadvantage, the degree of which depended on specific codon placement.
Synonymous Alleles Decreased Gene Expression, Protein Level, and Enzyme Activity
To determine what aspect of FAE expression or function was altered in our synonymous variants, we measured the levels of mRNA and FAE protein, as well as FAE activity in cell extracts. All synonymous variants exhibited less mRNA than WT, despite having the same promoter as the WT allele at the chromosomal locus (fig. 2B). However, the decrease in gene expression was fairly modest compared with the decrease in protein levels (fig. 2C), resulting in a very low protein/mRNA ratio in mutants (fig. 2D). Thus, mutants not only had less fae mRNA (apparently due to a process downstream of transcriptional initiation) but they also produced less FAE protein from each mRNA molecule. Note that our mRNA and protein measurements cannot distinguish between inefficient transcription (or translation) and rapid transcript (or protein) degradation.
The growth differences between strains CO, VA, and RN (each with 50% rare codons; discussed earlier) may be partially explained by the activity of FAE produced by each of these strains (fig. 2E). Although strains CO and RN produced similar amounts of FAE, protein extracts from strain CO showed significantly higher catalytic activity. Conversely, although strain VA produced only one-third the amount of protein as strain RN, the apparent higher activity of its protein allowed it to grow as fast as strain RN. This difference in enzyme activity despite equivalent enzyme levels may arise from mistranslation and/or altered cotranslational protein folding, both of which could change parameters such as binding of substrate to the active site. The enzyme activity data are therefore largely consistent with relative protein production and growth rate observed in mutants. Thus, the particular location of codons affected not only the quantity but also the amount of active enzyme produced.
Fitness Effects of fae Codon Variants as a Function of Transcript Properties
In an effort to determine the cause of low protein production in mutants, we analyzed mutant alleles for possible correlations between protein level and predicted transcript properties that may alter gene expression (Kudla et al. 2009). We found that protein production did not correlate with overall GC composition (r = 0.04, P = 0.94) or whole-transcript folding energy (r = −0.03, P = 0.94; table 2). Furthermore, in contrast with previous results for GFP expression in Escherichia coli (Kudla et al. 2009), we failed to find a significant association between protein levels and the energy of folding measured for the 5′ region or for 50-nucleotide windows (supplementary fig. S3, Supplementary Material online). It is also possible that the 5′ sequences of mutant alleles may have altered translation initiation rates leading to low protein production in mutants. However, we did not find a monotonic relationship between predicted translation initiation rate and protein production, nor could we identify known translation termination or RNA regulatory motifs within the fae alleles. Protein production was also uncorrelated with the average tRNA adaptation index (tAI; an estimate of translation speed; r = 0.14, P = 0.76; table 2). Finally, we do not see evidence for a 5′ “ramp” of slow codons in the WT or other alleles (supplementary fig. S4, Supplementary Material online), suggesting that differential ribosome sequestration or ribosomal collision frequency (Tuller et al. 2010) is unlikely to explain the observed patterns of FAE production. Thus, neither mRNA structure nor translatability appears to be a consistent cause of low protein production (and fitness) in our synonymous fae versions, although these mechanisms may be important for individual alleles. The diametrically opposite fitness of WT and AF despite very similar transcript properties means that this lack of support for various hypotheses cannot be ascribed to low statistical power to detect associations due to few variants.
Table 2.
Summary of Properties of Mutant fae Alleles.
| Allele | AC | AF | VA | RN | CO | AR | WT |
|---|---|---|---|---|---|---|---|
| Growth rate (h−1) | 0 | 0 | 0.08 | 0.09 | 0.12 | 0.12 | 0.23 |
| mRNA | 0.24 | 0.12 | 0.47 | 0.8 | 0.77 | 0.75 | 1 |
| Protein | 0 | 0.03 | 0.1 | 0.31 | 0.27 | 0.31 | 1 |
| Protein per mRNA | 0.01 | 0.26 | 0.21 | 0.39 | 0.34 | 0.41 | 1 |
| Enzyme activity | nd | nd | 0.24 | 0.14 | 0.35 | 0.17 | 1 |
| Number of rare codons | 18 | 0 | 83 | 81 | 83 | 162 | 13 |
| Rare codons (%) | 10.9 | 0 | 50.6 | 49.4 | 50.6 | 98.8 | 7.93 |
| Number of frequent codons | 146 | 164 | 81 | 81 | 81 | 0 | 118 |
| Frequent codons (%) | 89 | 100 | 49.4 | 49.4 | 49.4 | 0 | 71.9 |
| Intermediate codons (%) | 0 | 0 | 0 | 1.22 | 0 | 1.22 | 20.1 |
| GC (%) | 64.1 | 66.8 | 51.7 | 51.7 | 51.7 | 36.6 | 63.7 |
| mRNA folding energy (kcal/mol) | −80 | −78 | −50 | −63 | −64 | −31 | −75 |
| tAI | 0.28 | 0.29 | 0.24 | 0.24 | 0.24 | 0.2 | 0.29 |
Note.—To facilitate comparison, alleles are shown in increasing order of growth rate on methanol and the WT (with maximum fitness) are highlighted in gray. mRNA and protein amount and activity are shown relative to WT. For enzyme activity, “nd” denotes no data available due to very low protein production.
Next, we tested whether the presence of rare codon pairs could have caused the observed decrease in protein production in our mutants, as shown in previous studies (Coleman et al. 2008). Of the 246 codon pairs that are most rare in M. extorquens AM1 protein-coding genes (i.e., occur <100 times), only three are significantly correlated with protein production in our strains (P < 0.005). However, in all three cases the correlation is positive rather than negative (r = 0.92) and is driven by the single occurrence of the codon pair in the WT allele and its absence in all the mutants. We found only positive correlations even when we expanded the set of rare codon pairs to include pairs that occur less than 500 times in the genome. Therefore, it is unlikely that overrepresentation of rare codon pairs caused low protein production in our strains.
Finally, we examined our synonymous fae alleles for evidence of internal Shine-Dalgarno (SD)-like motifs that may affect gene expression in bacteria. Recent results using ribosomal footprinting showed that SD-like motifs in the transcript with very strong binding affinity to the anti-SD sequence in 16S rRNA may cause ribosomes to pause during translation (Li et al. 2012). Therefore, we tested whether the frequency of SD-like hexamer sequences in our synonymous variants could explain low protein production in our strains. We found that protein production was negatively correlated with the frequency of two hexamers: GAACAA (r = −0.92, P = 0.003) and TGGCCA (r = −0.77, P = 0.045; supplementary fig. S5, Supplementary Material online). However, these hexamers are unlikely to be the cause of low production in our strains for two reasons. First, both hexamers have very low affinity for the anti-SD sequence (0 and −1.3 kcal/mol, respectively), making it improbable that they could significantly slow down translation. Second, their frequency across alleles was inconsistent with the differences in protein production between strains (fig. 2C). GAACAA occurred once in all alleles except WT, which does not correspond to our observation of differential protein production across mutants. Similarly, strain AR produced far less protein than WT even though the hexamer TGGCCA was absent in both WT and AR (supplementary fig. S5, Supplementary Material online). Although these hexamers alone may not sufficiently decrease translation speed, it is possible that multiple high-affinity SD-binding sites in the mutant alleles were responsible for low protein production. Such high-affinity hexamers were not uncommon in mutant alleles (fig. 3), and therefore we pooled all hexamers with relatively high binding affinity to anti-SD. We found that their combined frequency was negatively correlated with FAE protein production (fig. 4; note that for affinity < −6 kcal/mol, the relationship is not significant without the WT allele: R2 = 0.016, P = 0.8; for affinity < −4 kcal/mol, the relationship remains significant even after removing the WT allele: R2 = 0.68, P = 0.04). Thus, although no single hexamer with high binding affinity can explain our data, it is plausible that ribosomal pausing due to multiple SD-like sequences resulted in low protein production and fitness in our synonymous mutants.
Fig. 3.

Frequency of hexamers in fae alleles with different predicted affinity to the anti-SD sequence. More negative values indicate stronger binding. Strains are colored as in table 1. The dashed line indicates maximum affinity to anti-SD observed in the WT allele. For clarity, hexamers that are absent in a given allele are omitted.
Fig. 4.

Protein production in mutant strains as a function of the total number of hexamers with high binding affinity to anti-SD, with two different thresholds for high affinity (more negative affinity indicates stronger binding). Points are colored and labeled as in table 1.
Insufficient Enzyme Activity Rather Than Excessive Cost Underlies Observed Fitness Effects
We sought to test the hypothesis that insufficient FAE activity, rather than excessive protein expression cost, underlies the fitness effects of our codon-altered variants. First, we found that none of the fae mutants produced detectable misfolded protein aggregates (supplementary fig. S6, Supplementary Material online). Second, we found that the variants decreased fitness only when FAE was necessary for growth. During growth on succinate, FAE production is unnecessary but still quite high (∼50% of the amount produced during growth on methanol; Okubo et al. 2007). If the fitness defects in our variants were a direct result of excessive costs of FAE production, we would expect that the fitness disadvantage during growth on succinate should simply be one-half of that seen on methanol. However, we found that none of the variants imposed a disadvantage during growth on succinate (fig. 5; ANOVA for effect of strain: P = 0.66). Together, these data provide two lines of evidence against protein expression costs as the driver of the large fitness differences during growth on methanol, when the enzyme product is necessary.
Fig. 5.

Growth rate (mean ± 2 SE [standard error]) of synonymous fae mutants (strains are labeled as in table 1) on permissive succinate medium (diamonds) and selective methanol medium (filled circles). All pairwise differences in growth rate on succinate are nonsignificant (Tukey–HSD correction, P > 0.7).
To directly test whether insufficient benefit from FAE catalysis was responsible for poor growth, we quantified the growth rate of strains carrying synonymous fae alleles on a plasmid with a strong, cumate-inducible promoter. If the mutant alleles decreased fitness because their products were costly (e.g., misfolded proteins), their growth rate would decrease with increasing inducer concentration. Instead, we found that except for strain AC (which remained below our limit of detection for both FAE protein and growth), fae overexpression increased the growth rate of all mutants (fig. 6). These data, in concert with those described earlier, firmly establish that the growth of codon-altered variants is limited by the amount of FAE enzyme, rather than overwhelming fitness costs of fae expression.
Fig. 6.
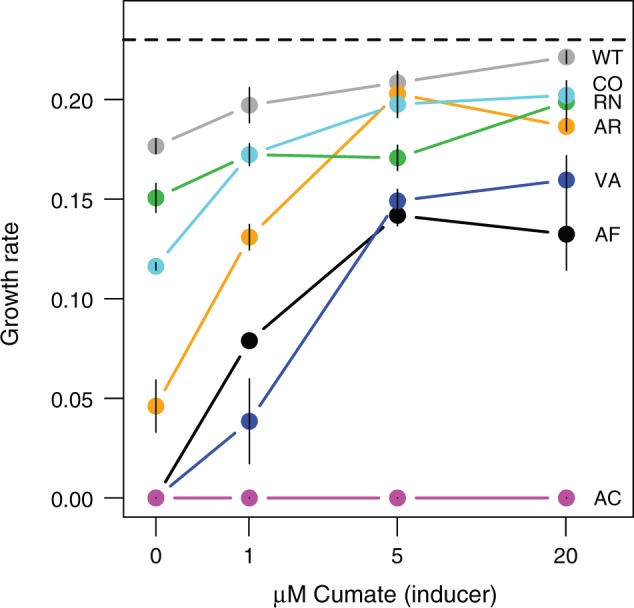
Fitness as a function of induced gene expression. Growth rate (mean ± SE; n = 3) of fae knockout strains carrying plasmid-borne fae alleles on a regulated promoter in methanol, as a function of inducer concentration. The strain carrying the WT allele shows growth even in the absence of cumate inducer, due to high background gene expression from the regulated plasmid (∼40% of native chromosomal level). The dashed line indicates the growth rate of the chromosomal mutant carrying the WT allele (from fig. 2A). Note that due to the cost of plasmid carriage (Chou and Marx 2012), none of the alleles—including the WT version—achieved the expression level of the chromosomal WT strain.
Discussion
We have demonstrated the deleterious consequences of altering the primary coding sequence of a metabolic enzyme-coding gene that is essential for growth in a specific environmental niche. Although our work complements previous experimental studies on codon usage in model systems such as E. coli and Saccharomyces cerevisiae, it presents significant advances and distinct conclusions. First, we use a functional gene that enables survival in a specific carbon niche, rather than a heterologous gene removed from its evolutionary and genomic context (e.g., GFP; discussed later). Our results corroborate those of Carlini (Carlini and Stephan 2003; Carlini 2004; Hense et al. 2010), who showed (in one of the rare examples of work with endogenous genes) that altering codon usage in the Adh gene decreased protein production and reduced ethanol tolerance in Drosophila melanogaster. A second advantage of our work is that our model organism (M. extorquens) is not closely related to commonly used laboratory models such as E. coli or S. enterica, and thus our results demonstrate the broader fitness effects of codon usage. Third, by focusing our efforts on several specific synonymous variants, we were able to quantify their proximate effects on gene expression, enzyme quantity, and activity in detail. This complements the strength of high-throughput studies that address distributions of effects for one or two phenotypes like gene expression or growth. Finally, by manipulating gene expression levels, we could specifically test (and reject) a major prediction of the translational selection hypothesis—that the effects of codon usage are amplified under high expression. Contrary to expectations of many mechanistic hypotheses, we found that the growth disadvantage of altering codon usage is mitigated under high gene expression. Together, these attributes have allowed us to explore various biological effects of each of our synonymous variants, in contrast to previous experiments where a large number of synonymous variants were studied without focusing on specific variants (Kudla et al. 2009; Lind, Berg, et al. 2010). Our experiments thus allow us to generate predictions for evolutionary dynamics following codon changes. For instance, mutations that increase expression of the synonymous fae variants (e.g., in the promoter region of fae) should be selectively favored in all cases except in strain AC (fig. 6). Note that increasing expression did not increase growth rate of variants to the same degree; thus, there appear to be additional problems with protein production that would require changes to the coding sequence itself. By allowing mutant strains to evolve in the laboratory, we are now testing these predictions.
Current understanding of codon bias evolution is greatly influenced by prior experimental work on the costs of heterologous protein production arising from nonoptimal codon usage. Heterologous gene expression is an important field of active research, and our data support previous results showing that matching a gene’s codon usage with host codon bias may not be universally optimal for protein expression (Maertens et al. 2010). These previous studies have demonstrated that codon usage can significantly alter the production and properties of proteins (Gustafsson et al. 2004; Welch et al. 2009), but the mechanistic basis for these effects appears to vary across genes. For instance, high GFP production in E. coli was maximized by low 5′ mRNA folding stability (Kudla et al. 2009), whereas the production of other heterologous proteins was maximized by the use of tRNAs that are robust to amino acid starvation (Welch et al. 2009). Qian et al. (2012) recently suggested that a close match between tRNA concentration and relative codon usage may be more important for translation than increasing the use of codons recognized by the most abundant tRNAs (which is the focus of most codon bias indices, e.g., the codon adaptation index). Together, these studies paint a complicated picture of mechanisms that underlie the dependence of protein expression on codon usage. However, their interpretation in light of codon bias evolution is limited because heterologous proteins are necessarily removed from the evolutionary history and ecological context of the organism. This context is important because genes evolve in concert with the rest of the genome rather than in isolation. Therefore, data on the fitness and physiological effects of codon usage in native genes are necessary to understand the evolution of biased codon usage. The evolutionary lineage of functional genes is also important to consider in light of frequent lateral gene transfer in prokaryotes, because codon usage can affect the outcome of horizontal gene transfer (Jain et al. 2003; Tuller et al. 2011). Greater understanding of the fitness effects of altering codon usage will thus be important to predict the nature and success of genetic exchange between species.
We found that the observed fitness decline in mutants was a direct result of loss of beneficial enzyme product (in quantity and/or quality), rather than increased costs of producing the protein. In our experiments, we can reject mechanistic hypotheses for low fitness that rely on the costs of gene expression (supplementary table S1, Supplementary Material online). None of the fae alleles imposed a growth defect with increasing gene expression or showed evidence of misfolded protein accumulation; all were selectively neutral when FAE activity was unnecessary. Furthermore, none of the hypotheses that postulate lowered benefits of gene expression are consistent with all our mutant fitness and protein expression data (supplementary table S1, Supplementary Material online). Both primary sequence (location of rare or frequent codons) and codon frequency can affect fitness. Some of the fitness differences are clearly site dependent, for example, between strains CO, VA, and RN, all of which have identical codon composition with 50% rare and 50% frequent codons. Our results corroborate those from previous comparative analyses, showing that codon order in protein coding genes is biased and probably evolves under selection (Cannarozzi et al. 2010). However, in general, using too many “frequent” codons was worse than using too many “rare” codons. Although previous experimental studies did not make explicit fitness measurements, they have shown parallel results: using only common codons does not always maximize protein expression (Kudla et al. 2009; Welch et al. 2009). Thus, the detrimental effects of using only frequent codons that we document may prove to be more generally applicable. In our experiments, even if only a few codons were responsible for the fitness differences between strains AR and AF, it would still imply that frequent codons could impose a stronger fitness disadvantage than rare codons, because each of these strains was composed of mutually exclusive codons. Regardless of the precise mechanism for low fitness (e.g., whether it is due to codon usage or transcript stability or structure), our results show that some frequent codons should be strongly disfavored in the context of their specific locations within fae.
Two recent studies offer independent hypotheses explaining why frequent codons may not always be better than their synonymous counterparts. Qian et al. (2012) proposed that the most optimal codon use is one that matches the relative proportions of tRNAs in a cell, rather than using as many of the frequent codons as possible. Thus, all synonymous codons are important for translation, and the degree of imbalance between proportional codon use and tRNA abundance should determine protein production. The model therefore predicts that protein production (and fitness) of strain AR codons should be less than AF codons, because the former has a stronger imbalance in proportional codon usage and tRNA abundance (supplementary fig. S7, Supplementary Material online). However, our experimental data show the opposite result: AR has higher fitness than AF (fig. 2A). Furthermore, overexpressing a gene with biased codons should exaggerate any fitness defects of codon usage-tRNA imbalances, which is also contrary to our observations (fig. 6). Thus, codon usage-tRNA imbalance cannot explain our results. However, selection against such imbalance may more generally explain why frequent codons are not fixed in populations.
A second study by Li et al. (2012) showed that hexamers that are similar to SD sequences could increase translation elongation times by binding ribosomes in the middle of the transcript. Although SD-like sequences could not explain all the ribosomal pausing observed in that analysis, the work shows that depending on the neighboring codons, frequently used codons could be detrimental if they result in an SD-like sequence. This hypothesis predicts that protein production should be negatively correlated with the frequency of SD-like sequences in the mRNA. In our experiments, the pooled frequency of SD-like sequences with high anti-SD affinity is the only factor that is correlated with our observed protein levels (fig. 4). Altering the local sequence context of frequent and rare codons may result in the creation of new ribosome-binding sites, indicating that frequent ribosomal pausing during translation is the major cause of low enzyme production in our synonymous variants.
Besides the direct effect on FAE levels, a secondary predicted consequence of such frequent anti-SD like hexamers is the sequestration of available ribosomes. However, as discussed earlier, the observed fitness rescue with increasing gene expression (fig. 6) runs counter to the hypothesis that ribosomal sequestration from the global pool is costly. We propose that this apparent contradiction may be resolved by recognizing that ribosomal pausing during translation may both decrease beneficial protein (in this case FAE) and incur costs due to ribosome sequestration. The relative importance of these effects as a function of gene expression will depend on the decrease in beneficial focal protein relative to the costs of global sequestration. With small increments in gene expression, the benefits of increased protein production are likely to outweigh global sequestration costs. With large increments in gene expression, however, ribosome sequestration may impose a significant cost. Such costs at very high expression levels may be the reason why some of the mutants cannot be completely rescued by fae overexpression. For instance, strains VA and AF have many hexamers with high affinity to anti-SD (fig. 4), and neither of them approach WT fitness even at high levels of gene expression (fig. 6).
More generally, although SD-like sequences seem to play an important role in affecting protein production in our experiments, the broader significance of SD-like sequences in governing codon usage remains unclear. SD sequences are short and very similar across bacteria (typically a variant of GGAGG). Hence, selection to avoid SD-like sequences could only explain avoidance of a few codons that resemble the specific SD sequence. Given that the “frequent” codons in strains AF and AC create problematic ribosomal-binding sites, their prevalence in the M. extorquens genome is very puzzling. If global codon usage were shaped by selection against anti-SD sites, these codons would not be expected to cause such strong fitness defects. Thus, we speculate that selection on SD-like sequences could shape codon usage only in conjunction with other factors that influence its evolution.
We propose that the observed lack of broad and consistent experimental support for most of the existing hypotheses (supplementary table S1, Supplementary Material online) arises because the hypotheses have largely been derived from genome-level correlations averaged across many genes. Such averaging cancels out gene-specific effects and enhances the appearance of more general effects, even if the latter are small relative to gene-specific effects. This discrepancy between apparent selection acting on individual genes versus genome-wide properties may also explain why codons that are generally highly enriched in protein-coding genes are not always selectively favored in fae. Therefore, we speculate that multiple (nonmutually exclusive) gene-specific mechanisms are required to explain why certain codons are used more often in protein-coding genes. Thus, we predict that the discordance between simple mechanistic models and experimental results—such as those we observe in our experiments—will also be observed for functional genes in diverse organisms. Our focal gene fae does not stand out as an outlier within the distribution of codon bias in highly expressed genes of M. extorquens (supplementary fig. S1, Supplementary Material online) or other species. Thus, it is likely that relatively few altered codons in each of our mutant alleles were responsible for a majority of the fitness effects that we observe. Tracking down these causal codons and the context in which they decrease fitness is not trivial, but is a promising avenue for further research.
Together with previous studies, our data help establish that synonymous mutations do not constitute a special “neutral” category. Although such mutations appear to face weaker selection than nonsynonymous mutations on average, their effects and evolutionary consequences may be more similar to those of nonsynonymous mutations than recognized previously. For instance, altered codon usage in a synthetic antibiotic-resistance gene decreased protein levels and reduced E. coli fitness, which was restored during experimental evolution via promoter mutations that increased gene expression (Amoros-Moya et al. 2010). Lind, Tobin, et al. (2010) also found that synonymous point mutations in the ribosomal subunit genes in S. enterica could have large impacts on fitness. Our results described here pinpoint an example where the selective benefit of accurately and rapidly expressing the protein is very high, so that our alterations to existing codon bias in both directions had large fitness effects. One caveat of our study is that we ignore intermediate-frequency codons that are found at a minority (20%) of sites in fae; these merit further investigation. Collectively, this growing body of work on codon usage bias in functional genes promises to provide novel insights into the evolution of codon bias.
Although many genes may not face the same selective pressures as fae, our experiments are an important step toward understanding the relevance of codon usage and selection acting upon it in bacterial evolution. In the end, the biggest rule in codon usage may be that each individual gene is an exception. Given that there are so many physiological levels at which “silent” mutations can affect expression, the most relevant mechanism(s) acting on any given gene may be different from the next one. This pluralist view suggests that to understand the role of codon usage in simultaneously optimizing expression of multiple genes, we need more such analyses on the fitness of altered codon usage in individual functional genes from diverse taxa.
Materials and Methods
Generating Mutant fae Alleles
Evolutionarily conserved amino acid residues evolve under strong purifying selection and tend to have a stronger bias in their codon usage. Our goal was to test the fitness effects of altering this biased codon usage. Therefore, we first identified evolutionarily conserved amino acid residues in M. extorquens AM1 with respect to a closely related strain M. extorquens DM4 (using sequences of 4,285 genes with >70% amino acid alignment, out of a total of 4,424 total ortholog pairs). We then identified those codons that are significantly enriched at conserved amino acid residues across the genome (relative to evolutionarily variable residues) in M. extorquens AM1. For each amino acid (except methionine and tryptophan both encoded by a single codon), we picked the most enriched codon (“frequent”) and the most scarcely used codon (“rare”; supplementary table S2, Supplementary Material online). Based on the list of frequent and rare codons, we synthesized six synonymous mutant fae alleles (table 1). For designing strains VA and CO, we used a multiple alignment of fae sequences across 26 species to identify the 50% most conserved and most variable residues within fae. All mutant strains used in the study were created on the background of an fae knockout strain (CM2563) retaining the native fae promoter. Details of the cloning steps used to create chromosomal and regulated promoter mutants can be found in the Supplementary methods, Supplementary Material online.
Media and Growth Conditions
We used a version of Hypho minimal medium composed of the following: 1) 50 mL/L of “P solution” (33.1 g/L K2HPO4.3H2O and 25.9 g/L NaH2PO4.H2O); 2) 50 mL/L of “S solution” (5 g/L (NH4)2SO4 and 2 g/L MgSO4.7H2O); 3) 1 mL/L of Z3 metal mix (0.177 g/L ZnSO4.7H2O, 1.466 g/L CaCl2.2H2O, 0.107 g/L MnCl2.4H2O, 2.496 g/L FeSO4.7H2O, 0.177 g/L (NH4)6Mo7O24.4H2O, 0.374 g/L CuSO4.5H2O, 0.238 g/L CoCl2.6H2O and 0.1 g/L Na2WO4.2H2O); 4) 900 mL/L dH2O; 5) carbon substrate as required (15 mM methanol or 3.5 mM sodium succinate); and 6) 2% w/v agar if solid media were required. All cultures were grown at 30°C.
Quantifying Fitness, Gene Expression, Protein Production, and Enzyme Activity
We measured growth rate of strains across 48 h, using automated hourly readings of optical density (OD600) of cultures growing in 48-well culture plates incubated on a shaker. For expression assays, we allowed cells to grow to mid-exponential phase in 50-mL flasks with succinate medium, and induced fae expression with methanol. This strategy of induction for a short time balanced the desire for steady-state fae expression with the fact that some of the variants are incapable of growth on methanol and fae mutants are known to suffer from “methanol-sensitivity” due to an inability to handle formaldehyde production (Marx et al. 2003). One hour after induction, we harvested cells for mRNA and protein extraction. We used quantitative real time polymerase chain reaction (PCR) to estimate fae mRNA copy number relative to an endogenous housekeeping ribosomal gene (rpsB). We quantified FAE protein (relative to an endogenous 64 kDa reference protein) using a denaturing gradient gel followed by a Western Blot probed with anti-FLAG antibody. To measure the activity of FAE, we used 300-mL cell cultures induced with methanol for 1 h, in a coupled assay with the enzyme MtdB (methylamine dehydrogenase) as described previously (Vorholt et al. 2000) (Supplementary methods, Supplementary Material online).
Supplementary Material
Supplementary methods, figures S1–S7, and tables S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Miki Lee and David Chou for experimental advice; Nigel Delaney for growth quantification software; David Robinson for help with designing mutant gene sequences; and Gene-Wei Li for sharing data on Escherichia coli hexamer-anti-SD binding energy. They thank members of the Marx lab, Arvind Subramaniam, Jue Wang, and Sergey Kryazhimskiy for insightful comments and discussion. This work was supported by the National Institutes of Health (grant GM078209 to C.J.M. and GM088344 to D.A.D.), and the National Institute of General Medical Sciences (grant F32GM096705 to N.C.M.-G.).
References
- Akashi H. Synonymous codon usage in Drosophila melanogaster—natural selection and translational accuracy. Genetics. 1994;136:927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoros-Moya D, Bedhomme S, Hermann M, Bravo IG. Evolution in regulatory regions rapidly compensates the cost of nonoptimal codon usage. Mol Biol Evol. 2010;27:2141–2151. doi: 10.1093/molbev/msq103. [DOI] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarozzi G, Schraudolph NN, Faty M, Rohr von P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y. A role for codon order in translation dynamics. Cell. 2010;141:355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- Carlini DB. Experimental reduction of codon bias in the Drosophila alcohol dehydrogenase gene results in decreased ethanol tolerance of adult flies. J Evolution Biol. 2004;17:779–785. doi: 10.1111/j.1420-9101.2004.00725.x. [DOI] [PubMed] [Google Scholar]
- Carlini DB, Stephan W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003;163:239–243. doi: 10.1093/genetics/163.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistoserdova L, Chen S-W, Lapidus A, Lidstrom ME. Methylotrophy in Methylobacterium extorquens AM1 from a genomic point of view. J Bacteriol. 2003;185:2980–2987. doi: 10.1128/JB.185.10.2980-2987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H-H, Marx CJ. Optimization of gene expression through divergent mutational paths. Cell Rep. 2012;1:133–140. doi: 10.1016/j.celrep.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazzo P, Cerveñansky C, Marín M, Reiss C, Ehrlich R, Deana A. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem Biophys Res Commun. 2002;293:537–541. doi: 10.1016/S0006-291X(02)00226-7. [DOI] [PubMed] [Google Scholar]
- Curran JF, Yarrus M. Rates of aminoacyl-tRNA selection at 29 sense codons in vivo. J Mol Biol. 1989;209:65–77. doi: 10.1016/0022-2836(89)90170-8. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold L. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu Rev Biochem. 1988;57:199–233. doi: 10.1146/annurev.bi.57.070188.001215. [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hense W, Anderson N, Hutter S, Stephan W, Parsch J, Carlini DB. Experimentally increased codon bias in the Drosophila adh gene leads to an increase in larval, but not adult, alcohol dehydrogenase activity. Genetics. 2010;184:547–555. doi: 10.1534/genetics.109.111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol. 2003;20:1598–1602. doi: 10.1093/molbev/msg154. [DOI] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Oh JM, Kim I-W, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the mdr1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- Komar AA. A pause for thought along the co-translational folding pathway. Trends Biochem Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-W, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Berg OG, Andersson DI. Mutational robustness of ribosomal protein genes. Science. 2010;330:825–827. doi: 10.1126/science.1194617. [DOI] [PubMed] [Google Scholar]
- Lind PA, Tobin C, Berg OG, Kurland CG, Andersson DI. Compensatory gene amplification restores fitness after inter-species gene replacements. Mol Microbiol. 2010;75:1078–1089. doi: 10.1111/j.1365-2958.2009.07030.x. [DOI] [PubMed] [Google Scholar]
- Maertens B, Spriestersbach A, Groll von U, et al. (11 co-authors) Gene optimization mechanisms: a multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Sci. 2010;19:1312–1326. doi: 10.1002/pro.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx CJ, Chistoserdova L, Lidstrom ME. Formaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J Bacteriol. 2003;185:7160–7168. doi: 10.1128/JB.185.23.7160-7168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Skovran E, Guo X, Sivam D, Lidstrom ME. Implementation of microarrays for Methylobacterium extorquens AM1. OMICS. 2007;11:325–340. doi: 10.1089/omi.2007.0027. [DOI] [PubMed] [Google Scholar]
- Parmley JL, Hurst LD. How do synonymous mutations affect fitness? Bioessays. 2007;29:515–519. doi: 10.1002/bies.20592. [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2010;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Yang J-R, Pearson NM, Maclean C, Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33:1141–1153. doi: 10.1093/nar/gki242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 2007;24:374–381. doi: 10.1093/molbev/msl166. [DOI] [PubMed] [Google Scholar]
- Sørensen MA, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207:365–377. doi: 10.1016/0022-2836(89)90260-x. [DOI] [PubMed] [Google Scholar]
- Sørensen MA, Pedersen S. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol. 1991;222:265–280. doi: 10.1016/0022-2836(91)90211-n. [DOI] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- Tuller T, Girshovich Y, Sella Y, Kreimer A, Freilich S, Kupiec M, Gophna U, Ruppin E. Association between translation efficiency and horizontal gene transfer within microbial communities. Nucleic Acids Res. 2011;39:4743–4755. doi: 10.1093/nar/gkr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt JA, Marx CJ, Lidstrom ME, Thauer RK. Novel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol. J Bacteriol. 2000;182:6645–6650. doi: 10.1128/jb.182.23.6645-6650.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol. 2008;25:568–579. doi: 10.1093/molbev/msm284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.