

Abstract
Over the past three decades the metabolism and functions of the polyamines have been actively pursued as targets for antineoplastic therapy. Interactions between cationic polyamines and negatively charged nucleic acids play a pivotal role in DNA stabilization and RNA processing that may affect gene expression, translation and protein activity. Our growing understanding of the unique roles that the polyamines play in chromatin regulation, and the discovery of novel proteins homologous with specific regulatory enzymes in polyamine metabolism, have led to our interest in exploring chromatin remodelling enzymes as potential therapeutic targets for specific polyamine analogues. One of our initial efforts focused on utilizing the strong affinity that the polyamines have for chromatin to create a backbone structure, which could be combined with active-site-directed inhibitor moieties of HDACs (histone deacetylases). Specific PAHAs (polyaminohydroxamic acids) and PABAs (polyaminobenzamides) polyamine analogues have demonstrated potent inhibition of the HDACs, re-expression of p21 and significant inhibition of tumour growth. A second means of targeting the chromatin-remodelling enzymes with polyamine analogues was facilitated by the recent identification of flavin-dependent LSD1 (lysine-specific demethylase 1). The existence of this enzyme demonstrated that histone lysine methylation is a dynamic process similar to other histone post-translational modifications. LSD1 specifically catalyses demethylation of mono- and di-methyl Lys4 of histone 3, key positive chromatin marks associated with transcriptional activation. Structural and catalytic similarities between LSD1 and polyamine oxidases facilitated the identification of biguanide, bisguanidine and oligoamine polyamine analogues that are potent inhibitors of LSD1. Cellular inhibition of LSD1 by these unique compounds led to the re-activation of multiple epigenetically silenced genes important in tumorigenesis. The use of these novel polyamine-based HDAC or LSD1 inhibitors represents a highly promising and novel approach to cancer prevention and therapy.
Introduction
Polyamines are naturally occurring polycationic alkylamines that are essential for eukaryotic cell growth. By virtue of their positively charged amine groups, polyamines interact with negatively charged DNA, RNA, proteins and phospholipids to change their structure and conformation. The enzymes controlling polyamine metabolism and intracellular concentrations are highly regulated and can rapidly react to changing environmental conditions. Intracellular polyamine levels and metabolism are frequently dysregulated in cancer and other hyperproliferative diseases, thus making polyamine function and metabolism attractive targets for therapeutic intervention [1,2]. The key polyamine biosynthetic enzyme, ODC (ornithine decarboxylase), has long been thought to be a marker of carcinogenesis and tumour progression [3]. Inhibiting polyamine biosynthesis by specifically targeting ODC as an anticancer strategy has yet to demonstrate significant clinical success, but it has demonstrated considerable promise as a strategy for cancer chemoprevention [4]. Recently, more focus has been directed towards the development of polyamine analogues designed to mimic the regulatory roles of natural polyamines but to have altered function. A number of synthetic polyamine analogues have exhibited encouraging effects against tumour growth in both cell culture and animal studies and several hold promise as chemotherapeutic agents [5].
There are considerable data demonstrating that chromatin is a major target for the natural polyamines and polyamine-based drugs [6–8]. Therefore we have attempted to use this property to advance the hypothesis that specific polyamine analogues could target the chromatin remodelling enzymes, including the HDACs (histone deacetylases) and the newly identified histone LSD1 (lysine-specific demethylase 1). These enzymes, among others, are responsible for normal gene regulation, and in a variety of disease processes their activity may lead to aberrant silencing of important tumour suppressor genes. As aberrant epigenetic silencing of tumour suppressor genes is a common occurrence in the development of cancer, this strategy holds considerable promise for the treatment of neoplastic disease, and the present chapter will discuss the most recent findings in the field [9].
Polyamine metabolism
Polyamines are critical for eukaryotic cell growth and thus maintenance of appropriate intracellular concentrations via a highly regulated interplay between biosynthesis, catabolism, uptake and excretion is required for normal function (Figure 1). Two major regulatory enzymes of polyamine biosynthesis are ODC and AdoMetDC (S-adenosylmethionine decarboxylase). The regulatory protein Az (antizyme) facilitates the degradation of ODC and down-regulates the transport of polyamines into the cell, and thus is considered to be dedicated principally to the feedback regulation of polyamine levels [10]. Polyamine catabolism was initially thought to be a two-step procedure regulated by the rate-limiting enzyme SSAT (spermidine/spermine N1-acetyltransferase), which provides substrate for the generally constitutively expressed APAO (N1-acetylpolyamine oxidase) [11]. After acetylation of spermine and spermidine by SSAT to form N1-acetylspermine and N1-acetylspermidine respectively, acetyl derivatives are then cleaved into 3-acetamidopropanal, the ROS (reactive oxygen species) H2O2, and spermidine and putrescine respectively, through the action of FAD-dependent APAO. However, recent studies from our laboratory and others have demonstrated that a variably spliced human SMO (spermine oxidase) efficiently uses unacetylated spermine as a substrate, and that this enzyme is inducible by specific polyamine analogues [12–15]. These findings indicate the existence of a more complex polyamine catabolic pathway in the regulation of polyamine homoeostasis than originally thought.
Figure 1. Polyamine metabolism in mammals.

The major regulatory enzymes of polyamine biosynthesis are ODC and AdoMetDC. ODC forms putrescine from L-ornithine and is the first rate-limiting step in polyamine biosynthesis. The regulatory protein Az can facilitate degradation of ODC and negatively regulate the eukaryotic polyamine transport system. AdoMetDC forms decarboxylated S-adenosylmethionine (DC-AdoMet) from S-adenosylmethionine, and DC-AdoMet acts as an aminopropyl donor. Spermidine synthase and spermine synthase transfer the aminopropyl group from DC-AdoMet to putrescine or spermidine for the synthesis of spermidine, spermine and MTA (5′-deoxy-5′-methylthioadenosine) respectively. After acetylation of spermine and spermidine by SSAT to form N1-acetylspermine and N1-acetylspermidine respectively, acetyl derivatives are then cleaved into 3-aceto-aminopropanal, the ROS H2O2 and spermidine and putrescine respectively, through the action of FAD-dependent APAO. SMO is a highly inducible FAD-dependent enzyme that directly oxidizes spermine to produce spermidine, 3-aminopropanal and H2O2.
Epigenetic regulation of gene expression
Epigenetic modification of chromatin is a major regulator of gene expression. Epigenetic modifications refer to heritable changes in chromatin/DNA that are not due to changes in primary sequence. These include a variety of modifications of the histone proteins and methylation of cytosine residues in DNA. The histone proteins are the major packaging proteins of eukaryotic DNA. The eukaryotic genome requires packaging of DNA both for structural purposes, as a means of fitting 2 m of DNA into the nucleus, and as one mechanism for the regulation of gene expression. Two of each of the core histone proteins, H2A, H2B, H3 and H4, are assembled into the nucleosome, around which 146 bp of DNA are wound. This nucleosome is the basic packaging unit of eukaryotic chromatin and the density of nucleosomes and the affinity of the individual nucleosomes on any stretch of DNA determine the accessibility of the gene to various factors, including the basic transcriptional machinery. Importantly, in cancer, aberrant epigenetic silencing of gene expression, including tumour suppressor genes, is a common occurrence [9].
There are numerous possible modifications of histone tails, including acetylation, methylation, ubiquitination, phosphorylation, SUMOylation and ribosylation, each of which can affect the expression of genes [16]. The specific modification of histones determines, in part, which regions of the genome are in an open and transcriptionally active confirmation and which are closed and transcriptionally inactive. The most studied of the histone modifications are acetylation/deacetylation and methylation. Acetylated histones are typically associated with active gene transcription. However, histone methylation can either be activating or inhibitory with respect to transcription, depending on the specific residue methylated. Histones may be methylated on either lysine (K) or arginine (R) residues, and the methylation status of key histone lysine residues, such as H3K4, H3K9, H3K27, H3K36, H3K79 and H4K20, represent specific epigenetic marks that are associated with transcriptional regulation. Although the dynamic nature of histone acetylation, mediated through the counterbalancing activity of HATs (histone acetyltransferases) and HDACs, has been known for some time [17], only recently has a similar dynamic regulation of histone methylation been demonstrated. Shi et al. [18] reported the discovery of the first enzyme able to specifically demethylate Lys4 of histone H3. The newly identified protein was therefore named LSD1 (lysine-specific demethylase; BHC110) [18]. Subsequent to the discovery of LSD1, a family of Jmj C (Jumonji C) domain-containing histone demethylases was also identified, thus expanding the number of known proteins involved in chromatin remodelling [19]. These findings firmly establish that histone methylation is a dynamic process under enzymatic control, similar to the other post-translational histone modifications, and suggest that this modifying enzyme may also be a rational target for therapeutic intervention [20].
Exploiting polyamine structure to target aberrantly silenced genes
The incorporation of a polyamine structure into the design of a putative drug offers several benefits as a targeting vehicle, particularly when chromatin is the target. A selective, energy-dependent polyamine transport system allows molecules resembling the general polyamine structure to be accumulated by cells, and the cationic nature of the polyamine backbone provides affinity to the negatively charged chromatin. Several investigators have used the polyamine structure as a backbone to which various active moieties have been conjugated, including alkylating agents, DNA intercalators and other antiproliferative agents [21,22]. Delcros et al. [23] have tested the limits of polyamine transport with respect to size and types of molecules that can be attached to the polyamine backbone and still be effectively transported. The findings of each of the above studies clearly indicate that the polyamine structure can be used effectively to transport specific active moieties into cells and in many cases, target chromatin. We recently attempted to build on this paradigm with a new generation of agents that target chromatin [25,26,36]. However, unlike previous attempts, our goal was not to damage chromatin, but rather to alter its regulation of gene expression.
Polyamine analogues as HDAC inhibitors
The growing interest in drugs that alter chromatin is based on the recognition that epigenetic regulation of gene expression plays a critical role in the aetiology and progression of cancer and, unlike gene mutations or loss, epigenetic changes are, in theory, reversible [9]. Aberrant silencing of tumour suppressors and other genes has been found in all cancers examined. One of the major regulators of chromatin conformation, and hence gene expression, is histone acetylation. As stated above, acetylation of histones is generally associated with transcriptionally active chromatin, and activity of HDACs lead to condensation of chromatin and inhibition of transcription. A number of class I/II HDAC inhibitors have been examined for their ability to re-express silenced genes [24]. Although several class I/II HDCA inhibitors have been synthesized and some are currently in clinical trial, virtually all of them have been designed to target primarily the zinc cofactor at the active site of the HDACs [24].
We have previously reported the use of various polyamine analogue structures combined with active-site-directed inhibitor moieties of the class I/II HDACs [25,26]. This strategy has the advantage of using known structures that inhibit the HDACs, the hydroxamic acids and benzamides, combined with the high chromatin affinity of the polyamine structure. Additionally, as tumour cells are known to transport and accumulate many of the polyamine analogues, the polyamine backbone offers the likelihood that these compounds would be readily transported into tumour cells. Specific members of the PAHA (polyaminohydroxamic acid) and PABA (polyaminobenzamide) polyamine analogues demonstrated potent inhibition of the class I/II HDACs in a cell-free system, and in treated leukaemia cells re-expression of the growth regulating CDK (cyclin-dependent kinase) inhibitor p21 was observed. Some of these analogues also significantly inhibited tumour cell growth in vitro.
An additional advantage of using the polyamines as a backbone for HDAC inhibitors is that it provides a scaffold upon which it is possible to design molecules that possess selectivity for the individual HDACs (see Chapter 6 by Woster). This possibility would be useful in the design of molecular tools to study the effects of inhibiting the individual HDACs, and may provide a therapeutic advantage over existing non-selective inhibitors. One promising compound that demonstrates some selectivity with respect to HDAC inhibition was the compound designated 17 (Figure 2) in Varghese et al. [26]. The class II HDAC6, in addition to having histone targets, is also capable of deacetylating α-acetyltubulin. Compound 17 demonstrated potent functional in situ inhibition of HDAC6 resulting in a substantial increase in α-acetyltubulin in treated cells. These data underscore the possibility of using the flexibility allowed by the polyamine structure to design selective inhibitors for each of the individual class I/II HDACs.
Figure 2. Chemical structures of polyamine analogues.

Compound 17 selectively inhibits HDAC6 activity and increases acetylated α-tubulin in HCT116 colorectal cancer cells. 1c and 2d are potent inhibitors of LSD1 activity and re-activate aberrantly silenced genes in tumour cells. PG-11144 and PG-11150 polyamine analogues contain ten amines and are a cis/trans pair with double bonds in the centre of their structure. Oligoamines competitively inhibit LSD1 activity and re-activate aberrantly silenced genes in colorectal cancer cells.
Although considerable work remains to be done, the initial analysis of the polyamine analogue HDAC inhibitors of both the PAHA and PABA families shows considerable promise.
Targeting LSD1 for gene re-expression
As stated above, the discovery of LSD1 and the Jmj C domain-containing demethylases indicated that histone methylation, like histone acetylation, is a dynamic process. Structural analysis demonstrates that LSD1 is highly conserved across species and consists of an N-terminal SWIRM (Swi3p/Rsc8c/Moira) domain, a central protruding tower domain and a C-terminal amine oxidase-like domain, containing a FAD-binding subdomain, which is highly homologous with MAOs (monoamine oxidases) and the polyamine oxidases, SMO and APAO [18,27] (Figure 3). LSD1 catalyses the demethylation of mono- or di-methylated Lys4 of histone H3 (H3K4) by cleavage of the α-carbon bond of the substrate through an oxidative process with the reduction of FAD. FADH2 is re-oxidized by oxygen to produce H2O2, leading to the generation of an imine intermediate. The imine intermediate is subsequently hydrolysed to generate formaldehyde and the amine of lysine (Figure 4). Despite the similarity of structure and chemistry between these amine oxidases, LSD1 demonstrates entirely different biological activity and cellular localization than do the MAOs (Table 1). MAOs, including two isoforms MAO A and MAO B, are bound to the outer mitochondrial membrane [28]. APAO is a peroxisomal enzyme, whereas SMO is found in both the cytoplasm and nucleus [14]. Both APAO and SMO are FAD-dependent enzymes that oxidize polyamine substrates and produce H2O2. The structural and catalytic similarities of these FAD-dependent oxidases has been instructive in understanding the basic biology of FAD-dependent oxidases, as well as in the search for effective inhibitors that can interact with LSD1.
Figure 3. Structure and domain organization comparison of LSD1 and polyamine Oxidases.

(A) LSD1 is 852 amino acids long and consists of an N-terminal SWIRM domain, a central protruding tower domain and a C-terminal AOL (amine oxidase-like) domain that contains a FAD-binding catalytic subdomain. Lys661 (K661) of LSD1 is a critical residue at the FAD-binding site that is associated with the N5 atom of FAD through a hydrogen bond. The primary splice variant of human SMO codes for a protein of 511 amino acids, and Lys367 (K367) at the C-terminal is the key residue for FAD association. The predominant human splice variant of APAO contains 551 amino acids, and Lys322 (K322) is important for FAD binding. (B) Structural comparison of the FAD-associated catalytic centre of LSD1 and polyamine oxidases. The catalytic domains of LSD1, SMO and APAO possess over 60% similarity in amino acid sequences.
Figure 4. Mechanism of histone demethylation by LSD1.
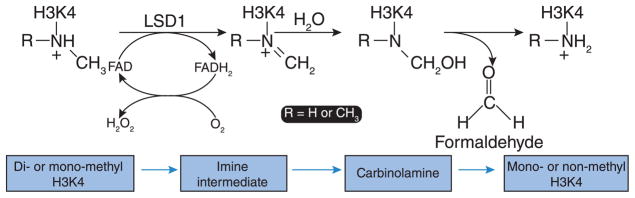
LSD1 catalyses the FAD-dependent demethylation of mono- (R=H) or di-methyl (R=CH3) Lys4 of histone 3 through transferring two hydrogen atoms from methylated H3K4 to FAD with the resultant reduction of oxygen to H2O2. The imine intermediate is then hydrolysed to an unstable carbinolamine that subsequently degrades to release formaldehyde.
Table 1.
Comparison of biological functions between amine oxidases and LSD1
| Characteristic | MAO | APAO | SMO | LSD1 |
|---|---|---|---|---|
| Subcellular localization | Mitochondrial membrane | Peroxisome | Cytoplasm, nucleus | Nucleus |
| Substrate | Arylalkyl amines | N1-acetyl spermine, N1-acetyl spermidine | Spermine, spermidine | Histone, proteins |
| FAD-dependent | Yes | Yes | Yes | Yes |
| H2O2 product | Yes | Yes | Yes | Yes |
| Aldehyde product | Yes | Yes | Yes | Yes |
| Cellular function | Neurological activities | Polyamine catabolism | Polyamine homoeostasis | Transcription regulation |
Linking LSD1-suppressed gene expression to the role of LSD1 in tumorigenesis
LSD1 is a member of a multiprotein co-repressor complex that includes CoRest, HDAC1/2 and BHC80. Several recent studies demonstrate that inhibition of LSD1 by siRNA (small interfering RNA) or pharmacological inhibition changes global and promoter-specific H3K4me2 (dimethylated H3K4) levels and increases expression of some known LSD1/CoREST-target genes. For example, Shi et al. [18] demonstrated an up-regulation of neuron-specific M4 AchR, SCN1A-3A and the CDK inhibitor p57KIP2 in LSD1 RNAi (RNA interference)-treated HeLa cells. Lee et al. [27] reported an increase in global H3K4me2, as well as transcriptional de-repression of two LSD1 target genes, Egr1 and the pluripotent stem cell marker Oct4, in P19 EC cells treated with a non-specific MAO inhibitor. A recent study demonstrated that knockout of LSD1 in embryonic stem cells induces progressive loss of DNA methylation and a decrease in DNA methyltransferase 1 stability and protein levels [29]. These interesting findings demonstrate a previously unknown mechanistic interaction between histone demethylase and DNA methyltransferase.
DNA promoter hypermethylation frequently acts in concert with histone modifications that result in decreased chromatin-activating marks, such as H3K4me (monomethylated H3K4), resulting in increased repressive marks such as H3K9me and H3K27me, and in the aberrant silencing of specific genes [30]. Since the discovery of LSD1 as an important demethylase of the key activating mark H3K4, the potential association of LSD1 activity with tumorigenesis has been intensively investigated. A number of recent studies indicate that an alteration in the function of LSD1 has a role in cancer. For example, high-level LSD1 expression has been linked to an increased risk of prostate cancer recurrence, suggesting a tumour-promoting role for LSD1 [31]. In another study, LSD1 was shown to possess a pro-oncogenic activity through its regulation of pro-survival gene expression and p53 transcription in human breast cancer cells [32]. Bradley et al. [33] reported that induction of LSD1 is one of the early responses to chemical carcinogens in HMECs (human mammary epithelial cells), and that it may affect the expression of multiple genes critical in early-stage mammary oncogenic transformation. Very recently, Schulte et al. [34], demonstrated that a non-specific MAO inhibitor is effective in inducing expression of genes associated with differentiation in neuroblastoma cells, and that the inhibitor can cause significant in vivo inhibition of tumour growth. These studies have established LSD1 as an important link to the development and progression of cancer and provide a rationale for developing LSD1 inhibitors as a target for therapeutic intervention.
Identification of polyamine analogues as effective LSD1 inhibitors
Human LSD1 shares 20% similarity in overall structure with that of other FAD-dependent amine oxidases. Specifically, the catalytic domains of LSD1 and SMO are over 60% similar in amino acid sequence (Figure 3). This similarity suggests that specific polyamine analogues may function as effective inhibitors of LSD1 (Table 2).
Table 2.
Inhibitory effects of polyamine analogues on the activity of recombinant polyamine oxidases, LSD1 and tumour cell growth
| Polyamine | APAO | SMO | LSD1 | Growth |
|---|---|---|---|---|
| 1c | ++ | +/− | +++ | + |
| 2d | +++ | +++ | +++ | ++ |
| PG-11144 | Unknown | +++ | ++ | +++ |
| PG-11150 | Unknown | +++ | ++ | +++ |
No effect; + to +++, modest to potent inhibition.
Although the natural polyamines or acetylpolyamines are not substrates of LSD1 [18], the strong association of polyamines with chromatin and the structural similarity between the polyamines and the lysine tails of histones suggest that polyamines and/or polyamine analogues may alter the activity of LSD1 and other chromatin modifiers. In our previous studies we discovered that specific polyamine analogues function as potent inhibitors of purified polyamine oxidases [15,35]. Since the active site of LSD1 is closely related to that of SMO and APAO, and because guanidines have been shown to inhibit the activity of structurally related polyamine oxidases, we sought to determine whether a small library of bisguanidine and biguanide polyamine analogues (Figure 2) functioned as effective inhibitors of LSD1. Most of these compounds were found to inhibit the demethylase activity of recombinant LSD1 by >50% at concentrations less than 1 μM [36]. The two most potent analogues, 1c [1,11-bis(N2,N3-dimethyl-N1-guanidino)-4,8-diazaundecane] and 2d (1,15-bis{N5-[3,3-(diphenyl)propyl]-N1-biguanido}-4,12-diazapentadecane), exhibited non-competitive inhibition kinetics, suggesting that these compounds probably bind to LSD1 at a site other than at the active site. The possible mechanisms of action include the fact that these LSD1 inhibitors may block FAD from associating with its binding site, or the fact that these inhibitors might induce a conformational change in LSD1, such that LSD1 is no longer able to bind to its histone lysine substrate correctly. It is, however, possible that the enzymatic kinetics obtained when using recombinant LSD1 and a short peptide as a substrate in a cell-free system may not be representative of the interaction of the inhibitor and the nucleosome in association with the LSD1–CoRest–HDAC complex in situ.
A class of long-chain polyamine analogues known as oligoamines has also been found to potently inhibit APAO or SMO (Table 2), suggesting that these analogues may also inhibit LSD1 [36]. Among others, we tested two cis/trans oligoamine isomers, PG-11144 and PG-11150 (Figure 2), for their ability to inhibit LSD1 [36] These oligoamines exhibit competitive-inhibition kinetics, suggesting that the oligoamines may directly compete with substrate at the active site in a manner different than seen for the bisguanidine and biguanide analogues. This new class of polyamine analogue LSD1 inhibitors offers another promising avenue of investigation.
It should be noted that the precise mechanism of inhibition of LSD1 by the polyamine analogues remains unclear. Ongoing crystallographic analysis should facilitate a better understanding of the molecular and structural basis of LSD1 inhibition by polyamine analogues, and perhaps provide insight into the design of more effective inhibitors [37].
Inhibition of LSD1 by novel polyamine analogues reactivates silenced genes in cancer cells
To determine whether the in vitro inhibition of LSD1 activity by polyamine analogues translated into a cellular response, the effects of polyamine analogues as LSD1 inhibitors on global H3K4me status were examined in multiple human cancer cell lines, including colorectal, breast, lung and leukaemia. Exposure of cancer cells to these polyamine analogues leads to increased global H3K4me2 and H3K4me1, with no change in H3K4me3 (trimethylated H3K4) and H3K9me2 [36,38]. The promoter region of H3K4me2 is usually associated with open chromatin and active transcription, and the occupancy of H3K4me2 was found to be at low levels in the promoters of a number of frequently DNA hypermethylated and epigenetically silenced genes important in tumorigenesis [30]. A number of such silenced genes have been identified in colorectal cancer cell lines, such as HCT116 and RKO. These genes include members of the WNT signalling pathway antagonists, SFRPs (secreted frizzle-related protein) family, GATA family transcription factors, the mismatch repair gene MLH1, the cell-cycle regulator CDKN2A, and the tissue invasion regulator TIMP3 (tissue inhibitor of metalloproteinase 3) [30]. Treatment of HCT116 cells with 1c or 2d for 48 h resulted in the re-expression of three members of the SFRP family, SFRP1, SFRP4 and SFRP5, as well as of the GATA5 transcription factor [36]. Similarly, treatment with PG-11144 or PG-11150 for 24 h led to the re-expression of SFRP1 and SFRP2 [38]. Leukaemia cells treated with 2d resulted in increased global H3K4me2 and the re-expression of E-cadherin, an important tumour suppressor gene [39].
Chromatin immunoprecipitation analysis demonstrated that polyamine analogue-induced gene re-expression in HCT116 cells is closely associated with increased active histone marks H3K4me1/me2 and acetyl-H3K9, and decreased repressive marks H3K9me1/me2 [36,38]. However, no change of H3K4me3 levels were detected after treatment with the polyamine analogues, suggesting that these compounds specifically target LSD1 rather than other histone demethylases such as the Jmj C histone demethylases that are capable of demethylating H3K4me3 [19]. Some other hallmarks of silenced chromatin, such as H3K9me3 and PcG (polycomb) group protein-mediated H3K27 methylation, remain unchanged in the promoter regions of the analogue-activated genes after treatment with LSD1 inhibitors. These results are similar to the chromatin state of HCT116 treated with the DNA methylation inhibitor DAC (5-aza-2′-deoxycytidine) [40].
As noted above, CpG island hypermethylation frequently acts in concert with abnormal histone mark activities in silencing genes. Interestingly, the reexpression of aberrently silenced genes by our unique polyamine analogues occurs without wholesale changes in the methylation status of CpG islands of the gene examined. Similar results were observed in silenced genes that were reactivated by inhibition of the class III HDAC, SIRT1 (sirtuin 1) [41]. These results indicate that treatment with LSD1 inhibitors alone is sufficient to produce gene re-expression, even when dense promoter DNA methylation is maintained.
It is possible that the polyamine analogues have effects other than inhibition of LSD1 that lead to gene re-expression. To address this issue, we compared the effects of the inhibition of LSD1 with the analogues 1c and 2d with those induced by an RNAi-mediated knockdown in LSD1 expression in HCT116 cells. LSD1 depletion by RNAi was accompanied by increased H3K4me2 at the promoters of re-activated genes, and resulted in modest reexpression of the examined genes [36]. We found that pharmacological inhibition of LSD1 with the analogues was more effective than was RNAi with respect to re-expression of silenced genes, which may reflect inherent differences in chromatin structure resulting from inhibitor–LSD1 complexes compared with RNAi-induced decreases in LSD1 protein.
Combination of LSD1 inhibitors with other agents targeting epigenetic regulation of gene expression
Combination chemotherapy is an important strategy in modern cancer treatment, frequently having the advantage of allowing for lower and better-tolerated doses of individual chemotherapeutic agents, while at the same time improving the overall response as compared with when the agents are used alone. The combination of DNA methyltransferase and HDAC inhibitors has demonstrated synergistic effects in re-expressing epigenetically silenced genes in cultured cancer cells, and has produced clinical responses in patients with leukaemias [42,43]. Our latest studies demonstrate that the combination of low-dose oligoamines with DAC results in synergistic expression of the SFRP2 gene [38]. This result suggests that the combination of LSD1 inhibitors and DNA methylation inhibitors can collaborate in the re-expression of specific silenced genes and may provide a useful strategy for the potential clinical utility of these two agents, each of which targets different epigenetic regulatory enzymes.
Conclusions
Increasing knowledge regarding the molecular mechanisms of polyamine metabolism in cancer cells that has accumulated over the last three decades has provided unique opportunities for the development of many promising therapeutic agents. Despite the somewhat disappointing results obtained from clinical trials of the early polyamine biosynthesis inhibitors, polyamine metabolism remains a rational target for cancer therapy, and opportunities still exist for development of novel agents with better specificity when targeting cancer cells and fewer side effects affecting normal tissues. The unique association of ‘superinduction’ of polyamine catabolic enzymes with a cytotoxic response for certain polyamine analogues has been well elucidated and noted. The interaction of polyamine or polyamine-based analogues with nucleic acids has received considerable attention, although the exact mechanisms of this phenomenon are still being defined. Epigenetic regulation of gene expression has emerged as an important target for drug development in cancer therapy. Specific polyamine analogues have been identified as potent inhibitors of the HDACs. LSD1 is a newly identified homologue of polyamine oxidase. Owing to its important activity in modifying chromatin and gene transcription, significant attention has been focused on developing LSD1 inhibitors as an effective strategy in treatment of cancer and other diseases. Our recent findings clearly suggest that novel polyamine analogues are powerful inhibitors of LSD1, both alone and in combination with other agents that target epigenetic silencing, and that they are capable of inducing re-expression of several aberrantly silenced genes important in tumorigenesis. The use of these compounds represents a new direction for drug development in cancer prevention and therapy.
Summary.
Polyamine effects on chromatin remodelling are important in the stabilization of DNA structure and in RNA processing and may affect diverse cellular functions including transcriptional regulation.
Drug design based on polyamine structure, when combined with active site-directed inhibitor moieties of the HDACs, led to the successful development of specific HDAC inhibitors of the PAHA and PABA families. These inhibitors demonstrate potent inhibition of HDAC in tumour cells.
The recent identification of flavin-dependent LSD1, which specifically demethylates mono- and di-methyl Lys4 of histone 3, plays critical roles in regulating gene transcription.
The structural and catalytic similarities of LSD1 and polyamine oxidases have facilitated the identification of a group of biguanide, bisguanidine and oligoamine polyamine analogues as potent inhibitors of LSD1.
Inhibition of LSD1 alone and in combination with other agents targeting epigenetic silencing are capable of inducing re-expression of a number of aberrantly silenced genes that are important in tumorigenesis.
References
- 1.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 2.Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem. 2006;281:14529–14532. doi: 10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- 4.Gerner EW, Meyskens FL., Jr Combination chemoprevention for colon cancer targeting polyamine synthesis and inflammation. Clin Cancer Res. 2009;15:758–761. doi: 10.1158/1078-0432.CCR-08-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seiler N. Pharmacological aspects of cytotoxic polyamine analogs and derivatives for cancer therapy. Pharmacol Ther. 2005;107:99–119. doi: 10.1016/j.pharmthera.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Basu HS, Smirnov IV, Peng HF, Tiffany K, Jackson V. Effects of spermine and its cytotoxic analogs on nucleosome formation on topologically stressed DNA in vitro. Eur J Biochem. 1997;243:247–258. doi: 10.1111/j.1432-1033.1997.0247a.x. [DOI] [PubMed] [Google Scholar]
- 7.Basu HS, Sturkenboom MC, Delcros JG, Csokan PP, Szollosi J, Feuerstein BG, Marton LJ. Effect of polyamine depletion on chromatin structure in U-87 MG human brain tumour cells. Biochem J. 1992;282:723–727. doi: 10.1042/bj2820723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato N, Ohtake Y, Kato H, Abe S, Kohno H, Ohkubo Y. Effects of polyamines on histone polymerization. J Protein Chem. 2003;22:303–307. doi: 10.1023/a:1025032906494. [DOI] [PubMed] [Google Scholar]
- 9.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coffino P. Regulation of cellular polyamines by antizyme. Nat Rev Mol Cell Biol. 2001;2:188–194. doi: 10.1038/35056508. [DOI] [PubMed] [Google Scholar]
- 11.Pegg AE. Spermidine/spermine-N1-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab. 2008;294:E995–E1010. doi: 10.1152/ajpendo.90217.2008. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Devereux W, Woster P, Stewart T, Hacker A, Casero R., Jr Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 2001;61:5370–5373. [PubMed] [Google Scholar]
- 13.Vujcic S, Diegelman P, Bacchi CJ, Kramer DL, Porter CW. Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem J. 2002;367:665–675. doi: 10.1042/BJ20020720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray-Stewart T, Wang Y, Goodwin A, Hacker A, Meeker A, Casero RA., Jr Nuclear localization of human spermine oxidase isoforms: possible implications in drug response and disease etiology. FEBS J. 2008;275:2795–2806. doi: 10.1111/j.1742-4658.2008.06419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster P, Casero R., Jr Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem Biophys Res Commun. 2003;304:605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 16.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 17.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Lan F, Matson C, Mulligan P, Whetstine J, Cole P, Casero R, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 19.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 21.Holley JL, Mather A, Wheelhouse RT, Cullis PM, Hartley JA, Bingham JP, Cohen GM. Targeting of tumor cells and DNA by a chlorambucil-spermidine conjugate. Cancer Res. 1992;52:4190–4195. [PubMed] [Google Scholar]
- 22.Phanstiel OI, Price HL, Wang L, Juusola J, Kline M, Shah SM. The effect of polyamine homologation on the transport and cytotoxicity properties of polyamine-(DNA-intercalator) conjugates. J Org Chem. 2000;65:5590–5599. doi: 10.1021/jo0002792. [DOI] [PubMed] [Google Scholar]
- 23.Delcros JG, Tomasi S, Duhieu S, Foucault M, Martin B, Le Roch M, Eifler-Lima V, Renault J, Uriac P. Effect of polyamine homologation on the transport and biological properties of heterocyclic amidines. J Med Chem. 2006;49:232–245. doi: 10.1021/jm050018q. [DOI] [PubMed] [Google Scholar]
- 24.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 25.Varghese S, Gupta D, Baran T, Jiemjit A, Gore SD, Casero RA, Jr, Woster PM. Alkyl-substituted polyaminohydroxamic acids: a novel class of targeted histone deacetylase inhibitors. J Med Chem. 2005;48:6350–6365. doi: 10.1021/jm0505009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varghese S, Senanayake T, Murray-Stewart T, Doering K, Fraser A, Casero RA, Jr, Woster PM. Polyaminohydroxamic acids and polyaminobenzamides as isoform selective histone deacetylase inhibitors. J Med Chem. 2008;51:2447–2456. doi: 10.1021/jm701384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee M, Wynder C, Schmidt D, McCafferty D, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Binda C, Mattevi A, Edmondson DE. Structure–function relationships in flavoenzyme-dependent amine oxidations: a comparison of polyamine oxidase and monoamine oxidase. J Biol Chem. 2002;277:23973–23976. doi: 10.1074/jbc.R200005200. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 30.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer: a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 31.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 32.Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- 33.Bradley C, van der Meer R, Roodi N, Yan H, Chandrasekharan MB, Sun ZW, Mernaugh RL, Parl FF. Carcinogen-induced histone alteration in normal human mammary epithelial cells. Carcinogenesis. 2007;28:2184–2192. doi: 10.1093/carcin/bgm100. [DOI] [PubMed] [Google Scholar]
- 34.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Hacker A, Murray-Stewart T, Frydman B, Valasinas A, Fraser AV, Woster PM, Casero RA., Jr Properties of recombinant human N1-acetylpolyamine oxidase (hPAO): potential role in determining drug sensitivity. Cancer Chemother Pharmacol. 2005;56:83–90. doi: 10.1007/s00280-004-0936-5. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Greene E, Murray-Stewart T, Goodwin A, Baylin S, Woster P, Casero R., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stavropoulos P, Hoelz A. Lysine-specific demethylase 1 as a potential therapeutic target. Expert Opin Ther Targets. 2007;11:809–820. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Murray Stewart T, Wu Y, Marton L, Woster P, Casero R. Novel oligoamine/polyamine analogues inhibit lysine-specific demethylase 1 (LSD1), induce re-expression of epigenetically silenced genes, and inhibit the growth of established human tumors in vivo. AACR 100th Annual Meeting; Denver, CO, U.S.A.. 18–22 April 2009; 2009. Abstract LB-173. [Google Scholar]
- 39.Murray-Stewart T, Huang Y, Woster P, Casero R. Polyamine analogue inhibition of lysine-specific demethylase 1 in human acute myeloid leukemia cell lines. Proc Am Assoc Cancer Res. 2008;49:2605. [Google Scholar]
- 40.McGarvey K, Fahrner J, Greene E, Martens J, Jenuwein T, Baylin S. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 41.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cameron E, Bachman K, Myohanen S, Herman J, Baylin S. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 43.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]