

Abstract
Polyketide 13 [=2-hydroxy-5-((6-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-2-propylchroman-4-one] and three related known compounds 7, 9 and 11 were obtained and structurally characterized from Streptomyces sundarbansensis strain, an endophytic actinomycete isolated from the Algerian marine brown algae Fucus sp. Compound 13 was obtained as the major metabolite from optimized culture conditions, by using Agar state fermentation. Due to tautomeric equilibrium, 13 in CD3OD solution was able to incorporate five deuterium atoms, as deduced by NMR and ESI-MS/MS analysis. The 2-hydroxy-γ-pyrone form was established for these metabolites based on the comparison of their experimental IR spectra with the DFT calculated ones, for both the corresponding 4-hydroxy-α-pyrone and 2-hydroxy-γ-pyrone forms. During antibacterial evaluation, compound 13 stood out as the most active of the series, showing a selective activity against the gram positive pathogenic methicillin-resistant S. aureus (MRSA, MIC = 6 μΜ), with a bacteriostatic effect.
Keywords: marine Streptomyces, antibacterial activity, phaeochromycins, density functional calculations, hydroxypyrone tautomerism
1. Introduction
Bacteria proved to be a particularly prolific resource with a surprisingly small group of taxa, which accounted for the vast majority of isolated compounds [1]. Among these taxa, actinomycetes are the most economically and biotechnologically valuable prokaryotes [2], responsible for the production of about half of the discovered bioactive secondary metabolites, including antitumor and immunosuppressive agents and antibiotics [3]. In particular, the genus Streptomyces accounts for about 80% of the actinomycete natural products reported to date [4]. Considerable attention is currently focused on the study of new actinomycetes coming from poorly researched habitats, with the aim of finding new bioactive products which are useful for biotechnological access to new potential drugs [5,6]. It has also been suggested that metabolites obtained from algae and invertebrates such as sponges, mollusks and tunicates are actually produced by microorganisms [7,8], hence marine organism-associated actinobacteria are attracting increasing interest as potential sources of natural products.
Polyketides represent a highly diverse structural class of products found also in bacteria, demonstrating varied biological functions [9]. Over the past decades, the isolation and structural characterization of shunt products from act mutants of Streptomyces coelicolor derived by classical mutagenesis led to the elucidation of several compounds, which typically show the presence of: (i) a methyl group, as in mutactin (1), dehydrated mutactin (2), SEK34 (3) and its dehydrated SEK34b (4) [10,11,12]; (ii) a phenyl group, in wailupemycin F (5) and G (6) isolated using mutational analysis [13]; (iii) a propyl chain in metabolites recently isolated from an actinomycete strain Streptomyces phaeochromogenes LL-P018, phaeochromycin A–E (7–11) [14]. A phaeochromycin E analogue, BSM1 (12) bearing methyl instead of the propyl group has also been reported [15] (Figure 1).
Figure 1.

Molecular structures of some polyketides. Arbitrary numbering is for convenience.
The aim of this work is the isolation of metabolites responsible for the antibacterial activities observed in the crude extract from a marine-derived Streptomyces sp., isolated from the algae Fucus sp., collected along the Bejaia coastline in Algeria. Pure compounds 7, 9, 11 and 13 were structurally characterized and their activities against five pathogenic bacteria were then evaluated. We also report on the tautomerism involved in the hydroxypyrone unit which is present in structures 7, 9 and 13, and we discuss a method capable of establishing the α- or γ-forms based on a comparison between experimental and DFT calculated IR spectra.
2. Results and Discussion
2.1. Production and Structural Characterization of Metabolites
A pure strain Streptomyces sp. WR1L1S8 was isolated from the inner tissue of the brown marine algae Fucus sp. collected along Bejaia coastline, Algeria and revealed 100% 16S rRNA genes sequence similarity to Streptomyces sundarbansensis MS1/7T. The representative specie was recently isolated from sediments of the Sundarbans mangrove forest in India, and reported to produce 2-allyloxyphenol [16].
A preliminary study was carried out in order to establish the influence of culture conditions on metabolites production of the strain WR1L1S8, by selecting a process of optimal nutrient medium and fermentation. According to the results, Starch Casein Agar (SCA) supplemented with 50% seawater was the best medium. Moreover, the Agar Surface Fermentation (ASF) process performed on SCA medium of the studied strain permitted a better growth inhibition of the test germs than the submerged fermentation method (SbF). The crude ethyl acetate extract of the strain culture showed promising antibacterial activities. A silica gel chromatography allowed isolation of compounds 7, 9, 11 (Figure 1) and 13 (Figure 2), in pure forms as seen by high-performance liquid chromatography (HPLC) analysis. The online HPLC-ESIMS injection of the ethyl acetate extract showed the presence of 13 as a major compound and 7, 9 and 11.
Figure 2.

Molecular structure of compound 13 and its derivative by incorporation of deuterium atoms. Numbering is for convenience.
The presence in ESI (+)-MS spectrum of the signal at m/z 353 attributable to [M + Na]+ and in ESI (−)-MS at m/z 329 corresponding to [M − H]−, suggested the molecular mass of compound 13 (Figure 2). Electron impact (EI)-MS spectrum showed an intense peak at m/z 312 corresponding to the loss of a water molecule in agreement with the ESI-MS fragmentation; high resolution experiment on m/z 312 gave the molecular composition C18H16O5.
The downfield region in 1H NMR spectrum recorded in CDCl3 exhibited three signals, each for one proton, at δH 7.39 (t, J = 8.2 Hz), 6.97 (s) and 6.91 (d, J = 8.2 Hz) assigned as belonging in a contiguous position of the same aromatic ring, and two singlets at δH 5.75 and 5.33, that the HMBC correlations observed for the latter one allowed to assign at H-4 and H-2, respectively (Table 1). A methylene group was detected by the presence of two doublets with the same geminal coupling constant (J = 16.4 Hz) at δH 4.28 and 4.20, on the same carbon at δC 38.6 and showing long range correlations with a pyrone ring (C-1 and C-4) and with an aromatic ring (C-8 and C-12). A second methylene unit was deduced by the presence of two doublets at δH 2.89 and 2.79, both HSQC correlated with the same carbon at δC 47.1 and HMBC correlated to C-12, C13, C-15 and the alkyl C-16 position. The last evidence gave indication also for the n-propyl chain at C-15 position, confirmed by the long range correlation of protons at C-16 (δH 1.88) with C-14 and C-16.
Table 1.
NMR spectroscopic data for compound 13 (400 MHz, in CDCl3 and CD3OD). δ in ppm, J in Hz.
| Position | δH (CD3OD) | δH (CDCl3) | δC, (CDCl3) | HMBC a |
|---|---|---|---|---|
| 1 | – | – | 165.4 | |
| 2 | – | 5.33, s | 89.9 | 3, 4, 5 |
| 3 | 171.3 | |||
| 4 | 5.65, s | 5.75, s | 101.2 | |
| 5 | 166.9 | |||
| 6 | 4.23, dd(16.2) | 4.20, d(16.4) | 38.6 | 1, 4, 8, 12 |
| 4.32, dd(16.2) | 4.28, d(16.4) | |||
| 7 | 133.5 | |||
| 8 | 6.99, d (8.6) | 6.99, s | 125.2 | |
| 9 | 7.48, t (8.1) | 7.39, t (8.2) | 135.4 | 7, 11 |
| 10 | 6.93, d (7.2) | 6.90, d(8.2) | 118.4 | 8, 11, 12 |
| 11 | 158.1 | |||
| 12 | 118.5 | |||
| 13 | 194.0 | |||
| 14 | – | 2.79, d (16.0) | 47.1 | 12,13,15, 16 |
| – | 2.89, d (16.0) | |||
| 15 | 102.3 | |||
| 16 | 1.89, m | 1.88, m | 43.1 | 14, 15, 17, 18 |
| 17 | 1.53, m | 1.57, m | 17.1 | 15, 16, 18 |
| 17 | 1.00, t (7.0) | 0.99 (t, 7.0) | 14.04 | 17, 16 |
a HMBC correlations are from protons(s) in the line with the indicated carbon(s).
If compared to the one recorded in CDCl3,1H NMR spectrum of 13 in CD3OD showed a fast disappearance of some signals, in detail the singlet assigned at H-2 and the two doublets for the protons at C-14 are lacking (Supplementary Figure S4). Otherwise, the ESI-MS spectrum recorded in positive ion mode detection for the same CD3OD solution of 13 showed a signal at m/z 358; comparing it with the corresponding signal at m/z 353 attributed to [M + Na]+ by direct injection of a methanol solution of 13, it was possible to deduce the incorporation of five deuterium atoms in the molecule (Figure 2). Fragmentation MS/MS experiments on m/z 358 gave diagnostic signals: (i) at m/z 338 for the loss of a D2O molecule (in agreement with the corresponding fragment at m/z 335 produced from m/z 353, Experimental Section) indicating the H/D exchange commonly occurring for the alcohol group at C-15, and for the protons at C-14 by a favored equilibrium with the enol form stabilized by conjugation with an aromatic ring; (ii) at m/z 245 deriving from the loss of a pyrone fragment bearing two deuterium atoms by the breakage in C5-C6 position ((Supplementary Figure S7). These results were confirmed by the data from ESI-MS spectrum in negative ion mode, where the signal at m/z 332 attributed to [M − D]− molecular ion for deuterated 13 was observed ((Supplementary Figure S8). This behavior of compound 13 is due to the possibility of hydroxypyrones to exist in two tautomeric forms: 4-hydroxy-α-pyrone (=2H-pyran-2-one) and 2-hydroxy-γ-pyrone (=4H-pyran-4-one), for which the deuterium exchange was observed [17]. Although compound 13 presented no appreciable optical activity, the weak Cotton effect observed by circular dichroic analysis spoke for the chiral property of the natural molecule.
Compound 13 is structurally similar to the previously reported polyketide SEK34 (3, Figure 1) by engineering biosynthesis [11]. NMR data of the two products showed that they differ only in the alkyl chain at the position of C-15, and on this basis, we can suggest the γ-pyrone form for this structure. Compound 13 represents the lacking member in the recently reported series of phaeochromycins A–E (7–11), which are the first polyketides bearing the n-propyl chain [14]. A strictly related compound corresponding to the 4-hydroxy α-pyrone form of 13 was reported with a poor structural elucidation, as a shunt product from the engineered system for the production of analogues of frenolicin B [18], so that 13 is the first example from marine derived actinomycetes.
Compound 7, structurally characterized by extensive NMR analysis and ESI-mass spectrometry (Experimental Section), was proven to be phaeochromycin B [14]. However, the assignment of a pyrone unit could not be unambiguously established by NMR correlations [14], so that further evidence avails; for this reason the method discussed below is proposed.
Similarly, structural analysis on compound 9 allowed for establishing that it was phaeochromycin C [14], even if the same ambiguity for the tautomeric pyrone assignment persisted.
Compound 11 resulted to be phaeochromycin E, through comparison of acquired NMR data, including HSQC and HMBC correlations, with these ones reported [14]. ESI-MS analysis, both in positive and negative ion mode, supported the molecular composition.
2.2. Hydroxypyrone Tautomerism
The reported γ-pyrone form for mutactin (1, Figure 1) was confirmed by X-ray analysis and by 2JC,H coupling constants observed for C-3 (δC 170 in acetone-d6) with both H-2 and H-4, whereas C-1 (δC 164.4) was coupled only to H-2 [10]. Successively, the same tautomeric form was established for the hydroxypyrone unit in SEK34 (3) structure, based on similar 13C NMR chemical shifts with mutactin [11]. Further reports of these molecular structures and new hydroxypyrone polyketides as phaeochromycins A–C (7–9, Figure 1) drew them in the α-pyrone form [14]. Based also on a general interest in the pyranone tautomerism [19], there is the need to look deeper into the structural study for this class of metabolites, with the aim of defining a method able to assign the right α- or γ-pyrone form.
Over the last decades, infrared spectroscopy has experienced a renewed role in the molecular structure elucidation of natural products, due to the availability of simulated spectra by density functional theory (DFT) calculations, used to assign the frequencies of chemical bonds through comparison with experimental data. IR spectra was calculated at a B3LYP/6-31G(d,p) level of theory for compounds 7, 9 and 13, both in 4-hydroxy-α-pyrone and in 2-hydroxy-γ-pyrone forms, and compared to the experimental FT-IR spectra ((Supplementary Figures S12–S17). In Table 2 the C=O values present in the structures of these molecules are reported.
Table 2.
DFT calculated stretching frequencies (in cm−1) for the carbonyl groups in α-pyrone (A) and γ-pyrone (B) forms and experimental values for 7, 9 and 13 and 4-hydroxy-6-methyl-2H-pyran-2-one as reference compound. Arbitrary numbering on A and B forms is for convenience.
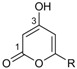 A |
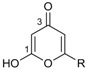 B |
||||
|---|---|---|---|---|---|
| νIRcalcd | |||||
| Compound | νIRexp. | C(1)=O | C(13)=O a | C(3)=O | C(13)=O a |
| 7 | 1637 | 1780 | 1639 | 1686 | 1642 |
| 1620 | |||||
| 1578 | |||||
| 1524 | |||||
| 9 | 1699 | 1780 | 1649 | 1683 | 1662 |
| 1645 | |||||
| 1607 | |||||
| 1598 | |||||
| 13 | 1689 | 1782 | 1678 | 1682 | 1698 |
| 1649 | |||||
| 1603 | |||||
| 1593 | |||||
| R=CH3b | 1738 | 1782 | – | 1692 | – |
| 1645 | |||||
| 1534 | |||||
It is evident that the simulated frequency for carbonyl stretching appears at 1780–1782 cm−1 in the α-pyrone form A and at 1682–1686 cm−1 in the γ-pyrone form B. The comparison with the experimental FT-IR spectra recorded for 7, 9 and 13 (showing no frequencies over 1700 cm−1) indicates that they may be described in the 2-hydroxy γ-pyrone form. In order to confirm that the method is plausible, commercial 4-hydroxy-6-methyl-2H-pyran-2-one was used. By comparing the calculated C=O frequencies for its two tautomeric forms with the experimental value (in agreement with the one reported [20]), it resulted in line with the known α-pyrone structure for this compound.
2.3. Antibacterial Evaluation
Promising activity against Gram positive S. aureus and methicillin resistant S. aureus (MRSA) and Gram negative E. coli and P. aeruginosa were observed by antimicrobial assays on the crude extract of the actinomycete WR1L1S8 strain, accounting for the evaluation on the pure isolated compounds 9, 11 and 13. They were tested preliminary on five bacterial test organisms, using the agar diffusion method. The compounds displayed similar activities toward the tested bacteria, with weak inhibition effects ((Supplementary Table S1), and a bacteriostatic activity against MRSA by compound 13. In a further investigation in order to define MIC values against MRSA, E. coli and P. aeruginosa, interestingly 13 showed selective antimicrobial activities (MIC = 2 μg/mL = 6.0 μM, Table 3) against MRSA, a bacterium responsible for human infections which is difficult to treat, showing resistance to penicillins and cephalosporins and mainly found among people with a greater risk of infection. Current findings are in line with the activities against S. aureus and MRSA bacteria, observed for the 4-hydroxy α-pyronepolyene, gombapyrone E [9]. It is noteworthy that compound 13 showed selective antibacterial activity against MRSA, but no growth inhibition against S. aureus ATCC 25923. This can be suggest that compound 13 could probably inhibit the production of Penicillin Binding Protein (PBP2a), encoded by the gene selectively involved in oxacillin or methicillin resistance S. aureus [21]. A similar behavior was reported for epicatechin gallate [22].
Table 3.
Minimum Inhibitory Concentration (MIC) values of compounds 9, 11 and 13 on some pathogenic strains.
| MIC (μg/mL) | |||
|---|---|---|---|
| Compounds | E. coli ATCC 25922 | MRSA ATCC 43300 | P. aeruginosa ATCC 27853 |
| 9 | – | >32 | >32 |
| 11 | >32 | >32 | >32 |
| 13 | 16 | 2 | >32 |
| Vancomycin | – | <2 | – |
| Gentamicin | <2 | <2 | – |
3. Experimental Section
3.1. General Experimental Procedures
All evaporations were carried out at room temperature under reduced pressure. Solvents (Merck, Milan, Italy) were used without purification. Flash chromatography (FC) was carried out on Merck Si-60 (15–25 μm) using hexane/ethyl acetate or dichloromethane/methanol gradient elution as mobile phase. TLC was performed on Merck silica gel 60 F254 and spots could be seen using UV light (λ 254 and 365 nm) and spraying with anisaldehyde/sulfuric acid reagent followed by heating. Preparative thin layer chromatography was carried out on 20 × 20 cm Merck Kiesel gel 60 F254 0.5 mm plates. Analytical HPLC-DAD-ELSD experiments were performed using a reversed phase C18 column (Hewlett Packard Hypersil BDS-C18, 250 × 4.00 mm). The mobile phase was applied as the linear gradient. Chromatographic parameters are the following: Detection—DAD 210 nm; mobile phase A acetonitrile, B water; 0–10 min 30% A–70% B, 10–30 min 100% A, flow rate 1 mL/min. Polarimetric data were obtained using a Bellingham & Stanley Limited ADP 440 apparatus, reporting [α]D in dm−1·deg mL g−1. Cotton effects were deduced by circular dichroic (CD) spectra, recorded by a Jasco J-710 spectropolarimeter. Infra-red spectra were recorded by using a FT-IR Tensor 27 Bruker spectrometer (Attenuated Transmitter Reflection, ATR configuration) at 1 cm−1 resolution in the absorption region 4000–1000 cm–1. A thin solid layer is obtained by the evaporation of a methanol solution of the sample. The instrument was purged with a constant dry air flux and clean ATR crystal as background was used. Spectra processing was made using Opus software package. NMR spectra were recorded with an Avance 400 Bruker spectrometer by using a 5 mm BBI probe; 1H at 400 MHz and 13C at100 MHz in CDCl3 (by previous treatment on basic alumina to avoid acidic traces), CD3OD or acetone-d6, δ values in ppm, relative to the solvent residual signals δH = 7.25 ppm for CDCl3, δH = 3.31 ppm for CD3OD, δH = 2.06 ppm for acetone-d6 where the solvent residual signals are relative to SiMe4 (=0 ppm); J values in Hz. Structural assignments are from Heteronuclear Single Quantum Correlation (HSQC) and Heteronuclear Multiple Bond Correlation (HMBC) experiments. NMR assignments were carried out following an arbitrary numbering (adopted for convenience), as reported in Figure 1 and Figure 2. Electron-impact (EI) mass spectra (m/z; rel.%) and HR-EI data were taken with a Kratos-MS80 mass spectrometer with home-made computerized acquisition software. ESI-MS data and tandem fragmentation spectra (MS/MS) were recorded by using a Bruker Esquire LC ion trap mass spectrometer, equipped with an ESI ion source in positive or negative ion mode as specified, through injection of the sample into the source from a methanol solution. MS conditions: source temp. 300 °C, nebulizing gas N2, 4 L/min, cone voltage 32 V, scan range m/z = 100–800. Fragmentation experiments were carried out by using helium to collisionally activate the selected primary ions.
3.2. Isolation and Identification of Streptomyces Strain WR1L1S8
Brown algae of the genus Fucus were collected in Bejaia coastline, Algeria (36°39′4.25″N; 5°25′3.88″E). Liquid tissue portions were prepared and used for the isolation of actinomycetes. Samples were spread on selective agar plates in triplicate Petri plates, within 24 h after collection, and were incubated at 28 °C for three weeks. Bacteria colonies were selected with sterile needles and purified by streaking them out onto Starch Casein Agar plates (SCA). After another seven days of growth, the only colony (strain WR1L1S8) that grew well and presented a relatively interesting antibacterial activity was transferred either to the same medium agar slants for short term storage at 4 °C and in 20% glycerol at −80 °C for long-term storage. The actinomycete strain was identified according to morphological physiological and chemotaxonomic studies. The molecular identification was determined by the partial 16S rRNA gene by direct sequencing of amplified 16S rDNA (DSMZ, Braunschweig, Germany).
3.3. Isolation and Structural Characterization of Compounds 7, 9, 11 and 13
Cultivation was carried out on 100 SCA plates using ASF process rather than liquid media (because of the apparently more abundant production of metabolites on agar media) and incubated at 28 °C for seven days. Mycelial mass together with the agar were cut into small pieces then mixed; they were thereafter sonicated in a sonication bath for 1 h and macerated overnight with 200 mL of ethyl acetate. The resulting solution was filtered using Whatman filter paper and the same maceration step with ethyl acetate was repeated. The resulting ethyl acetate broth extracts were combined and subsequently dried in a vacuum evaporator, to give the crude extract (500 mg), which was suspended in methanol (3 mL). The ethyl acetate extract was subjected to silica gel column chromatography using a gradient of hexane/ethyl acetate followed by dichloromethane/methanol, to afford 31 fractions.
Fraction 23 (36.5 mg), eluted with dichloromethane/methanol 8:2 (v/v) was purified further by preparative TLC using hexane/isopropanol 7:3 v/v as mobile phase to yield the violet compound 13 (13.6 mg; 2.72% from crude extract; Rf = 0.53) and 9 (5.6 mg; 1.12% from crude extract; Rf = 0.59). Their purity was confirmed by analytical RP-HPLC DAD at 245 nm using a gradient of acetonitrile/water 3:7 (tR = 10.6 and 15.5 min, respectively).
Combined fractions 24–25 (47.8 mg, eluted with dichloromethane/methanol 7:3 and 6:4, respectively), were further purified by preparative TLC on silica gel (ethyl acetate/isopropanol 7:3) to give pure compounds 11 (2.6 mg; 0.52% from crude extract ; Rf = 0.52) and 7 (3.7 mg; 0.74% from crude extract; Rf = 0.38). Their purity was confirmed by RP-HPLC DAD analysis at 245 nm (tR = 2.71 and 3.72 min, respectively).
Compound 7: Colorless amorphous glass. IR: 2962, 2959, 2900, 1637, 1620, 1578, 1524, 1468, 1408, 1373, 1358, 1242, 1157, 822 cm−1. NMR data (in acetone-d6): in agreement with the reported data [14]. ESI (+)-MS: m/z 353 [M + Na]+; MS/MS (353): m/z 335, 311, 241 and 222; ESI (−)-MS: m/z 329 [M − H]−, 659 [2 M − H]−; MS/MS (329): m/z 285, 267, 257; MS/MS (659): m/z 329.
Compound 9: Pale brown amorphous glass. IR: 2966, 2933, 2899, 1699, 1645, 1607, 1598, 1477, 1392, 1256, 1174, 968, 852 cm−1. NMR (in acetone-d6): in agreement with the reported data [14]. ESI (+)-MS: m/z 335 [M + Na]+, 313 [M + H]+; MS/MS (335): m/z 317, 293 and 249; MS/MS (313): m/z 295, 271, 254, 229, 201; ESI (−)-MS: m/z 311 [M − H]−; MS/MS (311): 296, 267, 249, 225, 201.
Compound 11: Pale brown amorphous glass. NMR (CD3OD): in agreement with the reported data [14]. ESI (+)-MS: m/z 269 [M + Na]+, m/z 291[M − H + 2 Na]+; ESI (−)-MS: m/z 311 [M − H]−.
Compound 13: [=2-hydroxy-5-((6-hydroxy-4-oxo-4H-pyran-2-yl) methyl)-2-propylchroman-4one]: pale violet amorphous glass. UV (MeOH) λmax(ε) 205(3240), 255 (940) and 285(800) nm; CD (MeOH, c = 3.6 × 10−3 M): Δε (230): +0.012, Δε (258): −0.012 IR: 2927, 2365, 1689, 1649, 1603, 1593, 1471, 1324, 1267, 1166, 1022, 850 cm−1. NMR data, see Table 1. ESI (+)-MS: m/z 353 [M + Na]+; MS/MS(353): m/z 335, 311, 293 and 241; ESI (−)-MS: m/z 351 [M + Na − 2 H]−, 329 [M − H]−, MS/MS (329): m/z 311, 285, 267 and 243; EI-MS: m/z (%): 330 (M+, 0.4), 312 (70), 295 (69), 268 (28), 253 (35), 226 (100); HREIMS m/z 312.0999 ± 0.0030 (calcd for C18H16O5, 312.0998); 295.0965 ± 0.0030 (calcd for C18H15O4, 295.0970).
3.4. Computational Details
Starting structures were generated by molecular mechanics minimization, using PC Model [23]. Quantum chemical calculations were performed using the Gaussian 03W revision E.01 package program set [24]. The standard basis set of choice was 6-31G (d,p) for all atoms. The gradient-corrected DFT with the three-parameter hybrid functional (B3) [25] for the exchange part and the Lee-Yang-Parr correlation function [26] were utilized. The simulations were made in vacuo. The optimized structural parameters were employed in the vibrational energy calculations at the DFT levels to characterize all stationary points as minima. For each optimized structure, no imaginary wavenumber modes were obtained, proving that a local minimum on the potential energy surface was actually found. The computed wavenumbers were scaled by factor 0.96, which is suggested for B3LYP/6-31G (d,p) calculations [27].
3.5. Antibacterial Assay
The antibacterial activities of 9, 11 and 13 were evaluated by agar dilution assay against several target organisms including Gram-negative bacteria Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853, as well as Gram-positive bacteria Staphylococcus aureus ATCC 25923, MRSA ATCC 43300 and Bacillus subtilis ATCC 6633. Erythromycin (15 μg/disc), gentamicin (10 μg/disc) and vancomycin (30 μg/disc) were used as positive controls.
The pure compounds were dissolved in DMSO (0.7 mg/mL for 9, 0.5 mg/mL for 11 and 1 mg/mL for 13). Briefly, assay plates were prepared by inoculating Mueller-Hinton agar medium with 24 h-old culture containing test organisms (107 CFU/mL). 100 μL of dissolved compounds were added to separate wells (6 mm diameter). The plates were placed at 4 °C for 2 h; zones of inhibition were recorded after 24 h of incubation at 37 °C. No growth inhibition was observed for the controls containing only the respective amount of solvent. Antimicrobial activities were assayed in triplicate.
For the purpose of MIC evaluation, each pure compound was dissolved in DMSO at concentrations mentioned above and diluted further to give required concentrations such as 2, 4, 8, 16 and 32 (μg/mL). An inoculum of 10 μL (107 CFU/mL) from culture of each test human pathogens, E. coli, MRSA and P. aeruginosa, were inoculated in each concentration plate. The plate’s cultures were incubated for 24 h at 37 °C.
MIC was defined as the lowest concentration of the purified compound showing no visible growth after overnight incubation. Vancomycin and Gentamicin were used as positive control for MRSA and E. coli, DMSO was used as negative control. Replicates were maintained for each test bacteria.
4. Conclusions
In summary, four polyketides including the major one with selective antibacterial activity against MRSA strain were isolated from the algal-derived endophytic actinomycete Streptomyces sundarbansensis, which represents the lacking member in the recently reported series of phaeochromycins A–E. We also proposed a method based on the comparison of experimental IR spectra with the DFT ones calculated in order to establish the 4-hydroxy α-pyrone or 2-hydroxy γ-pyrone tautomeric forms for these metabolites. The results indicated a γ-pyrone structure for these compounds, in analogy to the related polyketides mutactin and SEK34.
Acknowledgments
The activity of I.D. in Italy was supported by a PhD grant in the Erasmus Mundus “Averroes” project. The authors would like to thank Adriano Sterni and Damiano Avi, university of Trento for recording mass spectra and IR spectra, respectively. They are grateful to Paul Stratton, Trowbridge UK for the careful reading of the manuscript.
Supplementary Files
PDF-Document (PDF, 1078 KB)
Footnotes
Samples Availability: Available from the authors.
References
- 1.Jensen P.R., Mincer T.J., Williams P.G., Fenical W. Marine actinomycete diversity and natural product discovery. Antonie van Leeuwenhoek. 2005;87:43–48. doi: 10.1007/s10482-004-6540-1. [DOI] [PubMed] [Google Scholar]
- 2.Lam K.S. Discovery of novel metabolites from marine actinomycetes. Curr. Opin. Microbiol. 2006;9:245–251. doi: 10.1016/j.mib.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Berdy J. Bioactive microbial metabolites. J. Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- 4.Pimentel-Elardo S.M., Kozytska S., Bugni T.S., Ireland C.M., Moll H., Hentschel U. Anti-Parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs. 2010;8:373–380. doi: 10.3390/md8020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bredholt H., Fjærvik E., Johnsen G., Zotchev S.B. Actinomycetes from sediments in the Trondheim Fjord, Norway: Diversity and biological activity. Mar. Drugs. 2008;6:12–24. doi: 10.3390/md6010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stach J.E.M., Maldonado L.A., Ward A.C., Goodfellow M., Bull A.T. New primers for the class Actinobacteria: Application to marine and terrestrial environments. Environ. Microbiol. 2003;5:828–841. doi: 10.1046/j.1462-2920.2003.00483.x. [DOI] [PubMed] [Google Scholar]
- 7.De Carvalho C.C.C.R., Fernandes P. Production of metabolites as bacterial responses to the marine environment. Mar. Drugs. 2010;8:705–727. doi: 10.3390/md8030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen P.R., Fenical W. Marine bacterial diversity as a resource for novel microbial products. J. Ind. Microb. Biotechnol. 1996;17:346–351. doi: 10.1007/BF01574765. [DOI] [Google Scholar]
- 9.Park H.B., Yang H.O., Lee K.R., Kwon H.C. Gombapyrones E and F, new α-pyronepolyenes produced by Streptomyces sp. KMC-002. Molecules. 2011;16:3519–3529. [Google Scholar]
- 10.Zhang H.I., He X.G., Adefarati A., Galluci J., Cole S.P., Beale J.M., Keller P.J., Chang C.J., Floss H.G. Mutactin, a novel polyketide from Streptomyces coelicoor. Structure and biosynthetic relationship to Acinorhodin. J. Org. Chem. 1990;55:1682–1684. doi: 10.1021/jo00292a053. [DOI] [Google Scholar]
- 11.McDaniel R., Ebert-Khosla S., Hopwood D.A., Khosla C. Engineered biosynthesis of novel polyketides: ActVII and actIV genes encode aromatase and cyclase enzymes, respectively. J. Am. Chem. Soc. 1994;116:10855–10859. [Google Scholar]
- 12.McDaniel R., Ebert-Khosla S., Fu H., Hopwood D.A., Khosla C. Engineered biosynthesis of novel polyketides: Influence of a downstream enzyme on the catalytic specificity of a minimal aromatic polyketidesynthase. Proc. Natl. Acad. Sci. USA. 1994;91:11542–11546. doi: 10.1073/pnas.91.24.11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang L., Kalaitziz J.A., Nilsen G., Chen L., Moore B.S. Mutational analysis of the enterocinfavorskii biosynthetic rearrangement. Org. Lett. 2002;4:957–960. doi: 10.1021/ol0255155. [DOI] [PubMed] [Google Scholar]
- 14.Graziani E.I., Ritacco F.V., Bernan V.S., Telliez J.B. Phaeochromycins A–E, anti-inflammatory polyketides isolated from the soil actinomycete Streptomyces phaeochromogenes LL-P018. J. Nat. Prod. 2005;68:1262–1265. doi: 10.1021/np0500629. [DOI] [PubMed] [Google Scholar]
- 15.Kalaitziz J.A., Moore B.S. Heterologous biosynthesis of truncated hexaketides derived from the actinorhodin polyketide synthase. J. Nat. Prod. 2004;67:1419–1422. doi: 10.1021/np0499564. [DOI] [PubMed] [Google Scholar]
- 16.Arumugam M., Mitra A., Pramanik A., Saha M., Gachhui R., Mukherjee J. Streptomyces sundarbansensis sp. nov., an actinomycete that produces 2-allyloxyphenol. Int. J. Syst. Evol. Microbiol. 2011;61:2664–2669. doi: 10.1099/ijs.0.028258-0. [DOI] [PubMed] [Google Scholar]
- 17.Lord R.C., Phillips W.D. Exchange reactions of γ-pyrone and synthesis of deuterated pyrones. J. Am. Chem. Soc. 1952;74:2429–2430. doi: 10.1021/ja01129a082. [DOI] [Google Scholar]
- 18.Fitzgerald J.T., Ridley C.P., Khosla C. Engineered biosynthesis of the antiparasitic agent frenolicin band rationally designed analogs in a heterologous host. J. Antibiot. 2011;64:759–762. doi: 10.1038/ja.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goel A., Ram V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron. 2009;65:7865–7913. doi: 10.1016/j.tet.2009.06.031. [DOI] [Google Scholar]
- 20.Seixas de Melo J., Quinteiro G., Pina J., Breda S., Fausto R. Spectroscopic characterization of α- and γ-pyrones and their substituted 4-hydrxy and 4-methoxy derivative: An integrated infrared, photophysical and theoretical study. J. Mol. Struct. 2001;565–566:59–67. [Google Scholar]
- 21.Tenover F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006;119:3–10. doi: 10.1016/j.amjmed.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Gibbons S. Anti-Staphylococcal plant natural products. Nat. Prod. Rep. 2004;21:263–277. doi: 10.1039/b212695h. [DOI] [PubMed] [Google Scholar]
- 23.Gilbert K.E., Midland M.M. PCMODEL for Windows, version 7.00. Serena Software; Bloomington, IN, USA: 1999. [Google Scholar]
- 24.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Montgomery J.A., Vreven T., Jr., Kudin K.N., Burant J.C., et al. Gaussian Revision E. 01. Gaussian, Inc.; Wallingford, CT, USA: 2004. [Google Scholar]
- 25.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 26.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 27.Merrick J.P., Moran D., Radom L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A. 2007;111:11683–11700. doi: 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDF-Document (PDF, 1078 KB)