

Abstract
Due to its ability to regulate the growth, differentiation and apoptosis of cancer cells, retinoic acid (RA) is considered a signaling molecule with promising therapeutic potential in oncology. In this study, we show that RA is able to induce the intrinsic ability of breast cancer cells to recognize double-stranded RNA (dsRNA) through the upregulation of Toll-like receptor 3 (TLR3) expression. RA, co-administered with the dsRNA mimicker polyinosinic–polycytidylic acid (poly(I:C)), synergizes to mount a specific response program able to sense dsRNA through the concurrent upregulation of TLR3, the dsRNA helicases melanoma differentiation-associated antigen-5 (MDA-5) and RA-inducible gene-1 (RIG-1), and the dsRNA-activated protein kinase (PKR) expression, leading breast cancer cells to specifically express downstream transcriptional targets of dsRNA sensors, such as interferon-β (IFNβ), interleukin-8 (IL-8), chemokine (C-C motif) ligand 5 (CCL5), and C-X-C motif Chemokine 10 (CXCL10). A TLR3-dependent apoptotic program is also induced by RA and poly(I:C) co-treatment that correlates with the induction of the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and contributes to block breast cancer cell proliferation. The mechanisms of apoptosis induced by RA/poly(I:C) in breast cancer cells involve type I IFN autocrine signaling, caspase-8 and caspase-3 activation, as well as TRAIL signaling. Our results reveal important links among RA, TLR3 and TRAIL and highlight the combined use of RA and poly(I:C) as a potential effective tumor therapy by improving the apoptotic response of cancer cells with low sensitivity to the action of synthetic dsRNA.
Keywords: retinoic acid, polyinosinic–polycytidylic acid, apoptosis, Toll-like receptor 3, TNF-related apoptosis-inducing ligand (TRAIL)
Differentiation therapy by retinoic acid (RA) remains an important cancer treatment modality for the management of some types of cancer (e.g., acute promyelocytic leukemia (APL)). RA, the active form of vitamin A, signals through three RA receptors (RARα, -β, and -γ) and three retinoid-X-receptors (RXRα, -β, and -γ), which form RXR/RAR heterodimers. These receptors are the in vivo mediators of the RA-induced signaling and control a plethora of gene regulatory programs acting as ligand-dependent transcription factors.1 RA regulates cell growth, differentiation and death, and thus plays major roles in embryonic development and tissue remodeling.2 RA has been also shown to inhibit breast cancer cell growth and prevent mammary carcinogenesis in animal models, a process that generally involves the induction of apoptosis and cell-cycle arrest.3 Despite the remarkable efficacy of all-trans RA in APL,4 it has shown limited success in clinical trials of breast cancer.5, 6 Research focused on the actions of the RARs, on the regulation of their expression, and on the identification of their target genes is key to improve RA efficacy and to develop new therapies for cancer disease.
For the past 25 years, a special attention has been given to adjuvants or enhancers of immunity for cancer therapy. The Toll-like receptor (TLR) family recognizes pathogen-associated molecular patterns specific for microbial components.7, 8 Mainly expressed in innate immune cells, TLRs recognize these motifs and trigger innate immune activation and, subsequently, adaptive immunity. Several TLR agonists are currently being tested as adjuvants for anticancer vaccines and therapies.9 TLR3 acts as a critical sensor of double-stranded RNA (dsRNA) and has been found in endosomal compartments or at the cell surface of conventional dendritic cells, as well as in a variety of epithelial cells.8 In a similar way to natural dsRNA from viral origin, the synthetic dsRNA analog polyinosinic–polycytidylic acid (poly(I:C)) binds TLR3 leading to the activation of the transcription factors nuclear factor-κB (NF-κB) and interferon regulatory factor 3 (IRF3), and of kinases such as p38 and c-Jun N-terminal kinase (JNK), promoting the synthesis of proinflamatory cytokines, costimulatory molecules and type I interferons (IFNs). In fact, poly(I:C) was found to be a strong IFN inducer.10, 11 Additional cytosolic dsRNA receptors—RA-inducible gene-1 (RIG-1) and melanoma differentiation-associated antigen-5 (MDA-5)—have recently been identified.12 Because poly(I:C) promotes the activation of the innate immunity and thus of long-lasting T cells immunity,13 it is of great interest as a potential anti-cancer agent.14, 15, 16, 17 Importantly, it has recently been reported that TLR3 agonists, especially poly(I:C), can directly kill cancer cells through induction of apoptosis.14 Thus, TLR3 activation may cause tumors to regress by increasing apoptosis of cancer cells in a cell-autonomous manner and by directly or indirectly recruiting leukocytes, resulting in tumor lysis by natural killer and cytotoxic T-cells.
In this study, we report that RA-induced growth inhibition is accompanied by induction of TLR3 gene expression in breast cancer cells. We found that treatment with RA and poly(I:C) synergistically inhibited the growth and induced the apoptosis of breast cancer cells, which is accompanied by the concomitant upregulation of dsRNA receptor expression TLR3, RIG-1, MDA-5 and the dsRNA-activated protein kinase (PKR), and their downstream transcriptional target genes, interleukin-8 (IL-8), chemokine (C-C motif) Ligand 5 (CCL5), C-X-C motif Chemokine 10 (CXCL10) and IFNβ. Our results suggest that RA and poly(I:C) co-treatment induces cell death in a TLR3-dependent manner, which involves type I IFN autocrine signaling, caspase-8 and caspase-3 activation as well as the expression and activity of the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL).
Results
RA and poly(I:C) co-treatment inhibits breast cancer cell growth
Data obtained in our laboratory indicated that 9-cis-retinoic-acid (9cRA) induce the upregulation of TLR3 in SK-BR-3 cells (AMJ-L, unpublished results). In order to corroborate these data and to increase the relevance of the results, we decided to broaden the study and analyzed TLR3 expression in a panel of six human breast cancer cell lines after 9cRA treatment. 9cRA upgregulated TLR3 mRNA expression in all tested cell lines (Figure 1a). Since it is well recognized that RA and TLR3 agonists, on their own, can exert anti-proliferative effects in certain breast cancer cells, we decided to investigate the effect of a combined treatment of both. For this purpose, we performed dose-response experiments in several breast cancer cell lines treated with 9cRA in the presence or absence of poly(I:C). As expected, treatment of SK-BR-3 cells with 9cRA at the pharmacological concentration of 10−6 M displayed a weak growth inhibitory effects at 48 h, whereas no effects on cell proliferation could be seen after addition of poly(I:C) alone at any concentration used. Interestingly, 9cRA and 50 μg/ml poly(I:C) co-treatment exerted profound anti-proliferative effects in these cells (Figure 1b). At this concentration, 9cRA and poly(I:C) co-treatment inhibited cell growth in a time-dependent manner in SK-BR-3 cells, by about 60% and above 80% at 48 h and 72 h, respectively (Supplementary Figure S1). Similar results were obtained for H3396, ZR-75-1, and T47D cells. However in these cells, poly(I:C) alone was already able to inhibit growth, while 9cRA increased the sensitivity of the cells to the anti-proliferative action of poly(I:C). Taken together, these results suggest that the two signaling pathways can cooperate in the control of breast cancer cell proliferation.
Figure 1.

Regulation of TLR3 expression by 9cRA in breast cancer cells: 9cRA and poly(I:C) cooperate to inhibit the growth of breast cancer cells. (a) Quantitative RT-PCRs were performed with primers for TLR3 and β-actin mRNA using total RNA of different breast cancer cells treated with either vehicle (open bars) or 9cRA (black bars) for 48 h. Data represent the mean±S.D. of three different experiments. Asterisks denote statistically significant differences against the corresponding untreated cells (**P<0.01; ***P<0.001). (b) SK-BR-3, ZR-75-1, H3396, and T47D cells were treated with varying concentrations of poly(I:C) for 48 h in the presence or absence of 9cRA (10−6 M). Viability was measured by crystal violet. The results are shown as percentage of the values obtained in control conditions (cells grown in the presence of vehicle). All values are shown as the mean±S.D. of three different experiments
RA and poly(I:C) co-treatment induces caspase-dependent cell death
In order to find out whether cell death contributes to the anti-proliferative effect induced by 9cRA and poly(I:C) co-treatment in SK-BR-3 cells, we analyzed the sub-G1 cell-population as a measure of cell death. As shown in Figure 2a, neither the treatment with 9cRA nor with poly(I:C) alone had any noticeable effect on SK-BR-3 cell viability. However, 9cRA and poly(I:C) co-treatment dramatically induced cell death. Apoptosis induced by 9cRA and poly(I:C) co-treatment was also found in ZR-75-1 cells, although in these cells, poly(I:C) on its own clearly increased the percentage of apoptotic cells. In the human breast cancer cell lines Hs578T, H3396 and T47D, 9cRA and/or poly(I:C) failed to induce apoptosis (data not shown). However, the addition of cycloheximide (CHX) to the media was sufficient to render Hs578T sensitive to the pro-apoptogenic effects of 9cRA and poly(I:C) co-treatment, suggesting that an unstable inhibitor of 9cRA/poly(I:C)-induced apoptosis is expressed in this particular cell line that has to be degraded in order to allow apoptosis induction. In contrast, no effect of CHX could be detected in the other two cell lines (H3396 and T47D), suggesting that other mechanisms could also account for apoptosis resistance to 9cRA/poly(I:C).
Figure 2.

Induction of apoptosis in breast cancer cells by 9cRA and poly(I:C) treatment. (a) Breast cancer cells were treated with 9cRA (10−6M), poly(I:C) (50 μg/ml in all cell lines except for ZR-75-1 cells that were treated with 12.5 μg/ml), or the combination of both for 48 h, and cell death was assayed by PI staining and FACS analysis. In the case of Hs578T, H3396, and T47D cell lines, cells were pretreated with 9cRA for 24 h and cycloheximide (CHX) at a concentration of 2.5 μg/ml was added 2.5 h prior to stimulation with poly(I:C) or vehicle for other additional 24 h. Data represent the mean±S.D. of three different experiments. (b) SK-BR-3 cells were treated with vehicle (open bars) or 50 μg/ml of poly(I:C) (black bars) in the presence of increasing concentrations of 9cRA. Cell death was determined as in (a) after 48 h. Data represent the mean±S.D. of three different experiments. (c) SK-BR-3 cells were treated with vehicle (open bars) or with 50 μg/ml of poly(I:C) (black bars) in the presence of agonists specific for RAR (TTNPB), RXR (LG268) or both receptors (9cRA) for 48 h, followed by PI staining and FACS analysis. Data are shown as the mean±S.D. of three different experiments. (d) SK-BR-3 cells were treated for 48 h with vehicle, 9cRA (10−6M), 50 μg/ml of poly(I:C) or 100 μg/ml of poly(A:U) as indicated. The percentage of cells positive for PI staining was quantified as in (a). Data represent the mean±S.D. of three different experiments. (e) Immunoblot showing pro-caspase-8 cleavage and induction of active caspase-3 after treatment with 9cRA (10−6M), 50 μg/ml of poly(I:C) or the combination of both. The cleavage products of pro-caspase-8 are indicated as p43/41 and p18. β-Actin was used as loading control. The immunoblots show representative results from three independent experiments
Next, we decided to analyze more thoroughly the apoptotic pathway(s) activated in SK-BR-3 and ZR-75-1 cells consequently to 9cRA/poly(I:C) co-treatment. Our results show that 9cRA cooperates with poly(I:C) to induce apoptosis in a dose-dependent manner in SK-BR-3 cells (Figure 2b). Since 9cRA is a pan-RAR and -RXR agonist, we used specific agonists to test the contribution of each of these receptors to the cell death induction promoted by 9cRA and poly(I:C) co-treatment. As shown in Figure 2c, while TTNPB, an RAR-agonist, clearly cooperates with poly(I:C) to induce the apoptosis of SK-BR-3 cells, the specific RXR-agonist LG-100268 does not affect the viability of these cells, showing that RARs are the receptors involved. Cooperation between 9cRA and poly(I:C) to induce apoptosis seems specific for poly(I:C) since another dsRNA, polyadenylic–polyuridylic acid (poly(A:U)), does not synergize with 9cRA to induce apoptosis in SK-BR-3 cells (Figure 2d). Moreover, 9cRA and poly(I:C) co-treatment activates caspase-8 and caspase-3 as assessed by western blot in SK-BR-3 cells (Figure 2e). Taken together, these results suggest that an RAR-mediated caspase-dependent cell death mechanism contributes to the 9cRA/poly(I:C)-driven anti-proliferative effects in breast cancer cells.
RA and poly(I:C) co-treatment enhances the expression and signaling of dsRNA sensors
Since our observations show that TLR3 mRNA is upregulated by 9cRA in breast cancer cell lines, we next sought to investigate whether 9cRA and/or poly(I:C) regulate the expression of other dsRNA sensors in SK-BR-3 and ZR-75-1 cells that could act as potential mediators of 9cRA/poly(I:C)-driven apoptosis. As shown in Figure 3a, TLR3 mRNA was upregulated by 9cRA but not by poly(I:C) in SK-BR-3 cells. Importantly, 9cRA/poly(I:C) co-treatment synergistically induces the expression of TLR3 (Figure 3a). Similar results were seen at the protein level (Figure 3c). RIG-1 mRNA was also strongly upregulated by 9cRA alone, while no significant induction of MDA-5 and PKR expression by 9cRA was seen in SK-BR-3 cells. Note that, on its own, poly(I:C) did not induce the expression of these genes in this cell line. Interestingly, the combination of both 9cRA and poly(I:C) resulted in a synergistic induction of RIG-1, MDA-5 and PKR above the levels seen with either compound alone (Figure 3e). Similar results were obtained with ZR-75-1 cells albeit induction of these genes by 9cRA was lower than in SK-BR-3 cells, and poly(I:C) alone notably regulated the expression of all these genes. As it was observed for SK-BR-3 cells, 9cRA/poly(I:C) co-treatment synergistically induced the expression of these genes in ZR-75-1 cells (Figure 3b, d). These data suggest that 9cRA and poly(I:C) crosstalk to amplify an innate immune signaling pathway that drives breast cancer cells to apoptosis.
Figure 3.

9cRA and poly(I:C) cooperate to induce the expression of the dsRNA sensors TLR3, RIG-1, MDA-5, and PKR in SK-BR-3 and ZR-75-1 breast cancer cells. (a, b) Quantitative RT-PCR was performed with primers for TLR3 and β-actin mRNA using total RNA from SK-BR-3 (a) or ZR-75-1 (b) cells treated with vehicle or with poly(I:C) in the absence (open bars) or presence of 9cRA (black bars) for 24 h. Data represent the mean±S.D. of three different experiments. (c, d) TLR3 protein level was determined in SK-BR3 (c) or ZR-75-1 (d) cells after treatment with 9cRA, poly(I:C) or the combination of both for 48 h using β-actin as a loading control. The immunoblots show representative results from three independent experiments. (e, f) Quantitative RT-PCR was performed with primers for RIG-1, MDA-5, PKR, and β-actin mRNA using total RNA from SK-BR-3 (e) or ZR-75-1 (f) cells treated as described in (a). All values are shown as the mean±S.D. of three different experiments
Next, we decided to investigate whether 9cRA and poly(I:C) crosstalk produces an amplification of dsRNA receptor signaling by analyzing the expression of several downstream transcriptional targets in breast cancer cells such as IFNβ, and some interleukins and chemokines involved in the activation and chemoattraction of immune cells. As shown in Figure 4, the expression of IFNβ, IL-8, CCL5, and CXCL10 were synergistically induced after 9cRA/poly(I:C) co-treatment, but not with either treatment alone.
Figure 4.

9cRA and poly(I:C) synergize to induce IFNβ, IL-8, CCL5, and CXCL10 mRNA in SK-BR-3 breast cancer cells. Quantitative RT-PCR was performed with primers for IFNβ, IL-8, CCL5, CXCL10, and β-actin mRNA using total RNA from SK-BR-3 breast cancer cells treated with either vehicle or poly(I:C) in the absence (open bars) or presence of 9cRA (black bars) for 24 h. All values are shown as the mean±S.D. of three different experiments
Type I IFN signaling is involved in RA/poly(I:C)-induced apoptosis
As IFNβ has been described as a potent apoptotic inducer in several cell models, we sought to investigate whether that could be also the case for SK-BR-3 cells. Despite the dramatic induction of IFNβ by 9cRA/poly(I:C), recombinant IFNβ alone or in combination with 9cRA was a poor inducer of cell death in SK-BR-3 even at high doses. By contrast, recombinant IFNβ cooperated with poly(I:C) to elicit a dramatic induction of cell death in these cells (Figure 5a). To determine the role of type I IFNs in 9cRA/poly(I:C)-induced apoptosis, we assessed the neutralization of type I IFN receptor (IFNR) with specific monoclonal antibodies in 9cRA/poly(I:C)-treated cells. As shown in Figure 5b, the neutralization of type I IFNR significantly reduced 9cRA/poly(I:C)-induced apoptosis of SK-BR-3 cells, demonstrating that type I IFN signaling is involved in 9cRA/poly(I:C)-driven cell death. These results establish that type I IFN signaling is at least partially required for 9cRA/poly(I:C)-triggered citotoxicity.
Figure 5.
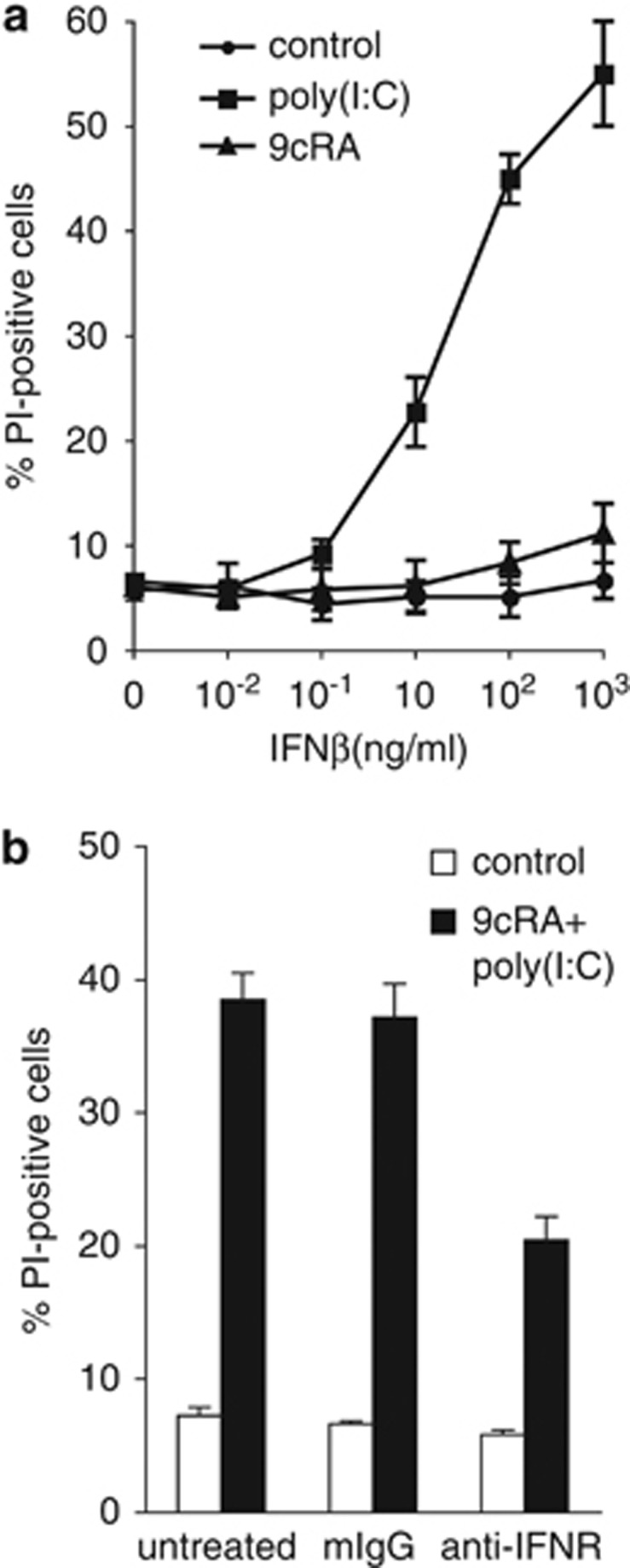
Apoptosis induced by 9cRA/poly(I:C) co-treatment in SK-BR-3 requires type I IFNR signaling. (a) SK-BR-3 cells were either untreated or treated with 9cRA or poly(I:C) in the presence of increasing doses of recombinant IFNβ for 48 h and cell death was assayed by PI staining and FACS analysis. The values represent the mean±S.D. of three different experiments. (b) SK-BR-3 cells were treated with or without a mixture of 9cRA and poly(I:C) for 48 h, and cell death was assayed by PI staining and FACS analysis. In all, 20 μg/ml of neutralizing antibodies for type I IFN receptor (anti-IFNR) or isotype control (mIgG) was added twice to the cell culture, 48 and 24 h before FACS analysis. Data represent the mean±S.D. of three different experiments
TRAIL expression is induced by RA/poly(I:C) co-treatment in breast cancer cells
The activation of caspase-8 implies the potential involvement of the apoptotic extrinsic pathway triggered by 9cRA/poly(I:C) co-treatment. TRAIL, an IFN target gene, is a death ligand that triggers cell death via the extrinsic pathway.18 Since RA induces apoptosis of cancer cells by inducing TRAIL expression19, 20 and TRAIL is involved in poly(I:C)-driven apoptosis in endothelial cells,21 we reasoned that TRAIL could be a mediator of 9cRA/poly(I:C) pro-apoptotic signaling in breast cancer cells. As expected, 9cRA induced TRAIL mRNA level (≈10-fold) in SK-BR-3 cells, and a similar induction was observed with poly(I:C) alone. Importantly, 9cRA/poly(I:C) co-treatment led to a synergistic upregulation of TRAIL mRNA (>1500-fold), as shown in Figure 6a. Protein immunoblotting revealed that synergistic upregulation of TRAIL by 9cRA/poly(I:C) co-treatment occurs in a time-dependent manner and correlates with caspase-3 and caspase-8 activation (Figure 6b). Similar results were obtained in ZR-75-1 cells with the only difference that poly(I:C), on its own, strongly induced both TRAIL mRNA and protein levels in these cells (Figure 6c and d). Therefore, these results indicate that 9cRA and poly(I:C) signaling can cooperate to synergistically induce TRAIL and its expression correlates with the activation of different apoptotic markers.
Figure 6.

9cRA and poly(I:C) synergize to induce the death ligand TRAIL expression in breast cancer cells. (a) Quantitative RT-PCR was performed with primers for TRAIL and β-actin mRNA using total RNA from SK-BR-3 breast cancer cells treated with vehicle or poly(I:C) in the absence (open bars) or presence of 9cRA (black bars) for 24 h. The data shown represent the mean±S.D. of three independent experiments. (b) Immunoblot showing TRAIL protein level, pro-caspase-8 cleavage, and induction of active caspase-3 in SK-BR-3 cells after treatment with 9cRA, poly(I:C) or the combination of both for different time periods in SK-BR-3 cells. β-Actin was used as loading control. The images shown are from one representative experiment performed three times with similar results. (c) TRAIL mRNA was determined in ZR-75-1 cells after 24 h treatment with 9cRA, poly(I:C) or the combination of both using β-actin as normalizer. The values represent the mean±S.D. of three experiments. (d) TRAIL protein levels were determined in ZR-75-1 cells after 48 h treatment with 9cRA, poly(I:C) or the combination of both using β-actin as loading control. The immunoblots show representative results from three independent experiments
TLR3 is required for RA/poly(I:C)-induced TRAIL upregulation and breast cancer cell apoptosis
In order to determine the involvement of TLR3 in 9cRA/poly(I:C)-driven cell death, TLR3 expression was suppressed through transfection of small interfering RNAs (siRNAs) (Figure 7b). Depletion of TLR3 protein levels virtually abrogated 9cRA/poly(I:C)-induced apoptosis, whereas transfection with scrambled-siRNAs had no effect (Figure 7a). To further confirm the above results, we compared the level of activation of caspase-8 and -3 by protein immunoblotting in the presence or absence of TLR3 siRNAs. While the levels of cleaved caspase-8 and -3 were induced by 9cRA/poly(I:C) in control cells, TLR3 depletion abolished caspase activation by 9cRA/poly(I:C) (Figure 7b). Moreover, 9cRA/poly(I:C)-driven TRAIL induction was completely abrogated by TLR3 siRNAs (Figure 7b). These results reveal that: (i) TLR3 is critically involved in 9cRA/poly(I:C)-driven apoptosis and (ii) TLR3 mediates the induction of the death ligand TRAIL by 9cRA/poly(I:C) in SK-BR-3 cells, reinforcing our hypothesis that TRAIL mediates 9cRA/poly(I:C)-driven apoptosis.
Figure 7.

Increased apoptosis and TRAIL expression after 9cRA and poly(I:C) co-treatment are TLR3-dependent. (a) SK-BR-3 cells were transiently transfected with scrambled (open bars) or TLR3-specific siRNAs (black bars). Seventy-two hours post-transfection, cells were treated with poly(I:C) in the absence or presence of 9cRA for 24 h as indicated and assayed for PI-positive cells. The values represent the mean±S.D. of three experiments. (b) Western blot showing caspase-8 activation, induction of active caspase-3, and TLR3 and TRAIL protein levels in the same siRNA transfected conditions as described in (a). The immunoblots show representative results from three independent experiments
TRAIL induction is critically involved in RA/poly(I:C)-induced apoptosis
To confirm that TRAIL induction is responsible for 9cRA/poly(I:C)-induced apoptosis, TRAIL protein was downregulated through transfection of siRNAs (Figure 8b). Interestingly, our results show that 9cRA/poly(I:C)-induced apoptosis was highly reduced when TRAIL levels were downregulated using siRNAs (Figure 8a). To further validate this, the levels of caspase-8 and -3 activation were compared in SK-BR-3 cells transfected with siRNAs against TRAIL versus scrambled siRNAs. While caspase-8 and -3 cleavage was induced by 9cRA/poly(I:C) in scrambled siRNA-transfected cells, downregulation of TRAIL significantly abolished caspase-8 and -3 activation by 9cRA/poly(I:C) (Figure 8b). These results suggest that TRAIL plays a main role in 9cRA/poly(I:C)-driven apoptosis in SK-BR-3 cells.
Figure 8.

TRAIL knockdown by siRNA transfection protects SK-BR-3 breast cancer cells against cell death induced by 9cRA/poly(I:C). (a) SK-BR-3 human breast cancer cells were transiently transfected with scrambled (open bars) or TRAIL-specific siRNAs (black bars). Seventy-two hours post-transfection, cells were treated with poly(I:C) in the absence and presence of 9cRA for 24 h as indicated, and assayed for PI-positive cells. The values represent the mean±S.D. of three experiments. (b) Western blot showing caspase-8 activation, induction of active caspase-3, and TRAIL protein level in the same conditions as described in (a). The immunoblots show representative results from three independent experiments
Discussion
In the present study, we provide evidence that RA is able to induce the expression of TLR3, allowing breast cancer cells to sense and respond to the synthetic dsRNA poly(I:C), and driving breast cancer cells to apoptosis. Apoptosis induced by this combination treatment is a caspase-, TLR3-, type I IFN- and TRAIL-signaling-dependent process. Our findings suggest that combined treatments with RA may improve the apoptotic cellular response in those cancer cells insensitive or with low sensitivity to the anti-tumor action of synthetic dsRNA.
The anti-tumor activity of poly(I:C) is well known since decades. Recent data have shown that poly(I:C) binding to TLR3 triggers apoptosis of human cancer cells.14, 22 In addition, poly(I:C) has indirect anti-tumor activities since it is a potent inducer of type I IFNs,23 which are known to exert anti-tumor actions on a variety of tumor cells. It has been shown that IFNs have anti-proliferative and pro-apoptotic effects in an extensive panel of malignant cells.24 Since the late 1960s, numerous studies have been performed using synthetic dsRNA to inhibit tumors in experimental animal models as well as in clinical trials. The anti-tumor activity of poly(I:C) is mainly due to its ability to induce IFNs, which either kill directly tumor cells or enhance the host immune response.
On the other hand, RA, due to its strong anti-proliferative and differentiative potential, has been widely used in both cancer therapy and cancer prevention.25 RA derivatives are currently used in the treatment of APL and cutaneous T-cell lymphomas, but they are also effective on B-cell malignancies.3 In addition to the induction of differentiation, RA is able to initiate several other biological programs that may contribute to its therapeutic potential. Indeed, it has been shown that RA induces apoptosis of APL cells and APL patient blasts through selective paracrine action of TRAIL.19 In breast cancer cells, previous data have shown that RA inhibits the proliferation of a large number of cell lines and xenograft models by modulating the expression of several genetic programs. In addition to directly target neoplastic cells, accumulated evidence indicate that RA may also exerts immunomodulatory effects. To a certain extent, the efficacy of RA in cancer treatment may reside in its ability to also modulate the function of immune effectors.26
Our findings show that RA induces TLR3 expression in several breast cancer cell lines, suggesting that TLR3 activation may play an important role in RA action in breast cancer cells. Importantly, RA regulation of TLR3 expression may be indirect or depend on intermediary short-lived protein(s), since incubation with CHX abrogates induction of TLR3 mRNA by RA (data not shown). It has been previously shown that the TLR3 promoter contains an ISRE/IRF-E and a STAT-binding site.27 These consensus sites might regulate the response of TLR3 to RA since it has been shown that RA can control the expression and activity of transcription factors such as IRF1 and STAT1 in cancer cells.20, 28, 29, 30, 31 Further studies will be required to know the molecular mechanism underlying the regulation of TRL3 expression by RA. Based on our finding that TLR3 expression is enhanced by RA, we hypothesized that RA could cooperate with TLR3 agonists to enhance their anti-cancer action. In our study, we found that (i) RA is able to sensitize breast cancer cells to the anti-proliferative effects of poly(I:C) and (ii) RA does not induce a noticeable cell death in breast cancer cells by itself but it synergizes with poly(I:C) to increase cell death. This synergy is specific for poly(I:C) since another TLR3 ligand such as poly(A:U) failed to cooperate with RA, even at the relative high concentration used in our study. Our results suggest that the increased levels of TLR3 obtained following treatment with RA are sufficient to allow signaling by poly(I:C) but not by poly(A:U). In this regard, previous reports have shown a similar activity for both TLR3 ligands but with a significant lower potency for poly(A:U) compared with poly(I:C).32, 33 Probably, differences in potency might be proportional to the ability of these ligands to mediate TLR3 multimerization and subsequent signaling. Alternatively, a different affinity of TLR3 for poly(I:C) and poly(A:U) could also account for the differences found between these two TLR3 ligands.
Notably, our results also show that RA and poly(I:C) synergistically induce the expression of TLR3, MDA-5, RIG-1, and PKR, thus amplifying the cellular response to poly(I:C). Increased expression of these dsRNA sensors could enhance the susceptibility of breast cancer cells to poly(I:C)-induced apoptosis as it occurs in other cell systems where ectopic overexpression of TLR3 increases the sensitivity of low TLR3-expressing neuroblastoma cells to poly(I:C).34 Thus, our results show that RA is able to modulate the expression of key innate immune players allowing breast cancer cells to sense and respond to mimickers of viral dsRNA such as poly(I:C). Given that knockdown of TLR3 completely abolished RA/poly(IC) induced apoptosis in SK-BR-3 cells, it is unlikely that RIG-1 or other dsRNA sensors play an important role in RA/poly(I:C)-induced apoptosis although we cannot discard that these cytoplasmic dsRNA receptors could be involved in other aspects of RA/poly(I:C) crosstalk.
In response to RA/poly(I:C) treatment, SK-BR-3 cells produce high amounts of IFNβ and TRAIL as well as the activation of caspase-3 and caspase-8. It has been shown that type I IFNs can directly contribute to tumor cell death in a caspase-dependent manner,35 or indirectly contribute to the upregulation of cytotoxic molecules such as TRAIL.36, 37 Molecular events involved in cell death induced by RA/poly(I:C) co-treatment include type I IFNR signaling that seems to be required but not sufficient for apoptosis, since recombinant IFNβ, per se, or in combination with RA does not recapitulate the proapoptogenic activity of RA/poly(I:C) in breast cancer cells. However, recombinant IFNβ and poly(I:C) co-treatment is able to induce apoptosis in breast cancer cells as it has been shown for other cell types. Thus, our findings suggest that, besides of type I IFN signaling, additional signals coming from poly(I:C) are required to induce a fully apoptotic program. It is likely that IFNβ contributes to TRAIL induction by RA/poly(I:C) in breast cancer cells, as it occurs in other cellular systems but that additional signals, apart from IFNβ, are required to sense and transduce TRAIL death signaling. Considering the contribution of type I IFNs in apoptosis induced by RA/poly(I:C), it is tempting to speculate that differences in the ability to produce and/or to respond to type I IFNs consequently to RA/poly(I:C) co-treatment could explain the observed variability in RA/poly(I:C)-induced cell death in different cellular contexts. On the other hand, it has been shown that RA can synergize with IFNs to induce several biological responses. In that sense, RA directly induces the expression of two transcription factors, STAT1 and IRF1, that play central roles in the IFN signal transduction pathways.20, 28, 29, 38, 39, 40, 41 In addition, it has been proposed that RA may indirectly activate IRF1 expression by a mechanism involving NF-κB.42 RA also enhances IFNβ-induced expression and phosphorylation of STAT1 in human leukemia cells43 explaining the RA/IFN synergy found in these cells. Therefore, it is possible that RA could be able to enhance poly(I:C) signaling in breast cancer cells by boosting type I IFN signaling.
Our findings also show that the activation of caspases and induction of TRAIL expression by RA/poly(I:C) require the engagement of TLR3, as depletion of TLR3 abrogates caspase-8 and caspase-3 activation as well as TRAIL induction. TRAIL selectively induces apoptosis of transformed and virally infected cells. Several studies have suggested that TRAIL may have a role as a tumor suppressor since TRAIL induces apoptosis selectively in cancer cells, while normal cells are largely insensitive to its apoptotic activity.44 Furthermore, TRAIL signaling pathway is critically involved in the immune surveillance against primary and metastatic tumors through natural killer cells.45 We have previously reported that TRAIL expression is remarkably enhanced in breast cancer cells after RA treatment.20 Our experiments are in agreement with our previous findings, and also show that RA can synergistically cooperate with poly(I:C) to induce TRAIL. Thus, in our study TRAIL has emerged as a likely candidate to mediate the apoptotic effect of RA/poly(I:C) combination in breast cancer cells. Notably, our results show that depletion of TRAIL abrogates apoptosis induced by RA/poly(I:C), showing that TRAIL is a main player in RA/poly(I:C)-induced apoptosis. Based on our results, we propose that RA/poly(I:C) treatment induces breast cancer cell apoptosis via TLR3-dependent mechanisms. RA/poly(I:C) stimulation leads to upregulation of IFNβ in breast cancer cells, which in turn may act in an autocrine manner to induce the expression of TRAIL, which is ultimately responsible for cell death induction by RA/poly(I:C) in breast cancer cells. Additionally, as poly(I:C) exerts direct cytostatic/cytotoxic effects against a wide spectrum of tumor cells, we would expect that any poly(I:C)-responsive tumor cell line that keeps an intact RA signaling axis and retains the ability to produce and respond to type I IFNs could be able to mount a synergistic anti-tumor response to a combination of RA and poly(I:C).
Materials and Methods
Cell lines and reagents
Hs578T, H3396, T47D, ZR-75-1, and BT-474 cells were cultured in RPMI (Life Technologies, Carlsbad, CA, USA) or Dulbecco's modified media (Sigma, St Louis, MO, USA), in the case of SK-BR-3 cells, containing red phenol with 10% (v/v) fetal calf serum (Life Technologies) and 2 mM glutamine (Life Technologies). For the T47D cell line, medium was supplemented with 0.6 μg/ml insulin (Sigma). All cells were incubated under standard culture conditions (37 °C, 5% CO2).
9-cis-RA (Sigma), the RAR-specific synthetic retinoid TTNPB (Sigma) and the RXR-specific rexinoid LG-100268 (Ligand Pharmaceuticals Inc., La Jolla, CA, USA) were dissolved in ethanol and used at 1 × 10−6 M unless otherwise indicated. Poly(I:C), high molecular weight, from Invivogen (San Diego, CA, USA), was dissolved in water and used at a concentration of 50 μg/ml, with the exception of ZR-75-1 cells where 12.5 μg/ml were used. Poly(A:U) (Invivogen) was dissolved in water and used at 100 μg/ml. Type I IFNR-blocking mAb was purchased from PBL Interferon Source. Recombinant IFNβ and CHX were obtained from Sigma.
Proliferation assays
Cells were fixed in 1% of glutaraldehyde for 10 min, washed with PBS 1 × , and stained with 0.1% (w/v) crystal violet staining solution for 30 min. After extensively rising with deionized water, the cells were air-dried, the dye was taken out in 10% (v/v) acetic acid and the optical density was measured at 590 nm.
Measurement of cell death
Sub-G1 cell-population was quantified by single staining (propidium iodide, PI), according to standard procedures. Briefly, the cells were trypsinized and 2.5 × 105 cells were washed with PBS 1 × , incubated overnight at 4 °C in a hypotonic buffer containing PI (0.1% (v/v) Triton X-100, 0.1% (w/v) sodium citrate and 50 μg/ml PI), and analyzed by flow cytometry (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Western blotting
Caspase-3, -8 (Cell Signaling, Danvers, MA, USA), β-actin-HRP (Sigma), TLR3 and TRAIL (R&D Systems, Minneapolis, MN, USA) antibodies were used to probe blots of extracts prepared using Triton Lysis buffer (10 mM Tris pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% (v/v) Triton X-100, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 2 mM sodium pyrophosphate, 10% glycerol, 1 mM PMSF, and protease inhibitors cocktail). Immune complexes were detected by chemiluminescence (GE Healthcare Biosciences, Pittsburgh, PA, USA).
Gene knockdown by siRNA
SK-BR-3 breast cancer cells were seeded 24 h prior to transfection with 100 nM ON-TARGETplus SMARTpool (Dharmacon, Thermo, Lafayette, CO, USA) for scrambled, TLR3 and TRAIL siRNAs using DharmaFECT-2 as transfection reagent following the manufacturer's instructions. After 16 h, siRNA–lipid complexes were removed and cells were allowed to recover for 72 h in complete medium before ligand treatment.
RNA extraction and quantification by RT-qPCR
Total RNA was isolated using Tri-Reagent (Sigma), as described by the manufacturer, and 1 μg of RNA was used in a reverse transcription reaction using iScript cDNA synthesis kit from Bio-Rad (Berkeley, CA, USA). Quantitative PCR was performed using the FS Universal SYBR Green Master Mix (Roche, Basel, Switzerland) on a Stratagene Mx3005P real-time PCR thermal cycler. Relative changes in mRNA levels of genes were assessed using the 2(−ΔΔCT) method and normalized to the housekeeping gene β-actin. The primers used are indicated in Supplementary Table 1.
Statistical analysis
Results are expressed as mean±S.D. Statistical significance was assessed by two-tailed unpaired Student's t-test using the Microsoft Excel software (version 11.5.4) (Redmond, WA, USA). Differences were considered significant when P<0.05. Statistical significance was expressed by asterisks (*P< 0.05; **P<0.01; ***P<0.001).
Acknowledgments
We thank I Yujnovsky, MJ Latasa, and N Cabrera-Poch for critical reading of the manuscript. This work was supported by funds from the MICINN (SAF2007-63634 and SAF2010-21195), CSIC (201120E105), and Fundación Médica Mutua Madrileña (2005 × 0584). ARBV was supported by funds from the MICINN. AMJ-L is a recipient of a grant from the Spanish MICINN (Ramón y Cajal Program). LG-100268 was a gift from Ligand Pharmaceuticals Inc., to whom we are greatly indebted. Suplementary Information accompanies the paper on Cell Death and Disease website.
Glossary
- APL
acute promyelocytic leukemia
- CCL5
chemokine (C-C motif) ligand 5
- CHX
cycloheximide
- CXCL10
C-X-C motif chemokine 10
- dsRNA
double-stranded RNA
- IFN
interferon
- IFNR
interferon receptor
- IL-8
interleukin-8
- IRF
interferon regulatory factor
- ISRE/IRF-E
interferon-stimulated response element/interferon regulatory factor binding site
- MDA-5
melanoma differentiation-associated antigen-5
- NF-κB
nuclear factor-κB
- PI
propidium iodide
- PKR
dsRNA-activated protein kinase
- poly(A:U)
polyadenylic–polyuridylic acid
- poly(I:C)
polyinosinic–polycytidylic acid
- RA
retinoic acid
- 9cRA
9-cis-retinoic-acid
- RAR
retinoic acid receptor
- RIG-1
retinoic acid-inducible gene-1
- RXR
retinoid X receptor
- siRNA
small interfering RNA
- TLR3
Toll-like receptor-3
- TRAIL
tumor necrosis factor (TNF)-related apoptosis-inducing ligand
- TTNPB
4-[(E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by Ciliberto
Supplementary Material
References
- Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- Wang Z, Sun G, Shen Z, Chen S, Chen Z. Differentiation therapy for acute promyelocytic leukemia with all-trans retinoic acid: 10-year experience of its clinical application. Chin Med J (Engl) 1999;112:963–967. [PubMed] [Google Scholar]
- Chiesa MD, Passalacqua R, Michiara M, Franciosi V, Di Costanzo F, Bisagni G, et al. Tamoxifen versus Tamoxifen plus 13-cis-retinoic acid versus Tamoxifen plus Interferon alpha-2a as first-line endocrine treatments in advanced breast cancer: updated results of a phase II, prospective, randomised multicentre trial. Acta Biomed. 2007;78:204–209. [PubMed] [Google Scholar]
- Singletary SE, Atkinson EN, Hoque A, Sneige N, Sahin AA, Fritsche HA, et al. Phase II clinical trial of N-(4-Hydroxyphenyl)retinamide and tamoxifen administration before definitive surgery for breast neoplasia. Clin Cancer Res. 2002;8:2835–2842. [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Gnjatic S, Sawhney NB, Bhardwaj N. Toll-like receptor agonists: are they good adjuvants. Cancer J. 2010;16:382–391. doi: 10.1097/PPO.0b013e3181eaca65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, et al. Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010;70:2595–2603. doi: 10.1158/0008-5472.CAN-09-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Wei H, Tian Z. IFN-producing killer dendritic cells contribute to the inhibitory effect of poly I:C on the progression of murine melanoma. J Immunother. 2008;31:555–562. doi: 10.1097/CJI.0b013e31817d8e75. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Trumpfheller C, Caskey M, Nchinda G, Longhi MP, Mizenina O, Huang Y, et al. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci USA. 2008;105:2574–2579. doi: 10.1073/pnas.0711976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 2010;17:942–951. doi: 10.1038/cdd.2009.190. [DOI] [PubMed] [Google Scholar]
- Dogusan Z, Garcia M, Flamez D, Alexopoulou L, Goldman M, Gysemans C, et al. Double-stranded RNA induces pancreatic beta-cell apoptosis by activation of the toll-like receptor 3 and interferon regulatory factor 3 pathways. Diabetes. 2008;57:1236–1245. doi: 10.2337/db07-0844. [DOI] [PubMed] [Google Scholar]
- Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, et al. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis. 2008;29:1334–1342. doi: 10.1093/carcin/bgn149. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- Altucci L, Rossin A, Raffelsberger W, Reitmair A, Chomienne C, Gronemeyer H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med. 2001;7:680–686. doi: 10.1038/89050. [DOI] [PubMed] [Google Scholar]
- Clarke N, Jimenez-Lara AM, Voltz E, Gronemeyer H. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. EMBO J. 2004;23:3051–3060. doi: 10.1038/sj.emboj.7600302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, et al. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha) J Biol Chem. 2011;286:15918–15928. doi: 10.1074/jbc.M110.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvalevsky E, Rivkin L, Rachmilewitz J, Galun E, Giladi H. TLR3 signaling in a hepatoma cell line is skewed towards apoptosis. J Cell Biochem. 2007;100:1301–1312. doi: 10.1002/jcb.21119. [DOI] [PubMed] [Google Scholar]
- Absher M, Stinebring WR. Toxic properties of a synthetic double-stranded RNA. Endotoxin-like properties of poly I. poly C, an interferon stimulator. Nature. 1969;223:715–717. doi: 10.1038/223715a0. [DOI] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Suh N. Chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer. 2002;2:537–543. doi: 10.1038/nrc844. [DOI] [PubMed] [Google Scholar]
- Montrone M, Martorelli D, Rosato A, Dolcetti R. Retinoids as critical modulators of immune functions: new therapeutic perspectives for old compounds. Endocr Metab Immune Disord Drug Targets. 2009;9:113–131. doi: 10.2174/187153009788452435. [DOI] [PubMed] [Google Scholar]
- Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, et al. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem. 2003;278:21502–21509. doi: 10.1074/jbc.M301476200. [DOI] [PubMed] [Google Scholar]
- Kolla V, Lindner DJ, Xiao W, Borden EC, Kalvakolanu DV. Modulation of interferon (IFN)-inducible gene expression by retinoic acid. Up-regulation of STAT1 protein in IFN-unresponsive cells. J Biol Chem. 1996;271:10508–10514. doi: 10.1074/jbc.271.18.10508. [DOI] [PubMed] [Google Scholar]
- Matikainen S, Ronni T, Hurme M, Pine R, Julkunen I. Retinoic acid activates interferon regulatory factor-1 gene expression in myeloid cells. Blood. 1996;88:114–123. [PubMed] [Google Scholar]
- Shang Y, Baumrucker CR, Green MH. The induction and activation of STAT1 by all-trans-retinoic acid are mediated by RAR beta signaling pathways in breast cancer cells. Oncogene. 1999;18:6725–6732. doi: 10.1038/sj.onc.1203084. [DOI] [PubMed] [Google Scholar]
- Gianni M, Terao M, Fortino I, LiCalzi M, Viggiano V, Barbui T, et al. Stat1 is induced and activated by all-trans retinoic acid in acute promyelocytic leukemia cells. Blood. 1997;89:1001–1012. [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Okahira S, Nishikawa F, Nishikawa S, Akazawa T, Seya T, Matsumoto M. Interferon-beta induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol. 2005;24:614–623. doi: 10.1089/dna.2005.24.614. [DOI] [PubMed] [Google Scholar]
- Chuang JH, Chuang HC, Huang CC, Wu CL, Du YY, Kung ML, et al. Differential toll-like receptor 3 (TLR3) expression and apoptotic response to TLR3 agonist in human neuroblastoma cells. J Biomed Sci. 2011;18:65. doi: 10.1186/1423-0127-18-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, et al. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21:1251–1262. doi: 10.1038/sj.onc.1205179. [DOI] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res. 2001;7:1821–1831. [PubMed] [Google Scholar]
- Choi EA, Lei H, Maron DJ, Wilson JM, Barsoum J, Fraker DL, et al. Stat1-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand and the cell-surface death signaling pathway by interferon beta in human cancer cells. Cancer Res. 2003;63:5299–5307. [PubMed] [Google Scholar]
- Matikainen S, Ronni T, Lehtonen A, Sareneva T, Melen K, Nordling S, et al. Retinoic acid induces signal transducer and activator of transcription (STAT) 1, STAT2, and p48 expression in myeloid leukemia cells and enhances their responsiveness to interferons. Cell Growth Differ. 1997;8:687–698. [PubMed] [Google Scholar]
- Pelicano L, Li F, Schindler C, Chelbi-Alix MK. Retinoic acid enhances the expression of interferon-induced proteins: evidence for multiple mechanisms of action. Oncogene. 1997;15:2349–2359. doi: 10.1038/sj.onc.1201410. [DOI] [PubMed] [Google Scholar]
- Chelbi-Alix MK, Pelicano L. Retinoic acid and interferon signaling cross talk in normal and RA-resistant APL cells. Leukemia. 1999;13:1167–1174. doi: 10.1038/sj.leu.2401469. [DOI] [PubMed] [Google Scholar]
- Pelicano L, Brumpt C, Pitha PM, Chelbi-Alix MK. Retinoic acid resistance in NB4 APL cells is associated with lack of interferon alpha synthesis Stat1 and p48 induction. Oncogene. 1999;18:3944–3953. doi: 10.1038/sj.onc.1202802. [DOI] [PubMed] [Google Scholar]
- Percario ZA, Giandomenico V, Fiorucci G, Chiantore MV, Vannucchi S, Hiscott J, et al. Retinoic acid is able to induce interferon regulatory factor 1 in squamous carcinoma cells via a STAT-1 independent signalling pathway. Cell Growth Differ. 1999;10:263–270. [PubMed] [Google Scholar]
- Chen Q, Ma Y, Ross AC. Opposing cytokine-specific effects of all trans-retinoic acid on the activation and expression of signal transducer and activator of transcription (STAT)-1 in THP-1 cells. Immunology. 2002;107:199–208. doi: 10.1046/j.1365-2567.2002.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–4765. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med. 2002;195:161–169. doi: 10.1084/jem.20011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.