

Abstract
Approximately 15% of gastrointestinal stromal tumors (GISTs) in adults and 85% in children lack mutations in KIT and PDGFRA and are known as wild type GISTs. Wild type GISTs from adults and children express high levels of insulin-like growth factor 1 receptor (IGF1R) and exhibit stable genomes compared to mutant GISTs. Pediatric wild type GISTs, GISTs from the multi-tumor Carney-Stratakis syndrome and the Carney triad share other clinico-pathological properties (e.g. early-onset, multifocal GISTs with epitheliod cell morphology) suggesting a common etiology. Carney-Stratakis is an inherited association of GIST and paragangliomas caused by germline mutations in succinate dehydrogenase (SDH) genes. The connection between defective cellular respiration and GIST pathology has been strengthened by the utilization of SDHB immunohistochemistry to identify SDH deficiency in pediatric GISTs, syndromic GISTs, and some adult wild type GISTs. SDHB and IGF1R expression was examined in 12 wild type and 12 mutant GIST cases. Wild type GISTs were screened for coding-region alterations in SDH genes and for chromosomal aberrations using genome-wide SNP and MIP arrays. SDHB-deficiency, identified in 11/12 wild type GIST cases, was tightly associated with over-expression of IGF1R protein and transcript. Biallelic inactivation of the SDHA gene was a surprisingly frequent event, identified in 5/11 SDHB-negative cases, generally due to germline point mutations accompanied by somatic SDHA allelic losses. As a novel finding, inactivation of the SDHC gene from a combination of a heterozygous coding-region mutation and hyper-methylation of the wild type allele was found in one SDHB-negative case.
Introduction
Gastrointestinal stromal tumors (GISTs) are mesenchymal neoplasms of the gastrointestinal tract that are thought to arise from the interstitial cells of Cajal or from less differentiated mesenchymal precursor cells (Kindblom et al., 1998; Sircar et al., 1999). Most GISTs express the type III receptor tyrosine kinase (RTK) KIT (CD117) (Medeiros et al., 2004; Tarn et al., 2005), and approximately 85-90% of sporadic adult GISTs possess gain-of-function mutations in KIT or in the related receptor PDGFRA (Hirota et al., 1998; Rubin et al., 2001; Tarn et al., 2005). Recently, activating mutations in the serine-threonine kinase BRAF have been identified in a small number of GIST cases (Agaram et al., 2008b; Agaimy et al., 2009; Belinsky et al., 2009; Hostein et al., 2010). Therapy with the tyrosine kinase inhibitor imatinib is initially effective for most KIT/PDGFRA mutant GISTs (Debiec-Rychter et al., 2006; Heinrich et al., 2008) but is generally less effective for the ~ 10-15% of GISTs that are “wild type” for these receptors (Heinrich et al., 2008; Janeway et al., 2009).
GISTs also occur in children, and ~ 85% of these tumors are wild type for KIT and PDGFRA (Janeway et al., 2007; Agaram et al., 2008a). Wild type GISTs from adults and children express higher levels of the insulin-like growth factor 1 receptor (IGF1R) than mutant GISTs (Prakash et al., 2005; Agaram et al., 2008a; Belinsky et al., 2008; Tarn et al., 2008; Pantaleo et al., 2009), and IGF1R may be a potential therapeutic target in wild type GIST (Tarn et al., 2008). Overexpression of IGF1R may be indicative of a more generalized gene expression profile common to wild type GISTs (Prakash et al., 2005; Agaram et al., 2008a; Pantaleo et al., 2011b), potentially characterized by expression of genes restricted to neural tissue (Pantaleo et al., 2011b). Wild type GISTs from adults and children also exhibit relatively stable genomes as compared to mutant GISTs (Janeway et al., 2007; Belinsky et al., 2009; Astolfi et al., 2010).
Several multitumor syndromes involving KIT/PDGFRA-wild type GIST have been described. Carney triad, a non-familial association of GIST with paragangliomas, pulmonary chondromas, or other tumors (Carney et al., 1977; Carney 1999; Zhang et al., 2010), affects mainly young females, and the GISTs present almost exclusively in the stomach as multi-focal growth with frequent lymph node metastasis. Carney-Stratakis, an inherited syndrome pre-disposing towards the development of multifocal gastric GIST and paragangliomas, is caused by germline mutations in component genes of the Krebs cycle complex succinate dehydrogenase (SDH) (McWhinney et al., 2007). SDH deficiency in tumorigenesis is thought to result in stabilization of hypoxia-inducible factor 1-alpha (HIF1A) and activation of pseudohypoxia signaling (Dahia et al., 2005; King et al., 2006; Koivunen et al., 2007). Several cases of SDHx gene mutations in non-Carney-Stratakis GISTs have recently been reported (Janeway et al., 2011; Pantaleo et al., 2011a; Pantaleo et al., 2011c), and a more generalized defect of the SDH pathway, indicated by a lack of immunohistochemical expression of the SDHB protein, has been identified in patients with the Carney triad or Carney-Stratakis syndrome, in sporadic pediatric GISTs, and in a small percentage of sporadic adult wild type cases (Gill et al., 2010; Gaal et al., 2011; Janeway et al., 2011; Miettinen et al., 2011).
In this study, the expression level of IGF1R and of several neural markers, as well as the presence of SDHB protein, were characterized in a series of 12 wild type and 12 mutant GIST cases. Wild type GIST cases were also evaluated for germline and somatic coding-region mutations in all SDH subunit genes. Chromosomal aberrations in these tumors were identified using genome-wide single-nucleotide polymorphism (SNP) and molecular inversion probe (MIP) arrays.
Materials and Methods
Preparation of Genomic DNA and Total RNA from Tumor Samples
All tumor samples and normal blood were obtained following informed consent under the Fox Chase Cancer Center Institutional Review Board guidelines. Tumor specimens obtained from surgically treated GIST patients were snap-frozen in liquid nitrogen and stored at -80°C until use. Frozen samples were embedded in optimal cutting temperature (OCT) medium, and sections cut and stained with hematoxylin/eosin to evaluate tumor cellularity. Genomic DNA was isolated from OCT sections or from patient blood using the Easy-DNA kit (Life Technologies), according to manufacturer's instructions. DNA concentration was determined using the NanoDrop 1000 (ThermoFischer Scientific), and agarose-gel electrophoresis used to check the integrity of DNA preparations. Total RNA was isolated from cut OCT sections using TRIzol® reagent (Life Technologies), quantified, and the RNA integrity verified using agarose-gel electrophoresis.
Immunohistochemical Analysis
Immunohistochemical (IHC) staining for KIT and IGF1R (Tarn et al., 2008) and SDHB (Gill et al., 2010b) was performed on 5 μm slides as described. All IHC evaluation was performed in a blinded manner by one author (DF). The following criteria were used to assess SDHB expression: positive score denotes discrete granular cytoplasmic staining (mitochondrial), whereas negative score indicates lack of mitochondrial staining with presence of internal positive controls (adjacent normal tissue, stromal cells, endothelial cells). IGF1R was scored based on distribution and intensity of positive tumor cell staining. Distribution: Absent tumor cell staining was scored as 0, <10% of positive tumor cells staining were scored 1, 10% to 50% of cells staining were scored as 2, 50% to 90% of cells staining were scored as 3, and >90 of cells staining were scored as 4. Intensity: Absent staining in tumor cells was scored as 0, equivocal was scored as 1, clearly positive was scored as 2, and strong positive staining was scored as 3. Scoring: The results for intensity and distribution were summed and a “score” assigned as follows: sum of 0, no staining (score 0); sum of 1 to 3, slight staining (score 1); sum of 4 to 5, moderate staining (score 2); and sum of 6 to 7, marked staining (score 3).
qRT-PCR Analysis
Random-primed cDNA was prepared from 2 μg total RNA using the High Capacity cDNA Reverse Transcription KIT (Life Technologies). RNA expression was measured by real-time PCR on an ABI PRISM 7900 HT Sequence Detection System using fluorescein phosphoramidite (FAM) primer/probe sets (Applied Biosystems). Primer-probe sets are described in Supplementary Table 2. RNA expression data for IGF1R, CDH2, ELAVL3, and SDH subunit genes were normalized to beta actin. For cases with more than one analyzed GIST, normalized expression values of the individual tumors were averaged for case reporting.
Mutational Analysis
Relevant exons and surrounding intronic regions were PCR-amplified and subjected to Sanger sequencing (Beckman Coulter Genomics). Mutational screening for KIT (exons 9, 11, 13, 17), PDGFRA (exons 12, 14, 18), and BRAF (exons 11,15) was as previously described (Belinsky et al., 2009). Primers for SDHA, designed to avoid amplification of pseudogenes on chromosomes 3 (SDHAP1, SDHAP2) and 5 (SDHAP3), have been previously described (Burnichon et al., 2010). Primers for SDHB, SDHC, and SDHD have been described (Wood et al., 2007). Primer sequences for amplification of cDNA regions of the SDHA and SDHC genes (Supplementary Table 3) were designed using Primer3 (Rozen and Skaletsky 2000). Mutation nomenclature conforms to the recommendations of the Human Genome Variation Society (den Dunnen and Antonarakis 2000).
SDHC Bisulfite Sequencing
Genomic DNA samples were bisulfite modified using the EZ DNA Methylation-Direct™ Kit (Zymo Research), as per manufacturer's instructions. Primers for amplification of bisulfite-modified DNA from the SDHC gene (Supplementary Table 3) were designed using MethPrimer (Li and Dahiya 2002). Amplicons were cloned into the vector pCR2.1 using a TA cloning kit (Life Technologies) and sequenced using vector-based primers. Universal Methylated DNA Standard (Zymo Research) and normal blood DNA were used as positive and negative controls, respectively, for methylation of CpG dinucleotides.
SNP and MIP Copy Number Analysis
Chromosomal copy number analysis and allele-specific genotyping using SNP arrays were performed as described (Belinsky et al., 2009). Briefly, genomic DNA isolated from macrodissected OCT-embedded tumor sections was digested with restriction endonucleases, adaptor-ligated and PCR-amplified. Purified PCR products were fragmented, biotin-labeled and hybridized to Affymetrix Genome-Wide Human SNP Array 6.0, or GeneChip® Human Mapping 50K Xba Arrays. Genotyping and copy number analysis for the 6.0 arrays was done using Affymetrix Genotyping Console 2.1 and CNAT 5.0, respectively, using default configurations. For the 50K arrays Affymetrix GeneChip® Genotyping Analysis Software 4.1 and CNAT 4.0, with default settings were used for genotyping and copy number analysis. For MIP analysis, genomic DNA was extracted from GIST samples and processed using OncoScan™ FFPE Express (Affymetrix, Inc., Santa Clara, CA). This assay utilizes MIP technology based on SNP microarrays and has been described previously in detail, including its ability to provide high quality copy number and loss of heterozygosity (LOH) data on formalin-fixed paraffin-embedded (FFPE) clinical samples (Wang et al., 2005; Absalan and Ronaghi 2007; Wang et al., 2007; Ji and Welch 2009; Wang et al., 2009; Wang et al., (in press)). The samples were prepared and run on this assay as previously described (Schiffman et al., 2009; Schiffman et al., 2010; Schiffman et al., 2011; Jahromi et al., in press). The data was analyzed with Nexus Copy Number v5.1 software (BioDiscovery, Inc., El Segundo, CA). Nexus settings for this analysis included Quadratic Wave Correction with SNP-FASST2 Segmentation, with the following settings: High gain 1.2, Gain 0.3, Normal 0, Loss -0.3, Big Loss -1.2 with a significance threshold of 5.0E-7 and call requirement including a minimum of 3 probes per segment. All probes were initially filtered prior to analysis to include only those with standard deviation < 0.2 and a call rate > 90%. All described regions for SNP and MIP assays are based on NCBI build 36.
Results
Clinical and Pathological Features of KIT/PDGFRA/BRAF Wild Type GISTs
12 KIT/PDGFRA/BRAF mutation-negative GIST cases and 12 mutant cases were examined in this study (Table 1). The 12 wild type cases included 11 adults with a median age of onset of 43 years (range 20-57) and a pediatric case (case 11). Three of the wild type cases (cases 7, 8 and the pediatric case) were Carney triad cases, having manifested extra-adrenal paragangliomas and/or pulmonary chondromas along with multiple GISTs. Nine of the wild type cases were females, 10 cases presented with gastric GIST, and 11 were of epitheliod or mixed epitheliod/spindle-cell morphology. One wild type GIST (case 12) originated in the small bowel and exhibited spindle-cell morphology. The kinase-mutant cases included 10 cases with KIT mutations and 2 with PDGFRA mutations. These 12 patients, including 7 males and 5 females, presented with a median age of onset of 64 years (range 37-86), with 7/12 of gastric and 3/12 small bowel origin, and with spindle-cell morphology in 10/12. All cases were Fox Chase cases except wild type cases 4 and 5, which correspond to cases 38 and 48, respectively, from a recent clinical trial from the Radiation Therapy Oncology Group (Rink et al., 2009).
Table 1.
Demographic and clinical description of GIST cases
| Case | Kinase genotypea | Age | Sex | Primary Site | Morphology |
|---|---|---|---|---|---|
| 1 | wild type | 39 | F | stomach | epitheliod/spindle |
| 2 | wild type | 52 | F | stomach | epitheliod/spindle |
| 3 | wild type | 33 | F | stomach | epitheliod/spindle |
| 4 | wild type | 43 | F | stomach | epitheliod/spindle |
| 5 | wild type | 48 | M | not available | epithelioid |
| 6 | wild type | 53 | M | stomach | epithelioid |
| 7b | wild type | 51 | F | stomach | epitheliod |
| 8b | wild type | 37 | M | stomach | epithelioid |
| 9 | wild type | 35 | F | stomach | epithelioid |
| 10 | wild type | 20 | F | stomach | epithelioid |
| 11b | wild type | 12 | F | stomach | epithelioid |
| 12 | wild type | 57 | F | small bowel | spindle |
| 13 | KIT: p.Asn567_Leu576delinsLysGlu | 71 | M | colon | spindle |
| 14 | KIT: p.Met552_Tyr553del | 66 | F | small bowel | spindle |
| 15 | KIT: p.Ala502_Tyr503dup | 43 | M | small bowel | spindle |
| 16 | KIT: pGln556_Lys558delinsAsn | 60 | F | stomach | spindle |
| 17 | KIT: p.Trp557_Val559delinsPhe | 63 | M | rectum | spindle |
| 18 | KIT: p.Tyr823Asp | 81 | F | stomach | mixed |
| 19 | KIT: p.Lys642Glu | 47 | M | small bowel | spindle |
| 20 | PDGFRA: p.[Asn659Tyr(;) p.Tyr849Cys] | 37 | M | stomach | epitheliod |
| 21 | KIT: p.Val559Gly | 70 | F | stomach | spindle |
| 22 | KIT: p.Val560Asp | 56 | F | stomach | spindle |
| 23 | KIT: p.Tyr823Asp | 84 | M | stomach | spindle |
| 24 | PDGFRA: p. Asp842Val | 86 | M | stomach | spindle |
Amino acid numbering from NP_000213.1 (KIT) and NP_006197.1 (PDGFRA)
Carney triad cases
IGF1R is Over-expressed in Wild Type SDHB-negative GISTs
IHC for both SDHB and IGF1R was performed to investigate the relationship between IGF1R expression and expression of SDHB in wild type and mutant GISTs. All GISTs were also stained for KIT expression. Representative IHC results are shown in Figure 1 and summarized in Table 2. All wild type and mutant GISTs were KIT-positive by IHC. IGF1R expression level was evaluated using a scoring system combining distribution and intensity of staining (Tarn et al., 2008). Eleven of 12 wild type GISTs exhibited marked staining for IGF1R (score = 3), while all 12 mutant cases exhibited slight or moderate staining (score = 1-2, Table 2). SDHB protein staining, on the other hand, was absent in wild type cases 1-11 but clearly present in all mutant GISTs and in wild type case 12. Tumors were denoted SDHB-negative when tumor cell negativity was accompanied by positive staining in internal controls such as adjacent normal tissue or endothelial cells (upper and middle panels in Figure 1). IGF1R expression varied strongly either as a function of kinase genotype (P ~ 9.6 × 10-6, Fisher exact probability, two-tailed) or SDHB status (P ~ 4.0 × 10-7).
Figure 1.

KIT, IGF1R and SDHB expression in GIST specimens. IHC expression in an adult wild type (top panel), pediatric wild type (middle panel) and mutant (bottom panel) GIST. Primary antibodies used include KIT (Dako), IGF-1R (Cell Signaling) and SDHB (Abcam). Positive KIT staining is evident throughout tumor tissue in all cases. Strong staining for IGF1R is seen in the wild type GISTs, while SDHB staining is evident in the mutant GIST and in the adjacent normal tissue and epithelial cells in the wild type cases, but absent in wild type GISTs.
Table 2.
IGF1R expression and SDHB status in GISTs
| Case | IGF1R score | SDHB status |
|---|---|---|
| 1 | 3 | negative |
| 2 | 3 | negative |
| 3 | 3 | negative |
| 4 | 3 | negative |
| 5 | 3 | negative |
| 6 | 3 | negative |
| 7 | 3 | negative |
| 8 | 3 | negative |
| 9 | 3 | negative |
| 10 | 3 | negative |
| 11 | 3 | negative |
| 12 | 2 | positive |
| 13 | 2 | positive |
| 14 | 1 | positive |
| 15 | 2 | positive |
| 16 | 2 | positive |
| 17 | 1 | positive |
| 18 | 2 | positive |
| 19 | 1 | positive |
| 20 | 2 | positive |
| 21 | 2 | positive |
| 22 | 2 | positive |
| 23 | 2 | positive |
| 24 | 2 | positive |
IGF1R expression was further quantified using qRT-PCR. SDHB-negative wild type GISTs expressed significantly higher levels of IGF1R RNA (~ 69-fold higher, P < 0.0001) as compared to SDHB-positive GISTs (Figure 2). Transcript levels of two markers of neural tissue, CDH2 (cadherin-2, type 1, neuronal) and ELAVL3 (embryonic lethal, abnormal vision, Drosophila-like 3) were also examined. Both genes were over-expressed in SDHB-negative wild type GISTs, although differences in expression were not as great as for IGF1R (CDH2~ 18-fold, ELAVL3 ~ 28-fold). In contrast, RNA expression analysis for SDHA, B, C and D revealed no significant differences in expression of the SDH subunits between SDHB-negative and SDHB-positive GISTs (data not shown).
Figure 2.

RNA expression of IGF1R, CDH2, and ELAVL3 in SDHB-negative and SDHB-positive GISTs as determined by qRT-PCR. Expression levels were normalized to beta-actin. Statistical significance as calculated by the Mann-Whitney test was as follows: IGF1R: P < 0.0001; CDH2: P ~ 0.002; ELAVL3: P ~ 0.003.
Germline Mutations and Somatic Inactivation of SDHA may be Common in Adult SDHB-negative Wild Type GIST
All 12 wild type GISTs were examined for defects in SDH subunit genes by sequencing all exons and bordering intronic regions in the SDHA, SDHB, SDHC, and SDHD genes. Tumor DNA was sequenced in all cases and in patient blood DNA when available. Genome-wide copy-number analysis and allele-specific genotyping was carried out using genome-wide single-nucleotide polymorphism (SNP) arrays. As tumor samples for case 10 were available only as formalin-fixed paraffin-embedded (FFPE) tissue, these samples were evaluated for chromosomal aberrations using molecular inversion probe (MIP) technology, which provides high quality results on FFPE specimens. Chromosomal copy number changes have previously been reported for some of the GIST cases (Belinsky et al., 2009) as noted in Supplementary Table 1.
Surprisingly, germline and/or somatic mutations in SDHA were identified in 5/11 SDHB negative cases (1-5), while an SDHC mutation was identified in case 6. Chromatograms illustrating these mutations are shown in Figure 3 and summarized in Table 3, along with relevant findings from the SNP analysis. Case 1 harbored a germline heterozygous missense mutation in SDHA exon 7 (SDHA c.818C>T) and a somatic missense mutation in exon 10 (SDHA c.1357G>A) (Figure 3A). The resulting amino acid changes (Table 3) are predicted to be pathological based on amino acid conservation and functional annotation (Calabrese et al., 2009; Adzhubei et al., 2010). Case 2 harbors a heterozygous 3 base deletion (SDHA c.457-2_c457delAGC) in the germline that spans the IVS4/exon 5 junction; this deletion is nearly homozygous in tumor DNA (Figure 3B). Sequencing of this region in cDNA prepared from the patient's GIST reveals the use of a cryptic splice acceptor site within exon 5 and the deletion of 4 ribonucleotides, which would predict a frame-shift and a resulting stop-codon downstream of the deletion (Figure 3B, Table 3). The SNP array analysis identified copy number loss at 5p15.33 in the tumor, a region that includes the SDHA gene (Table 3, Supplementary Figure 1). Case 3 harbors a heterozygous germline nonsense mutation (SDHA c.91C>T, p.Arg31*) in exon 2. The germline mutation is accompanied by copy-number neutral loss-of-heterozygosity (LOH) throughout most of the short arm of chromosome 5 in the GIST; as a result the C>T mutation is nearly homozygous in the tumor (Figure 3C, Table 3). Two homozygous nonsense mutations were identified in cases 4 and 5: SDHA p.Arg31* in exon 2 and SDHA p.Glu491* in exon 11 (Figure 3D,E; Table 3). Although germline DNA was not available for these cases, the SNP analysis suggested loss of the wild type allele in these tumors via chromosome 5p LOH (case 4) and copy-number loss (case 5) (Table 3, Supplementary Figure 1). Case 6 was the only case harboring an SDHC mutation, a heterozygous p.Gly75Asp (c.224G>A) mutation in exon 4. Interestingly, although no additional SDHC gene mutations or allelic losses were detected, sequencing through this region in the tumor cDNA suggests that only the mutant form is expressed (Figure 3F). To investigate the mechanism of inactivation of the wild type SDHC allele in this tumor, primers for amplification of bisulfite-modified DNA (Supplementary Table 4) were designed for a previously described CpG island in the 5’ region of the SDHC gene (Huang et al., 2009). Amplified DNA from the GIST (along with control methylated DNA and un-methylated DNA) was cloned into a plasmid vector (see Materials and Methods). DNA sequencing identified clones from the GIST that were either nearly fully methylated at Cpg nucleotides, or fully unmethylated in this region (Figure 4), presumably corresponding to the silenced wild type isoforms and the expressed mutant isoforms, respectively. Of 10 sequenced clones, 5 displayed methylation at 10 or 11 of 12 CpG di-nucleotides, while 5 displayed methylation on 0/12 CpGs (data not shown). Clones derived from the positive and negative controls (n = 5 each) exhibited CpGs that were either fully methylated or un-methylated (data not shown).
Figure 3.

Partial sequence chromatograms from amplified genomic DNA from patient blood, and from genomic DNA and cDNA from GISTs. Nucleotide numbering is from the coding regions for SDHA (NM_004168.2) and SDHC (NM_003001.3).
Table 3.
SDH mutationsa and allelic losses
| Case | SDH mutations (DNA) | SDH mutations (protein) | Germline DNA | GIST DNA | SDH allelic losses in GIST |
|---|---|---|---|---|---|
| 1 |
SDHA c.818C>T SDHA c.1357G>A |
SDHA p.Thr2731Ile SDHA p.Gly453Arg |
Heterozygous Absent |
Heterozygous Heterozygous |
None identified |
| 2 | SDHA c.457-2_c457delAGC | SDHA p.Leu153Lysfs*71 | Heterozygous | Homozygous | arr 5p15.33(152,441-3,564,616)x1 |
| 3 | SDHA c.91C>T | SDHA p.Arg31* | Heterozygous | Homozygous | arr 5p15.33.p13.2(261,369-93,515,804)x2 hmz |
| 4 | SDHA c.91C>T | SDHA p.Arg31* | Not available | Homozygous | arr 5p15.33.p15.2(173,508-23,417,765)x2 hmz |
| 5 | SDHA c.1471G>T | SDHA p.Glu491* | Not available | Homozygous | arr 5p15.33(134,437-1,173,438)x1 |
| 6 | SDHC c.224G>A | SDHC p.Gly75Asp | Not available | Heterozygous | None identified |
Nucleotide and amino acid numbering from the coding regions for SDHA (NM_004168.2) and SDHC (NM_003001.3)
Figure 4.
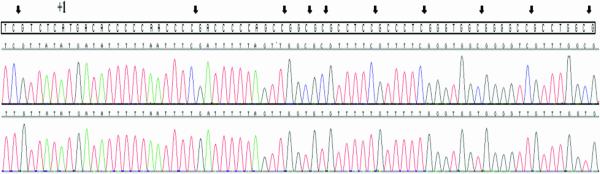
Partial chromatogram of cloned PCR products from the 5’ end of the SDHC gene. Tumor genomic DNA from case 6 was bisulfite-treated prior to amplification and cloning. Arrows show the location of 10 (of 12) CpG dinucleotides (seen in the boxed reference sequence) in the predicted CpG island. The upper chromatogram shows a cloned product with methylation of 11/12 CpGs (the third CpG is un-methylated), while the lower panel shows a fully un-methylated cloned product. The ATG site is indicated as +1.
Genome-wide chromosomal copy number changes were determined in 29 individual tumors from the 24 GIST cases (Supplementary Table 1). Wild type GIST samples exhibited a remarkably low-level of genomic instability, with most cases displaying 0-3 regions of copy number change. The exception to this is case 12, which exhibits a high degree of genomic instability. This case is the single SDHB-positive wild type case, and other properties such as small bowel location, spindle-cell morphology and a lower IGF1R IHC score distinguish this case from the other wild type cases in this study. The kinase-mutant cases, many of which have been previously reported (Belinsky et al., 2009), exhibit 2-19 regions of chromosomal aberrations and show the usual propensity towards copy-number loss at chromosomes 1p (58%), 14q (83%), 15q (50%), and 22q (42%).
Discussion
12 wild type and 12 mutant GISTs were investigated for IGF1R and SDHB expression, and the wild type tumors screened for mutations in SDHx genes. Notably, 11/12 wild type tumors exhibited complete lack of SDHB staining. SDHB-negative GIST includes GISTs from the Carney-Stratakis syndrome (in association with SDHx mutation), Carney triad GISTs, as well as some apparently sporadic wild type GISTs (Gill et al., 2010; Gaal et al., 2011; Janeway et al., 2011; Miettinen et al., 2011). The current study included 3 Carney triad cases and 8 sporadic SDHB-negative GISTs. Immunological SDHB deficiency in sporadic GISTs has a predilection for younger patients (Janeway et al., 2011; Miettinen et al., 2011). In support of this observation the 8 sporadic SDHB-negative GIST cases in this study presented with an average age of ~ 40 years, 20 years younger than the median age of a GIST diagnosis. The SDHB-negative cases were mostly females (8/11) and presented with gastric GISTs with epitheliod or mixed cell morphology, consistent with what has been described for “SDHB-negative” GISTs (Gill et al., 2010).
Strong immunohistochemical staining for IGF1R was seen exclusively in the 11 SDHB-negative GISTs, in agreement with a recent report (Chou et al., 2012). The one SDHB-positive wild type sample in this analysis (case 12) showed both weaker IGF1R protein staining and lower IGF1R RNA expression than the other wild type samples. The small bowel presentation, spindle-cell morphology, and multiple chromosomal aberrations of this GIST also distinguish this case from the other wild type cases. While no KIT, PDGFRA, or BRAF mutations were identified in case 12, we do not rule out the possibility that this case harbors a mutation in another kinase. Strong IGF1R staining in the present study correlates well with high steady-state IGF1R RNA levels. Over-expression of IGF1R RNA may be part of a global RNA expression profile for wild type GIST (Prakash et al., 2005; Agaram et al., 2008a; Pantaleo et al., 2011b). The study by Pantaleo et al describes high expression of a set of neural markers (including IGF1R) in a small group of wild type GISTs. Over-expression of two of these markers (CDH2 and ELAVL3) was validated in the wild type cases in the current study. The phenomenon of IGF1R over-expression in SDH-negative wild type GIST may reflect derivation from an earlier stage of ICC development (Pantaleo et al., 2011b). Alternatively, IGF1R over-expression as a potential consequence of stress due to SDH deficiency and mitochondrial dysfunction has also been suggested (Chou et al., 2012). Whether IGF1R over-expression in wild type GIST is a consequence of the observed SDH deficiency or is a feature of the un-identified cell of origin for these GISTs, IGF1R remains an attractive therapeutic target for wild type GIST.
This study convincingly demonstrates that germline mutations in SDHA accompanied by somatic allelic losses may be a common occurrence in sporadic SDHB-negative wild type GIST. Germline mutations in the SDHB-D genes, originally reported in Carney-Stratakis families (McWhinney et al., 2007; Pasini et al., 2008), are notably absent in non-familial Carney triad cases, although chromosomal losses in the region surrounding the SDHC gene (1q23.3) as well as 1p losses surrounding the SDHB gene have been described in Carney triad GIST (Matyakhina et al., 2007). In a panel of 34 patients with apparently sporadic wild type GIST, several mutations were found in SDHB (n = 3) and in SDHC (n = 1), all in pediatric and young adult cases (Janeway et al., 2011). In a panel of 66 SDHB-negative gastric GISTs, no mutations in a limited panel of exons from the SDHB, SDHC, and SDHD genes were reported (Miettinen et al., 2011). For the most part these studies did not focus on SDHA gene sequencing. SDHA was originally implicated as a tumor suppressor gene in paraganglioma (Burnichon et al., 2010), and, very recently, inactivating mutations in SDHA were reported in sporadic GISTs from three adults and a pediatric case (Pantaleo et al., 2011a; Pantaleo et al., 2011c). In the current study, biallelic inactivating SDHA gene mutations were identified in 5/11 SDHB-negative GIST cases, a finding that buttresses the case for SDHA as a tumor suppressor gene in GIST. The predominant scenario appears to be the presence of a germline SDHA point mutation or micro-deletion accompanied by somatic loss of the wild type allele, although in case 1 the second hit was a point mutation as well. In contrast, compound heterozygous point mutations were the mechanism of inactivation in 3 of 4 previously described GIST cases with SDHA-mediated SDH deficiency (Pantaleo et al., 2011c). Finally, the finding of a genetic lesion in an SDHC allele accompanied by epigenetic inactivation of the second allele adds a novel layer of complexity to the model of GIST tumorigenesis through SDH inactivation. The identification of additional mutations in SDH genes underscores the need for patients with wild type GIST to undergo genetic counseling for assessment of germ line SDHx mutations.
Supplementary Material
Supplementary Figure 1: Graphs of SNP log2 ratio, smoothed signal, and allele differences for cases 2, 4, and 5 (Genotyping Console Browser, Affymetrix. Corporation). Graphs cover a portion of chromosome 5p (bottom of figure) encompassing the SDHA gene (upper left, panels A-C). The segments at the bottom of each panel delineate the regions of 5p copy number loss in cases 2 and 5, and 5p copy number-neutral LOH in case 4.
Acknowledgements
We would like to acknowledge Drs. Joseph R. Testa and Jianming Pei of the Fox Chase Cancer Center Genomics Facility for processing and analysis of genome-wide SNP arrays. We would also like to acknowledge the FCCC Biosample Repository, and the FCCC Histopathology, Genotyping and Real-Time PCR, and DNA Sequencing Facilities for work contributing to this manuscript.
Supported by: This work was supported in part by grants from the National Cancer Institute (R01 CA106588, M.v.M. & A.K.G.; NIH R21 CA150831-02, M.v.M. & M.G.B.), an award from the Sarcoma Foundation of America (M.G.B.), and an award from the GIST Cancer Research Fund (M.v.M.).
References
- Absalan F, Ronaghi M. Molecular inversion probe assay. Methods Mol Biol. 2007;396:315–330. doi: 10.1007/978-1-59745-515-2_20. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foerster A, Hartmann A, Bihl MP. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol. 2009;62:613–616. doi: 10.1136/jcp.2009.064550. [DOI] [PubMed] [Google Scholar]
- Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, Maki RG, DeMatteo RP, Besmer P, Antonescu CR. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008a;14:3204–3215. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008b;47:853–859. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astolfi A, Nannini M, Pantaleo MA, Di Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu A, Saponara M, Lolli C, Di Scioscio V, Formica S, Biasco G. A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number. Lab Invest. 2010;90:1285–1294. doi: 10.1038/labinvest.2010.110. [DOI] [PubMed] [Google Scholar]
- Belinsky MG, Rink L, Cai KQ, Ochs MF, Eisenberg B, Huang M, von Mehren M, Godwin AK. The insulin-like growth factor system as a potential therapeutic target in gastrointestinal stromal tumors. Cell Cycle. 2008;7:2949–2955. doi: 10.4161/cc.7.19.6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky MG, Skorobogatko YV, Rink L, Pei J, Cai KQ, Vanderveer LA, Riddell D, Merkel E, Tarn C, Eisenberg BL, von Mehren M, Testa JR, Godwin AK. High density DNA array analysis reveals distinct genomic profiles in a subset of gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2009;48:886–896. doi: 10.1002/gcc.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296:1517–1518. doi: 10.1056/NEJM197706302962609. [DOI] [PubMed] [Google Scholar]
- Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543–552. doi: 10.4065/74.6.543. [DOI] [PubMed] [Google Scholar]
- Chou A, Chen J, Clarkson A, Samra JS, Clifton-Bligh RJ, Hugh TJ, Gill AJ. Succinate dehydrogenase-deficient GISTs are characterized by IGF1R overexpression. Mod Pathol. 2012;25:1307–1313. doi: 10.1038/modpathol.2012.77. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, Hodin R, Heitritter S, Moore F, Dluhy R, Sosa JA, Ocal IT, Benn DE, Marsh DJ, Robinson BG, Schneider K, Garber J, Arum SM, Korbonits M, Grossman A, Pigny P, Toledo SP, Nose V, Li C, Stiles CD. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, Hagemeijer A, Judson I. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Gaal J, Stratakis CA, Carney JA, Ball ER, Korpershoek E, Lodish MB, Levy I, Xekouki P, van Nederveen FH, den Bakker MA, O'Sullivan M, Dinjens WN, de Krijger RR. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol. 2011;24:147–151. doi: 10.1038/modpathol.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, Tobias V, Samra J, Goldstein D, Smith C, Sioson L, Parker N, Smith RC, Sywak M, Sidhu SB, Wyatt JM, Robinson BG, Eckstein RP, Benn DE, Clifton-Bligh RJ. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol. 2010;34:636–644. doi: 10.1097/PAS.0b013e3181d6150d. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, Bertagnolli MM, Fletcher JA. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, Bringuier PP, Scoazec JY, Coindre JM. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010;133:141–148. doi: 10.1309/AJCPPCKGA2QGBJ1R. [DOI] [PubMed] [Google Scholar]
- Huang KT, Dobrovic A, Fox SB. No evidence for promoter region methylation of the succinate dehydrogenase and fumarate hydratase tumour suppressor genes in breast cancer. BMC Res Notes. 2009;2:194. doi: 10.1186/1756-0500-2-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi MS, Putnam AR, Drusgal C, Wright J, Spraker H, Kinsey M, Zhou H, Boucher K, Randall RL, Jones KB. Molecular Inversion Probe analysis detects novel copy number alterations in Ewing Sarcoma. Cancer Genet. 2012;205:391–404. doi: 10.1016/j.cancergen.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084–9088. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Albritton KH, Van Den Abbeele AD, D'Amato GZ, Pedrazzoli P, Siena S, Picus J, Butrynski JE, Schlemmer M, Heinrich MC, Demetri GD. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52:767–771. doi: 10.1002/pbc.21909. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O'Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Welch K. Molecular inversion probe assay for allelic quantitation. Methods Mol Biol. 2009;556:67–87. doi: 10.1007/978-1-60327-192-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Matyakhina L, Bei TA, McWhinney SR, Pasini B, Cameron S, Gunawan B, Stergiopoulos SG, Boikos S, Muchow M, Dutra A, Pak E, Campo E, Cid MC, Gomez F, Gaillard RC, Assie G, Fuzesi L, Baysal BE, Eng C, Carney JA, Stratakis CA. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. Journal of Clinical Endocrinology & Metabolism. 2007;92:2938–2943. doi: 10.1210/jc.2007-0797. [DOI] [PubMed] [Google Scholar]
- McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA, Fletcher CD. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28:889–894. doi: 10.1097/00000478-200407000-00007. [DOI] [PubMed] [Google Scholar]
- Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–1721. doi: 10.1097/PAS.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo MA, Astolfi A, Di Battista M, Heinrich MC, Paterini P, Scotlandi K, Santini D, Catena F, Manara MC, Nannini M, Maleddu A, Saponara M, Lolli C, Formica S, Biasco G. Insulin-like growth factor 1 receptor expression in wild-type GISTs: a potential novel therapeutic target. Int J Cancer. 2009;125:2991–2994. doi: 10.1002/ijc.24595. [DOI] [PubMed] [Google Scholar]
- Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, Gnocchi C, Santini D, Catena F, Formica S, Martelli PL, Casadio R, Pession A, Biasco G. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011a;103:983–987. doi: 10.1093/jnci/djr130. [DOI] [PubMed] [Google Scholar]
- Pantaleo MA, Astolfi A, Nannini M, Ceccarelli C, Formica S, Santini D, Heinrich MC, Corless C, Dei Tos AP, Paterini P, Catena F, Maleddu A, Saponara M, Di Battista M, Biasco G. Differential expression of neural markers in KIT and PDGFRA wild-type gastrointestinal stromal tumours. Histopathology. 2011b;59:1071–1080. doi: 10.1111/j.1365-2559.2011.04071.x. [DOI] [PubMed] [Google Scholar]
- Pantaleo MA, Nannini M, Astolfi A, Biasco G. A distinct pediatric-type gastrointestinal stromal tumor in adults: potential role of succinate dehydrogenase subunit A mutations. Am J Surg Pathol. 2011c;35:1750–1752. doi: 10.1097/PAS.0b013e318230a523. [DOI] [PubMed] [Google Scholar]
- Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere JJ, Rustin P, Gimenez-Roqueplo AP, Eng C, Carney JA, Stratakis CA. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- Prakash S, Sarran L, Socci N, DeMatteo RP, Eisenstat J, Greco AM, Maki RG, Wexler LH, LaQuaglia MP, Besmer P, Antonescu CR. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol. 2005;27:179–187. doi: 10.1097/01.mph.0000157790.81329.47. [DOI] [PubMed] [Google Scholar]
- Rink L, Skorobogatko Y, Kossenkov AV, Belinsky MG, Pajak T, Heinrich MC, Blanke CD, von Mehren M, Ochs MF, Eisenberg B, Godwin AK. Gene expression signatures and response to imatinib mesylate in gastrointestinal stromal tumor. Mol Cancer Ther. 2009;8:2172–2182. doi: 10.1158/1535-7163.MCT-09-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- Schiffman JD, Wang Y, McPherson LA, Welch K, Zhang N, Davis R, Lacayo NJ, Dahl GV, Faham M, Ford JM, Ji HP. Molecular inversion probes reveal patterns of 9p21 deletion and copy number aberrations in childhood leukemia. Cancer Genet Cytogenet. 2009;193:9–18. doi: 10.1016/j.cancergencyto.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–519. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman JD, Lorimer PD, Rodic V, Jahromi MS, Downie JM, Bayerl MG, Sanmann JN, Althof PA, Sanger WG, Barnette P, Perkins SL, Miles RR. Genome wide copy number analysis of paediatric Burkitt lymphoma using formalin-fixed tissues reveals a subset with gain of chromosome 13q and corresponding miRNA over expression. Br J Haematol. 2011;155:477–486. doi: 10.1111/j.1365-2141.2011.08883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar K, Hewlett BR, Huizinga JD, Chorneyko K, Berezin I, Riddell RH. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am J Surg Pathol. 1999;23:377–389. doi: 10.1097/00000478-199904000-00002. [DOI] [PubMed] [Google Scholar]
- Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, Eisenberg B, Birbe R, Patchefsky A, Dunbrack R, Arnoletti JP, von Mehren M, Godwin AK. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11:3668–3677. doi: 10.1158/1078-0432.CCR-04-2515. [DOI] [PubMed] [Google Scholar]
- Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008;105:8387–8392. doi: 10.1073/pnas.0803383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Moorhead M, Karlin-Neumann G, Falkowski M, Chen C, Siddiqui F, Davis RW, Willis TD, Faham M. Allele quantification using molecular inversion probes (MIP). Nucleic Acids Res. 2005;33:e183. doi: 10.1093/nar/gni177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Moorhead M, Karlin-Neumann G, Wang NJ, Ireland J, Lin S, Chen C, Heiser LM, Chin K, Esserman L, Gray JW, Spellman PT, Faham M. Analysis of molecular inversion probe performance for allele copy number determination. Genome Biol. 2007;8:R246. doi: 10.1186/gb-2007-8-11-r246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Carlton VE, Karlin-Neumann G, Sapolsky R, Zhang L, Moorhead M, Wang ZC, Richardson AL, Warren R, Walther A, Bondy M, Sahin A, Krahe R, Tuna M, Thompson PA, Spellman PT, Gray JW, Mills GB, Faham M. High quality copy number and genotype data from FFPE samples using Molecular Inversion Probe (MIP) microarrays. BMC Med Genomics. 2009;2:8. doi: 10.1186/1755-8794-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cottman M, Schiffman JD. Molecular Inversion Probes: A novel microarray technology and its application in cancer research. Cancer Genet. 2012;205:314–355. doi: 10.1016/j.cancergen.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Zhang L, Smyrk TC, Young WF, Jr., Stratakis CA, Carney JA. Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol. 2010;34:53–64. doi: 10.1097/PAS.0b013e3181c20f4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Graphs of SNP log2 ratio, smoothed signal, and allele differences for cases 2, 4, and 5 (Genotyping Console Browser, Affymetrix. Corporation). Graphs cover a portion of chromosome 5p (bottom of figure) encompassing the SDHA gene (upper left, panels A-C). The segments at the bottom of each panel delineate the regions of 5p copy number loss in cases 2 and 5, and 5p copy number-neutral LOH in case 4.