

Abstract
The synthesis and characterization of two generation-4 polyamidoamine (PAMAM) dendrimers with S-nitrosothiol exteriors are reported. The hyperbranched macromolecules were modified with either N-acetyl-D,L-penicillamine (NAP) or N-acetyl-L-cysteine (NACys) and analyzed via 1H and 13C-NMR, UV absorption spectroscopy, MALDI-TOF mass spectrometry, and size exclusion chromatography. Treatment of the dendritic thiols with nitrite solutions yielded the corresponding S-nitrosothiol nitric oxide (NO) donors (G4-SNAP, G4-NACysNO). Chemiluminescent NO detection demonstrated that the dendrimers were capable of storing ~2 μmol NO·mg−1 when exposed to triggers of S-nitrosothiol decomposition (e.g., light and copper). The kinetics of NO-release were found to be highly dependent on the structure of the nitrosothiol (i.e., tertiary vs. primary) and exhibited similar NO-release characteristics to classical small molecule nitrosothiols reported in the literature. As demonstration of utility, the ability of G4-SNAP to inhibit thrombin-mediated platelet aggregation was assayed. At equivalent nitrosothiol concentrations (25 μM), the G4-SNAP dendrimer resulted in a 62% inhibition of platelet aggregation, compared to only 17% for the small molecule NO donor. The multivalent NO storage, the dendritic effects exerted on nitrosothiol stability and reactivity, and the utility of dendrimers as drug delivery vehicles highlight the potential of these constructs as clinically useful S-nitrosothiol-based therapeutics.
Introduction
S-nitrosothiols (RSNO) are ubiquitous in human physiology and are involved in a multitude of cell signaling cascades.1 Endogenously, nitrosothiols are formed either via the reaction of free thiols with N2O3, the reactive nitrogen species formed following the auto-oxidation of nitric oxide (NO), or through a host of redox mechanisms involving metabolites of NO and transition metal centers.1 Nitrosation of cysteine residues on serum albumin and the nitrosation/nitrosylation of deoxyhemoglobin account for a large portion of the 1.8 μM nitrosothiol concentration in blood.2, 3 As the main carriers of NO in vivo, nitrosothiols regulate several biological processes including vasodilation, platelet activation, neurotransmission, and tissue inflammation.1, 4, 5 Synthetic nitrosothiol NO donors such as S-nitroso-N-acetyl-DL-penicillamine (SNAP) have been employed to better understand the many complicated roles of NO in regulatory biology,6 with the most notable instance including the discovery by Ignarro and co-workers that NO is the vascular endothelial derived relaxation factor.7
Numerous reports have outlined the therapeutic potential of nitrosothiol adducts, including their toxicity toward cancerous cells,8, 9 antimicrobial activity,10–13 and cardioprotective effects during ischemia/reperfusion injury.14–16 Perhaps the most documented clinical application of S-nitrosothiols is as anti-platelet agents, whereby RSNOs inhibit platelet aggregation without affecting vascular tone.17–22 Despite the promising therapeutic potential of nitrosothiols, the inability to target NO to a specific site of action and the rapid systemic clearance associated with small drug molecules has seriously hindered the clinical development of nitrosothiol therapeutics. In response, several macromolecular nitrosothiol NO donors have been synthesized to store NO and expand RSNO delivery options. For example, S-nitrosocysteine polymer conjugates have been utilized as materials to aid in the prevention of thrombosis/restenosis23, 24 and for the delivery of RSNOs in topical biomedical applications.25 Meyerhoff and co-workers have employed nitrosothiol-modified fumed silica particles as polymer dopants and demonstrated tunable NO-release from hydrophobic polymers as a function of light.26 Protein-based nitrosothiol delivery vehicles have also been explored as a strategy to extend the systemic half-lives of exogenous RSNO donors and to aid in the treatment of circulation disorders.27–30
The use of polymeric and protein drug conjugates have led to significant strides in NO delivery. Unfortunately, the NO storage capacity (pmol – nmol NO·mg−1) and/or the number of sites for covalent attachment of additional functionalities remain limited. Previous work from our laboratory has described the synthesis of diazeniumdiolate-modified polypropylenimine dendrimer conjugates as macromolecular NO donors capable of storing up to 5.6 μmol NO·mg−1 with NO-release kinetics dependent on dendrimer structure.31 Dendrimers represent attractive vehicles for NO loading because of their multivalent exterior and its ability to not only to store large amounts of NO, but its capacity for additional functionalities for increasing drug specifity.31,32, 33 Herein, we report the synthesis and characterization of two thiol-derivatized generation 4 polyamidoamine (PAMAM) dendrimers capable of storing up to 2 μmol NO·mg−1 dendrimer when converted to the corresponding nitrosothiol NO donors (Figure 1). S-nitrosothiols have received considerable attention as the primary NO carriers in blood and their decomposition mechanisms represent alternative strategies for controlled NO-release, albeit under conditions distinct from diazeniumdiolates.31 The effect of SNAP-modified G4 dendrimer on platelet aggregation is explored and compared to SNAP, the analogous small molecule nitrosothiol, to confirm the retention of biological activity of dendrimer bound RSNOs.
Figure 1.
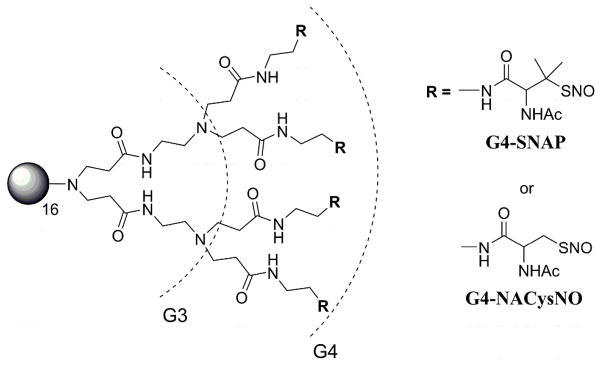
Generation 4 PAMAM containing a completely modified exterior (64 thiols) of S-nitroso-N-acetyl-D,L-penicillamine (G4-SNAP) or S-nitroso-N-acetylcysteine (G4-NACysNO).
Results and Discussion
Synthesis of Thiol Modified Dendrimers
G4-NAP (2)
Previously, Wang and co-workers developed a strategy toward increasing the cellular uptake of S-nitrosothiols in human prostate cancer cells by first reacting amino sugars with the cyclized thiol, 3-acetamido-4,4-dimethylthietan-2-one.9, 34 Utilizing a similar approach to modify the exterior amines of a generation 4 PAMAM dendrimer, N-acetyl-D,L-penicillamine was cyclized in low yields (29%) to 1 (3-acetamido-4,4-dimethylthietan-2-one) with acetic anhydride and pyridine.35 The G4-PAMAM dendrimer was then modified with thiol in one step via the reaction of the surface primary amines with thiolactone (1) (Scheme 1). Following dialysis and lyophilization, the extent of G4-NAP (2) modification was assessed via 1H NMR and the Ellman’s assay36, 37 to quantify the number of free thiols attached to the dendrimer scaffold. The integrated intensity of the protons on the geminal methyl substituents adjacent to the terminal thiols (C(CH3)SH) was compared to the interior methylene protons inside the dendrimer to confirm attachment of the N-acetyl-D,L-penicillamine groups (NMR spectra provided in Supporting Information). The number of immobilized thiol groups was calculated to be 63 thiols (NMR), correlating well with number of free thiols assayed on the dendrimer surface 62.1 ± 2.2 (Ellman’s assay).
Scheme 1.

Synthesis of S-nitrosothiol modified generation 4 PAMAM dendrimer, G4-SNAP (3).
Size exclusion chromatography (SEC) was employed to assess both the molecular weight and purity of the dendrimer conjugate. SEC revealed negligible low molecular weight impurities following the synthesis and isolation of product. However, a significant population of high molecular weight species was observed (Supporting Information). Although the N-acetyl-D,L-penicillamine-modified dendrimer contained 20% multimer by weight, the majority of the sample was the fully reduced 23,700 MW G4-NAP. Multimer formation was attributed to the presence of intra- and/or inter-dendrimer disulfide crosslinks but dendrimer conjugates were not stored/prepared under reducing conditions as such chemistry proved incompatible with subsequent S-nitrosothiol formation. High resolution MALDI-TOF mass spectrometry of 2 prepared in a sinapinic acid matrix indicated that the molecular weight of G4-NAP was 23,000 g/mol, further validating the SEC data. A summary of dendrimer characterization data is provided in Table 1.
Table 1.
Characterization data for generation 4 PAMAM thiol conjugates G4-NAP and G4-NACys and NO storage efficiency once converted to the corresponding S-nitrosothiol NO donors.
| Dendrimer | # thiols/mol | Molecular Weight | NO Storage Efficiency | |||||
|---|---|---|---|---|---|---|---|---|
| Ellman’s | 1H NMR | MALDI | SEC | % multimer | Mw/Mn | μmol NO·mg−1 | % yield | |
| G4-NAP | 62 ± 2 | 63 | 23,000 | 23,700 | 20% | 1.03 ± 0.15 | 1.7 ± 0.2 | 79% |
| G4-NACys | 49 ± 4 | 58 | 21,200 | 20,630 | 16% | 1.04 ± 0.11 | 2.1 ± 0.2 | 87% |
G4-NACys (6)
Although the synthetic procedure described above yields G4-NAP thiol-modified dendrimer in two steps, the approach results in a 100% thiol-modified exterior surface that may hinder the synthesis of multifunctional NO delivery vehicles. The opening of the thiolactone ring results in the formation of highly reactive thiols. Further conjugation chemistry to attach additional functionalities on the dendrimer would necessitate binding to the free thiols, reducing NO storage capacity. Thus, protected S-trityl-N-acetyl-L-cysteine was employed as a precursor to nitrosothiol formation allowing for future orthogonal coupling of ligands (e.g., folic acid, fluorescent dyes) to the dendritic exterior, followed by deprotection of the thiol which may then be converted to the RSNO donor. A similar approach was described previously to synthesize cysteine-modified poly(propyleneimine) dendrimers to allow native chemical ligation of both oligopeptides and recombinant proteins.38 N-acetyl-L-cysteine as trityl-protected using a mixture of triphenyl methanol and BF3-etherate in concentrated acetic acid (Scheme 2).26 Upon neutralization with saturated sodium acetate and extraction in ether, the white solid 4 was isolated (85% yield). Following activation to the succinimidyl ester 5, the S-trityl-N-acetyl-L-cysteine-protected thiol was coupled to G4 PAMAM by reaction with the dendrimer’s exterior amines. The trityl protected dendrimer conjugate was then treated with trifluoroacetic acid to remove the trityl protecting groups. The dendrimer product was dissolved in water, washed with ether, and lyophilized to yield 6 (G4-NACys) as a white flaky solid (67%).
Scheme 2.

Synthesis of G4-NACysNO, S-nitrosothiol modified generation 4 PAMAM dendrimer.
Size exclusion chromatography (SEC) of G4-NACys resulted in similar chromatograms as described above for G4-NAP, whereby 16% high molecular weight species was detected arising from dendrimers crosslinked via disulfide bridges (Supporting Information). The molecular weight of the higher molecular weight impurity calculated from light scattering data was 44,550 g/mol, approximately double the mass of the dominate G4-NACys single dendrimer (20,630 g/mol), suggesting dimer formation. MALDI-TOF mass spectrometry confirmed that the molecular weight of the prominent G4-NACys species was 21,200 g/mol. consistent with molecular weight observed in SEC. This protection/deprotection strategy employed herein may be useful in the future synthesis of multifunctional NO-releasing dendrimers as it allows for orthogonal coupling of multiple agents to the dendrimer scaffold without interfering with the thiol structure.
Conversion to S-nitrosothiol dendrimers
To convert the thiols to S-nitrosothiols, acidified nitrite solutions were employed. When compared to their alkyl nitrite counterparts, such solutions are faster and more efficient at nitrosothiol formation.39 The tertiary thiol (G4-NAP) and primary thiol (G4-NACys) dendrimers provide macromolecules with different thiol stabilities and chemical properties when converted to the corresponding NO donors. Dendrimer 2 (G4-NAP) was dissolved in a 50:50 mixture of MeOH/1M HCl and treated with NaNO2 on ice. The acidified nitrite solution rapidly turned green indicating S-nitroso-N-acetyl-D,L-penicillamine (SNAP) formation. Following removal of the solvent under reduced pressure at 0 °C, the nitrosothiol-modified dendrimer was reconstituted in cold ethanol and removed from the salt byproducts via filtration. The ethanol was again removed at 0 °C to yield 3 as a green polymer film in 92% yield. Low temperatures and shielding from light were maintained throughout the isolation procedure to prevent NO release and the formation of insoluble aggregates.
Primary nitrosothiol adducts (e.g., S-nitroso-cysteine) have been historically difficult to isolate following nitrosothiol formation due to poor RSNO stability.40 Similarly, the acidified nitrite approach described above proved troublesome when working with the G4-NACys dendrimer. Removal of the solvent led to an insoluble polymerized network of thiol dendrimers. As an alternative strategy to convert free thiols of 6 to S-nitrosothiol NO donors, the dendrimer was dissolved in a methanol/water solution, added to 200 mL of stirring toluene, and treated with isopentyl nitrite.26 After reaction with the organic nitrite source for 2 h, a pink precipitate was observed on the walls of the flask. The precipitate was removed via filtration and dried under vacuum to yield 7 (G4-NACysNO, 75%). As S-nitrosothiol dendrimer conjugates proved susceptible to thermal decomposition, low temperatures were employed during all isolation steps to minimize NO donor degradation and prevent dendrimer disulfide cross linking.
G4-SNAP and G4-NACysNO nitrosothiol dendrimer conjugates possessed characteristic absorption maxima governed by the tertiary or primary thiol structures (Figure 2). The G4-SNAP dendrimer had an intense broad band at 341 nm (π→π*) and a weaker absorption maxima at 596 nm (n→π*) giving the anti conformer of the tertiary thiol its greenish color.41, 42 The G4-NACysNO conjugate had a similar π→π* transition at 320 nm and a blue shifted n→π* transition at 545 nm.
Figure 2.

UV-Vis absorption spectra of G4-SNAP and G4-NACysNO dendrimer conjugates (2 mg/mL in EtOH solutions).
Characterization of NO Storage through RSNO Decomposition
G4-SNAP and G4-NACysNO provide two contrasting macromolecular nitrosothiol donors with NO-release properties dependent on NO donor structure. Chemiluminescent detection of NO released following RSNO decomposition was employed to characterize the NO-loading efficiency of these new NO release scaffold and study the effects of nitrosothiol structure on NO release.
Copper mediated NO-release
A major mechanism of S-nitrosothiol decomposition is mediated via the Cu+/Cu2+ redox couple and copper containing enzymatic metal centers.5, 42, 43 Previous work has confirmed that the breakdown reaction in solution proceeds via the reduction of Cu2+ by trace thiolate anion to yield Cu+ that subsequently reduces RSNO compounds to initiate NO release.43,44 This reaction does not proceed through a thiyl radical intermediate (Scheme 3A). Aqueous solution containing copper bromide (CuBr2) was utilized to decompose the S-nitrosothiol dendrimer conjugates to NO and corresponding dendritic disulfides. All copper mediated NO-release testing was performed in phosphate buffered saline (PBS, pH = 7.4) at physiological temperature (37 °C) and in the dark.
Scheme 3.
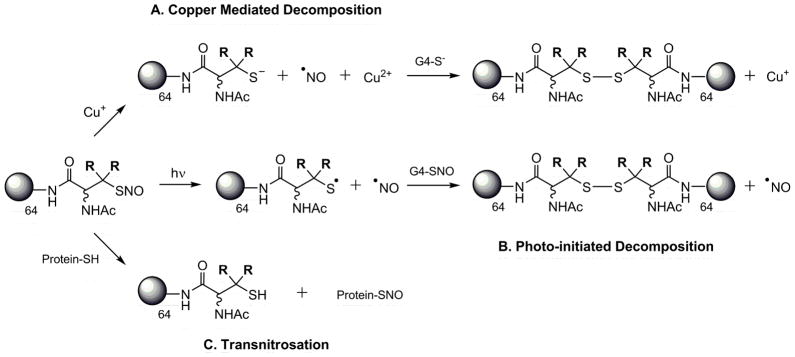
Mechanisms of nitrosothiol decomposition and NO release
The real-time NO-release profiles for copper initiated NO release from G4-SNAP (3) and G4-NACysNO (7) are shown in Figure 3 (A and B). The tertiary S-nitrosothiols of G4-SNAP exhibited a more rapid release of NO as evidenced by the maximum intensity of NO release, [NO]m. For example at 1 mM Cu2+, the [NO]m for G4-SNAP was 92,100 ppb/mg whereas the [NO]m for G4-NACysNO was only 26,200 ppb/mg. The kinetics of G4-SNAP dendrimer decomposition exhibited small changes as a function of Cu2+ concentration with 0.2 and 0.6 mM Cu2+ possessing nearly identical NO-release profiles. However, [NO]m of the primary S-nitrosothiol terminated dendrimer G4-NACysNO scaled with copper concentration (Figure 3B, Table 2) but never reaching the same intensity as the G4-SNAP NO-release. The full set of kinetic parameters including the time required to reach the maximum flux (tm) and half life of NO-release (t1/2) are given in Table 2. Despite the variable NO-release kinetics, the same level of total NO released (t[NO], Table 1) was achieved regardless of the copper concentration corresponding to 79% NO-storage efficiency for the synthesis of the G4-SNAP dendrimer and 87% for G4-NACysNO.
Figure 3.

A) NO-released from 3 (G4-SNAP) exposed to 200 μM (---), 600 μM (…...), and 1 mM (—) Cu2+ in PBS buffer at 37 °C. Additionally, two parameters used to anotate the NO-release kinetics are defined: maximum flux of NO-release ([NO]m) in ppb/mg and time required to reach the maximum (tm). B) NO-released from 7 (G4-NACysNO) exposed to 200 μM (---), 600 μM (…...), and 1 mM (—) Cu2+ in PBS buffer at 37 °C. C) Total NO-release curves of G4-SNAP and G4-NACysNO illustrating the kinetic difference between tertiary and primary nitrosothiol decomposition at 200 μM Cu2+. Two parameters used to describe NO-release data are also defined graphically: total NO released (t[NO]) in μmol NO/mg dendrimer and half-life (t1/2) of NO-release. (Data is truncated at 12 h, despite the increase of G4-NACysNO-release for up to 15 h.)
Table 2.
Kinetic parameters for NO-release from generation 4 PAMAM nitrosothiol conjugates G4-SNAP and G4-NACysNO as a function decomposition trigger and concentration.
| G4 Dendrimer | Trigger
|
t1/2 (min) | [NO]m (ppb/mg) | tm (min) | |
|---|---|---|---|---|---|
| type | [conc] | ||||
| G4-SNAP | Cu2+ | 0.2 mM | 2.3 | 63000 | 1.1 |
| 0.6 mM | 2.2 | 58700 | 1.3 | ||
| 1 mM | 1.5 | 92100 | 0.9 | ||
| light | 60 W | 200 | 1990 | 7.5 | |
| 100 W | 97 | 2360 | 9.5 | ||
| 200 W | 34 | 5800 | 2.0 | ||
| 37 °C | -- | ND | 158 | 6.4 | |
|
| |||||
| G4-NACysNO | Cu2+ | 0.2 mM | 106 | 2510 | 2.6 |
| 0.6 mM | 16 | 15900 | 3.1 | ||
| 1 mM | 7 | 26200 | 2.7 | ||
| light | 60 W | 97 | 1480 | 10.1 | |
| 100 W | 90 | 1790 | 24.4 | ||
| 200 W | 49 | 2210 | 27.4 | ||
| 37 °C | -- | ND | 116 | 8.8 | |
In the development of S-nitrosothiol therapeutics, copper-mediated NO-release holds minimal promise due the lack of free-circulating Cu2+. Ascorbate has often been mistaken as another trigger to initiate S-nitrosothiol decomposition as its concentration in the blood is appreciable (81 μM).45 However, ascorbate mediates NO release only through the reduction of trace metal ions in solution which then promote NO release.46 In the presence of EDTA, the dendritic nitrosothiols exhibited no increase in the rate of NO release upon exposure to concentrations of ascorbate up to 2 mM, suggesting that ascorbate is not a viable initiator of S-nitrosothiol dendrimer decomposition (data not shown). Of note, this does not rule out the possibility of other agents capable of facilitating the one-electron reduction to release NO for future therapeutic applications.5
Photoinitiated NO-release
Photoinitiated NO-release is another well-characterized mechanism of RSNO decomposition.47 Irradiation at either RSNO absorption maximum has been shown to result in homolytic cleavage of the S-N bond to yield NO and a thiyl radical in a process that is approximately first-order.47 Electron paramagnetic resonance spectra of spin-trapped thiyl radicals have been reported as concrete evidence for the phototriggered homolysis of the S-N bond.48 Photolysis of dendrimer nitrosothiol conjugates with a broad spectrum white light source also yielded a dendritic thiyl radical and NO (Scheme 3B). The reaction is further propagated when the resulting thiyl radical reacts with a neighboring RSNO branch to produce a disulfide and additional NO (Scheme 3B). Consistent with previously reported observations for nitrosothiol decomposition,26 the NO-release from G4-SNAP and G4-NACysNO dendrimers was dependent on light intensity and followed first order decay (Table 2). The nitrosothiol-modified dendrimers were also extremely responsive to the light source and exhibited a clear on/off behavior when the light was removed. The behavior of G4-SNAP exposed to repeat cycles of 2 min light irradiation followed by 3 min of darkness is shown in Figure 4A. When the 200 W light source was removed, the signal rapidly returned to baseline. The intensities of [NO]m slowly declined during subsequent irradiations, as expected with the depletion of RSNO. Homolytic cleavage of the S-N bond may also result via thermal decomposition of RSNO donors, albeit at a much slower rate. The difference between the slow thermal decomposition of G4-SNAP dendimer (9.31 pmol NO·mg−1·s−1, indicated by the asterisk) and the rate of NO-release upon exposed to 200 W light is shown in Figure 4B.
Figure 4.

A) Phototriggered on/off behavior of G4-SNAP dendrimer conjugate (3) at 200 W. Sample was irradiated at 2 min intervals followed by 3 min of darkness B) Permanent irradiation of G4-SNAP at 200 W illustrating the long first order decay over several hours. The * signifies thermal NO-release at 37 °C prior to exposing the dendrimer to the light source. (Inset: Total NO release curve for G4-SNAP.)
Overall, G4-SNAP dendrimer was more susceptible to changes in light intensity than G4-NACysNO. Both dendrimers resulted in similar half-lives and [NO]m at 200W (Table 2) but the most significant kinetic variation between the two conjugates was observed at early periods (<30 min). The initial rates of phototriggered decomposition for the G4-NACysNO dendrimer were also slower than the G4-SNAP conjugate as indicated by the time required to reach the maximum flux (tm) (Table 2). This initial delay and lower intensity of G4-NACysNO release is explained by a “cage effect” imposed by the G4-NACysNO dendrimer conjugate whereby a more compact solution structure results in the increased frequency of geminate radical pair recombination and slows the initial rates of NO release.49,50 Indeed, Shishido et al. have demonstrated that photogeneration of NO from S-nitroso-N-acetylcysteine in a PEG matrix resulted in a 22-fold decrease in the intial rate of RSNO decomposition when compared to normal aqueous media.50 Additionally, the PEG microenvironment reduced the rates of thermal decomposition at room temperature. The increased stability of the macromolecular G4-NACysNO structure suggests a “dendritic effect” on phototriggered NO release, whereby the G4-NACysNO solution structure may be more compact that G4-SNAP and thus lead to increased rates of radical recombination. This “cage effect” may be manipulated for future biomedical applications by increasing dendrimer size (G5–G8) or through the attachment of poly(ethylene glycol) functionalities to the dendrimer exterior leading to increased storage and handling of S-nitrosothiols.
S-Nitrosothiol Inhibition of Platelet Aggregation
Free nitric oxide inhibits platelet aggregation via a guanylyl-cyclase dependent mechanism.17 However, the signaling actions of RSNOs are not completely dependent on the release of free NO and the mechanism by which they deliver NO to platelets is much debated.51 Transnitrosation5 between RSNO and membrane bound protein thiols provides a mechanism for direct cellular communication and transfer of NO that does not proceed through a thiyl radical intermediate. Transnitrosation can be defined as the transfer of the nitroso functional group from a nitrosothiol donor to a free thiol as shown in Scheme 3C. Indeed, transnitrosation reactions between exogenous NO donors and proteins on the platelet surface are well known.52–54
The efficacy of the G4-SNAP dendrimer conjugate at inhibiting thrombin-catalyzed platelet aggregation was examined to compare the biological activity of the RSNO-modified dendrimers to free small molecule SNAP. The platelet aggregation of small molecule SNAP and G4-SNAP dendrimer as monitored by the loss of turbidity in solution using an aggregometer is shown in Figure 5. A dose-dependent inhibition of platelet aggregation was observed for the SNAP NO donor with 75% inhibition observed at 100 μM SNAP (Figure 5A). Solutions prepared with equimolar RSNO functionalites on G4-SNAP dendrimer (estimated at 64 RSNO per mol dendrimer) exhibited a pronounced inhibition of thrombin-mediated aggregation regardless of dendrimer concentration (Figure 5B). At 25 μM SNAP functionality on the G4-SNAP conjugate (390 nM dendrimer), a 62% reduction in aggregation was observed compared to only a 17% inhibition for 25 μM small molecule SNAP. The abundance of RSNO donors localized on the dendritic structure was far more effective than small molecule SNAP at reducing platelet aggregation. We speculate that the dendritic structure may facilitate interaction with the platelet surface effectively creating a greater local concentration of RSNO moieties and thereby lowering the effective dose of SNAP required to elicit the same therapeutic effect as free SNAP in solution. Control G4-NAP (2) and N-acetyl-D,L-penicillamine without NO were also evaluated at concentrations up to 100 μM and showed minimal effects on platelet aggregation. The inactivity of the thiol controls and the absence of SNAP decomposition triggers in these experiments (e.g., light, copper, etc.) indicate that the observed inhibition of platelet aggregation was mediated via transnitrosation and any resulting nitrogen species derived from transnitrosation. 52–54
Figure 5.

Human thrombin-initiated platelet aggregation in the presence of A) SNAP small molecule or B) G4-SNAP dendrimer conjugate. Turbity changes of the 0–100 uM RSNO donors were normalized to the DMSO signal (100%). C) Normalized percent aggregation for SNAP (gray) and G4-SNAP dendrimer (white). Control represents aggregation in 100 μM thiol control (N-acetyl-D,L-penicillamine or G4-NAP. Error is represented as standard error of the mean for two independent blood samples tested n=3 times each.
Conclusions
The synthesis of thiol-modified G4-PAMAM dendrimers and their conversion to S-nitrosothiol NO donors represents a new class of NO-releasing dendrimer constructs. The dendrimer scaffold enables the enhanced storage of RSNO donors relative to previously reported nitrosothiol delivery vehicles. For example, G4-PAMAM (MW 14,215 g/mol) has 64 surface sites for NO loading (Figure 1) compared to only 14 for a fully reduced serum albumin conjugate (MW ~66,000g/mol).27 The G4-SNAP and G4-NACysNO dendrimers effectively stored and released 1.7 ± 0.2 and 2.1 ± 0.2 μmol NO·mg−1, respectively, when using known triggers of nitrosothiol decomposition such as Cu2+ or light. S-nitrosothiol modified dendrimers serve as large storage reservoirs of NO and may thus prove useful for polymeric topical wound dressing formulations, for example. When exposed to a broad spectrum white light source, the rapid burst of NO from such dressings may release NO at concentrations necessary to kill bacteria that cause infection.10,11 Indeed, NO has recently been shown to kill even the most robust bacteria, including methicillin-resistant S. aureus (MRSA).10
The ability to undergo transnitrosation reactions with biological substrates (e.g., platelets) allows for careful control of NO-release previously unattainable with diazeniumdiolate-modified dendrimers.31 Via transnitrosation, the anti-platelet activity of G4-SNAP dendrimer was 45% more effective at preventing platelet aggregation at 25 μM than the corresponding small molecule NO donor SNAP. These RSNO dendrimers may serve as stable carriers of NO in vivo but targeting NO to specific tissues or cell types is challenging. The biggest advantage of the dendritic scaffold over other potential RSNO drug delivery vehicles is the ease in which multiple surface functionalities may be appended to the dendrimer exterior. For example, Baker and co-workers have synthesized dendrimers containing the methotrexate (an anti-cancer drug), fluorescent probes for imaging, and folic acid residues for targeting the delivery vehicle to cancerous tissue in vivo.55 Work is underway in our laboratory to synthesize multi-functional dendrimer conjugates that integrate the recent advancements in nanoparticle drug delivery with the current mechanistic understanding of nitrosothiols in physiology to create highly specialized NO-releasing therapeutics.
Experimental Section
General
Generation 4 polyamidoamine dendrimer (G4-PAMAM), N-acetyl-L-cysteine, trityl chloride, triphenylmethanol, boron trifluoride diethyl etherate (BF3-etherate), isopentylnitrite, L-glutathione, ethylenediamine tetraacetate (EDTA), N-hydroxy succinimide, dicyclohexylcarbodiimide, 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), and triethylamine were purchased from the Aldrich Chemical Company (Milwaukee, WI). N-acetyl-D,L-penicillamine and anhydrous sodium bisulfate were purchased from Fluka. Sodium nitrite and anhydrous pyridine were purchase from Acros Organic. All other common laboratory salts and solvents were purchased from Fisher Scientific. Water was purified using a Millipore Milli-Q gradient A-10 purification system (Bedford, MA). Spectra/Por® Float-A-Lyzers® were purchased from Spectrum Laboratories Inc. (Rancho Dominguez, CA). Absorption spectra were recorded on a Perkin Elmer Lambda 40 UV-Vis spectrophotometer (Norwalk, CT). Nuclear magnetic resonance (NMR) spectra were collected in CDCl3 and D2O (Sigma-Aldrich) using a 400-MHz Bruker Nuclear Magnetic Resonance spectrometer. Mass spectra were acquired on a Kratos MALDI AXIMA CFR plus instrument in linear mode (2 GHz), power 160. Dendrimer samples for mass analysis were prepared at a concentration of 2 mg/mL in a sinapinic acid matrix, 1:1 CH3CN/TFA (0.05%). Size exclusion chromatography was performed on a Wyatt DAWN EOS light scattering instrument interfaced with an Amersham Biosciences Akta FPLC system equipped with a Wyatt Optilab refractometer and Wyatt dynamic light scattering module. Nitric oxide release was measured using a Sievers 280i Chemiluminesce Nitric Oxide Analyzer (Boulder, CO) as described previously.56
Synthesis and characterization of dendrimer conjugates
Complete synthetic details for nitrosothiol modified dendrimer conjugates and all characterization data may be found in Supporting Information.
NO-release testing of nitrosothiol containing dendrimers
Chemiluminescence57 was employed to quantify the NO released from the S-nitrosothiol dendrimer conjugates upon exposure to various triggers previously identified to initiate nitrosothiol decomposition in the literature (e.g., copper, light, ascorbate and temperature). Total NO release (t[NO], μmol NO/mg) was used to quantify storage efficiency of the thiol-modified dendrimers. The percent yield values in Table 1 were calculated based on 64 theoretical amines on the dendrimer starting material, the successful conversion of these amines to 64 surface thiols, and their modification to 64 S-nitrosothiol moieties. Aliquots (10 μL) of the dendrimer products in 5 mM EDTA in water for G4-NACysNO or EtOH for G4-SNAP were added to a reaction flask containing 10 mM phosphate buffered saline (PBS, pH=7.4) with 5 mM EDTA to chelate trace copper in the media. A large excess of EDTA was required to completely remove initial bursts of NO-release attributed to Cu2+ ion impurities in solution. Chelator was not employed during Cu2+-mediated NO-release testing. All NO-release testing was conducted at physiological temperature (37 °C) except when testing the rates of thermal decomposition. Dendrimer concentrations in the 30 mL reaction flask ranged from 300–700 nM. The chemiluminescence analyzer was calibrated with NO gas (24.1 ppm). The value for converting instrument response (ppb) to moles of NO was obtained via the conversion known concentrations of nitrite standards to NO in a 0.1 M KOH/H2SO4 solution (1.34 × 10 −13 moles NO/ppb). During photo-initiated NO release experiments 60, 100, or 200 W bulbs were employed (Crystal Clear, General Electric). The light source was positioned ~5 inches from the sample. Copper solutions were prepared by dissolving CuBr2 in the running buffer (200 –1000 μM) prior to degassing the sample.
Chemiluminescence data for the NO-releasing dendrimers were represented in two graphical forms or plots: 1) chemiluminescence response in ppb NO/mg dendrimer versus time; and, 2) the total amount of NO-release (t[NO], μmol NO/mg) versus time. Kinetic parameters including the maximum flux of NO release ([NO]m) and the time required to reach that maxima (tm) were obtained from plot 1 while the half-life (t1/2) of NO release was determined from plot 2.
Platelet rich plasma isolation
Fresh platelets were prepared from venous blood drawn from healthy volunteers in accordance with institutional guidelines. Whole blood (42.5 ml blood to 7.5 ml anticoagulant) was drawn into a syringe containing citrate (3.2% by weight, pH = 7.4). Platelet rich plasma (PRP) was obtained by centrifugation (200 × g, 20 minutes) at 25 °C. The platelet rich plasma was centrifuged at 1000 × g for 10 min at room temperature to obtain a platelet pellet. The platelet pellet was suspended in the original volume of Tyrode’s buffer (without divalent cations, 137 mM NaCl, 3 mM KCl, 3 mM sodium phosphate, 2 mM sodium bicarbonate, 5 mM glucose, 10 mM Hepes) and centrifuged as in the last step. The final pellet was suspended in Tyrode’s buffer at 200,000 platelets/μL, as measured with a Heska hematological analyzer.
Platelet aggregation studies
Aliquots of platelets in Tyrode’s buffer at 200,000 platelets/μl were placed in the optical cuvettes in the aggregometer (Chrono-Log Whole Blood Aggregometer) and equilibrated to 37 °C while stirring. DMSO stocks of NO donor compounds (or neat DMSO as a carrier control) were diluted 1/100 into the stirred platelet suspensions and allowed to incubate for 15 min. Aggregation reactions were initiated by adding 100 × stocks of CaCl2 for a final calcium concentration of 10 mM and 100 NIH unit/ml stocks of human α-thrombin for a final concentration of 1 NIH unit/ml. The degree to which test agents inhibited thrombin-mediated platelet aggregation was estimated from the final extent of aggregation (turbidity change) at 25 min. The turbidity changes of each sample were normalized to the turbidity observed for the DMSO carrier. Dose response curves represent the average of two independent sets of measurements on different days with different blood donors tested n=3 times each. The error bars represent standard error of the mean between the two sets of measurements.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH EB000708) and the North Carolina Biotechnology Center (20006-MRG-1116). An Analytical Graduate Fellowship from Pfizer is gratefully acknowledged (NAS). We also thank Kenyon Evans-Nguyen and Ashutosh Tripathi for product analysis.
Footnotes
Supporting Information Available: Experimental details on the synthesis and characterization of PAMAM dendrimer conjugates.
References
- 1.Hogg N. Annu Rev Pharmacol Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 2.Tyurin VA, Tyurina YY, Liu S-X, Bayir H, Hubel CA, Kagan VE. Methods in Enzymology. 2002;352:347–360. doi: 10.1016/s0076-6879(02)52032-2. [DOI] [PubMed] [Google Scholar]
- 3.Stamler JS. Circ Res. 2004;94:414–417. doi: 10.1161/01.RES.0000122071.55721.BC. [DOI] [PubMed] [Google Scholar]
- 4.Al-Sa’doni HH, Ferro A. Curr Med Chem. 2004;11:2679–2690. doi: 10.2174/0929867043364397. [DOI] [PubMed] [Google Scholar]
- 5.Hogg N. Free Radical Biol Med. 2000;28:1478–1486. doi: 10.1016/s0891-5849(00)00248-3. [DOI] [PubMed] [Google Scholar]
- 6.Gref R, Minamitake Y, Paracchia MT, Trubetskoy V, Torchilin VP, Langer R. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 7.Ignarro LJ. Angew Chem Int Ed. 1999;38:1882–1892. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 8.Babich H, Zuckerbraun HL. Toxicol in Vitro. 2001;15:181–190. doi: 10.1016/s0887-2333(01)00006-6. [DOI] [PubMed] [Google Scholar]
- 9.Hou Y, Wang J, Andreana PR, Cantauria G, Tarasia S, Sharp L, Braunschweiger PG, Wang PG. Bioorg Med Chem Lett. 1999;9:2255–2258. doi: 10.1016/s0960-894x(99)00363-7. [DOI] [PubMed] [Google Scholar]
- 10.de Souza GFP, Yokoyama-Yasunaka JKU, Seabra AB, Miguel DC, de Oliveira MG, Uliana SRB. Nitric Oxide. 2006;15:209–216. doi: 10.1016/j.niox.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Fang FC. Nat Rev Microbiol. 2004;2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 12.Mannick JB. Proc Am Thorac Soc. 2006;3:161–165. doi: 10.1513/pats.200505-048BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schapiro JM, Libby SJ, Fang FC. Proc Natl Acad Sci U S A. 2003;100:8496–8501. doi: 10.1073/pnas.1033133100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell RM, Maddock HL, Yellon DM. Cardiovasc Res. 2003;57:405–415. doi: 10.1016/s0008-6363(02)00675-2. [DOI] [PubMed] [Google Scholar]
- 15.Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. J Pharmacol Exp Ther. 1995;274:200–206. [PubMed] [Google Scholar]
- 16.Schulz R, Kelm M, Heusch G. Cardiovasc Res. 2004;61:402–413. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Mellion BT, Ignarro LJ, Myers CB, Ohlstein EH, Ballot BA, Hyman AL, Kadowitz PJ. Mol Pharmacol. 1983;23:653–664. [PubMed] [Google Scholar]
- 18.Radomski MW, Rees DD, Dutra A, Moncada S. Br J Pharmacol. 1992;107:745–749. doi: 10.1111/j.1476-5381.1992.tb14517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Belder AJ, MacAllister R, Radomski MW, Moncada S, Vallance PJT. Cardiovasc Res. 1994;28:691–694. doi: 10.1093/cvr/28.5.691. [DOI] [PubMed] [Google Scholar]
- 20.Radomski MW, Moncada S. Adv Molec Cell Biol. 1997;18:367–381. [Google Scholar]
- 21.Molloy J, Martin JF, Baskerville PA, Fraser SCA, Markus HS. Circulation. 1998;98:1372–1375. doi: 10.1161/01.cir.98.14.1372. [DOI] [PubMed] [Google Scholar]
- 22.Salas E, Langford EJ, Marrinan MT, Martin JF, Moncada S, Debelder AJ. Heart. 1998;80:146–150. doi: 10.1136/hrt.80.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohl KS, West JL. Biomaterials. 2000;21:2273–2278. doi: 10.1016/s0142-9612(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 24.Bohl-Masters KS, Lipke EA, Rice EEH, Liel MS, Myler HA, Zygourakis C, Tulis DA, West JL. J Biomater Sci Polymer Edn. 2005;16:659–672. doi: 10.1163/1568562053783722. [DOI] [PubMed] [Google Scholar]
- 25.Seabra AB, da Silva R, de Oliveira MG. Biomacromolecules. 2005;6:2512–2520. doi: 10.1021/bm050216z. [DOI] [PubMed] [Google Scholar]
- 26.Frost MC, Meyerhoff ME. J Biomed Mater Res, Part A. 2005;72:409–419. doi: 10.1002/jbm.a.30275. [DOI] [PubMed] [Google Scholar]
- 27.Ewing JF, Young DV, Janero DR, Garvey DS, Grinnell TA. J Pharmacol Exp Ther. 1997;283:947–954. [PubMed] [Google Scholar]
- 28.Katsumi H, Nishikawa M, Ma SF, Yamashita F, Hashida M. J Pharm Sci. 2004;93:2343–2352. doi: 10.1002/jps.20147. [DOI] [PubMed] [Google Scholar]
- 29.Katsumi H, Nishikawa M, Yamashita F, Hashida M. J Pharmacol Exp Ther. 2005;314:1117–1124. doi: 10.1124/jpet.105.087429. [DOI] [PubMed] [Google Scholar]
- 30.Marks DS, Vita JA, Folts JD, Keaney JF, Jr, Welch GN, Loscalzo J. J Clin Investig. 1995;96:2630–2638. doi: 10.1172/JCI118328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stasko NA, Schoenfisch MH. J Am Chem Soc. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 32.Boas U, Heegaard PMH. Chem Soc Rev. 2004;33:43–63. doi: 10.1039/b309043b. [DOI] [PubMed] [Google Scholar]
- 33.Frechet JMJ. J Polym Sci, Part A: Polym Chem. 2003;41:3713–3725. [Google Scholar]
- 34.Hou Y, Wang J-Q, Ramirez J, Wang PG. Methods in Enzymology. 1999;301:242–249. doi: 10.1016/s0076-6879(99)01088-5. [DOI] [PubMed] [Google Scholar]
- 35.Moynihan HA, Roberts SM. J Chem Soc Perkin Trans. 1994;1:797–805. [Google Scholar]
- 36.Ellman GL. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 37.Van Horn JD, Bulaj G, Goldenberg DP, Burrows CJ. J Biol Inorg Chem. 2003;8:601–610. doi: 10.1007/s00775-003-0454-7. [DOI] [PubMed] [Google Scholar]
- 38.van Baal I, Malda H, Synowsky SA, van Dongen JLJ, Hackeng TM, Merkx M, Meijer EW. Angew Chem, Int Ed. 2005;44:5052–5057. doi: 10.1002/anie.200500635. [DOI] [PubMed] [Google Scholar]
- 39.Patel HMS, Williams DLH. J Chem Soc Perkin Trans. 1990;2:37–42. [Google Scholar]
- 40.Askew SC, Barnett DJ, McAninly J, Williams DLH. J Chem Soc, Perkin Trans. 1995;2:741–745. [Google Scholar]
- 41.Bartberger MD, Houk KN, Powell SC, Mannion JD, Lo KY, Stamler JS, Toone EJ. J Am Chem Soc. 2000;122:5889–5890. doi: 10.1021/ja0109390. [DOI] [PubMed] [Google Scholar]
- 42.Williams DLH. Acc Chem Res. 1999;32:869–876. [Google Scholar]
- 43.Dicks AP, Swift HR, Williams DLH, Butler AR, Al-Sa’doni HH, Cox BG. J Chem Soc Perkin Trans. 1996;2:481–487. [Google Scholar]
- 44.Singh RJ, Hogg N, Goss SPA, Antholine WE, Kalyanaraman B. Arch Biochem Biophys. 1999;372:8–15. doi: 10.1006/abbi.1999.1447. [DOI] [PubMed] [Google Scholar]
- 45.Frei B, England L, Ames BN. Proc Natl Acad Sci U S A. 1989;86:6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scorza G, Pietraforte D, Minetti M. Free Radic Biol Med. 1997;22:633–642. doi: 10.1016/s0891-5849(96)00378-4. [DOI] [PubMed] [Google Scholar]
- 47.Sexton DJ, Muruganandam A, McKenney DJ, Mutus B. Photochem Photobiol. 1994;59:463–467. doi: 10.1111/j.1751-1097.1994.tb05065.x. [DOI] [PubMed] [Google Scholar]
- 48.Singh RJ, Hogg N, Joseph J, Kalyanaraman B. J Biol Chem. 1996;271:18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- 49.Lu J-M, Wittbrodt JM, Wang K, Wen Z, Schlegel HB, Wang PG, Cheng J-P. J Am Chem Soc. 2001;123:2903–2904. doi: 10.1021/ja000384t. [DOI] [PubMed] [Google Scholar]
- 50.Shishido SM, de Oliveira MG. Photochem Photobiol. 2000;71:273–280. doi: 10.1562/0031-8655(2000)071<0273:pgmrtr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 51.Wanstall JC, Homer KL, Doggrell SA. Curr Vasc Pharmacol. 2005;3:41–53. doi: 10.2174/1570161052773933. [DOI] [PubMed] [Google Scholar]
- 52.Walsh GM, Leane D, Moran N, Keyes TE, Forster RJ, Kenny D, O’Neill S. Biochemistry. 2007;46:6429–6436. doi: 10.1021/bi0620712. [DOI] [PubMed] [Google Scholar]
- 53.Root P, Sliskovic I, Mutus B. Biochem J. 2004;382:575–580. doi: 10.1042/BJ20040759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bell SE, Shah CM, Gordge MP. Biochem J. 2007;403:283–288. doi: 10.1042/BJ20061146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JE., Jr Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 56.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mater. 2003;15:4193–4199. [Google Scholar]
- 57.Beckman JS, Conger KA. Methods. 1995;7:35–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.