

Abstract
While splicing differences between tissues, sexes and species are well documented, little is known about the extent and the nature of splicing changes that take place during human or mammalian development and aging. Here, using high-throughput transcriptome sequencing, we have characterized splicing changes that take place during whole human lifespan in two brain regions: prefrontal cortex and cerebellum. Identified changes were confirmed using independent human and rhesus macaque RNA-seq data sets, exon arrays and PCR, and were detected at the protein level using mass spectrometry. Splicing changes across lifespan were abundant in both of the brain regions studied, affecting more than a third of the genes expressed in the human brain. Approximately 15% of these changes differed between the two brain regions. Across lifespan, splicing changes followed discrete patterns that could be linked to neural functions, and associated with the expression profiles of the corresponding splicing factors. More than 60% of all splicing changes represented a single splicing pattern reflecting preferential inclusion of gene segments potentially targeting transcripts for nonsense-mediated decay in infants and elderly.
Keywords: alternative splicing, brain, development, human, RNA-seq
Introduction
Alternative splicing (AS) is a process that allows a single gene to produce multiple transcripts and, consequently, multiple proteins, by utilizing different splice sites in pre-mRNA (Maniatis, 1991). Splicing is mediated by the spliceosome, a dynamic complex containing both RNA and proteins, which catalyzes intron excision and joining of exons. This process may be affected by numerous splicing factors (SFs), proteins that bind to specific binding motifs (SFBMs) on pre-mRNA and affect spliceosome preferences for splicing sites (Black, 2003). Transcriptome-wide splicing studies conducted using microarrays, EST data analysis and, more recently, high-throughput sequencing have revealed extensive splicing differences among tissues, sexes, and species (Calarco et al, 2007; Blekhman et al, 2010). Particularly, AS is widespread in higher eukaryotes, affecting up to 95% of human multi-exon genes (Johnson et al, 2003; Pan et al, 2008; Wang et al, 2008; Takeda et al, 2010).
Transcriptome-wide studies of AS conducted to date have mainly focused on splicing differences among tissues, rather than splicing changes with age. On the individual gene level, changes in transcript splicing have been shown to have important roles in many human developmental processes, including neuronal cell migration (Blencowe, 2006), synapse formation (Ule et al, 2005), and establishing neurotransmitter receptor specificity (Dredge et al, 2001). In human aging, AS is associated with age-related disorders, such as Alzheimer’s disease (Buee et al, 2000), amyotrophic lateral sclerosis (Dredge et al, 2001), and cancer (Venables et al, 2009). Accordingly, the AS changes found in human aging and neurodegeneration were shown to overlap (Tollervey et al, 2011). Further, altered splicing of lamin A/C (LMNA) has been linked to a human premature aging phenotype (De Sandre-Giovannoli and Levy, 2006). At the transcriptome-wide level, however, splicing changes taking place in human development and aging have not been investigated. Here, we took advantage of high-throughput sequencing to assess the extent, functional significance, and potential regulation of splicing changes taking place in two brain regions, prefrontal cortex (PFC) and cerebellum, over the entire human lifespan.
Results
Transcriptome sequencing
To assess changes in gene splicing in the human brain over the course of lifespan, we sequenced the poly-A+ transcriptome in a total of 35 individuals, with ages ranging from 2 days to 98 years, using high-throughput transcriptome sequencing (RNA-seq) on the Illumina platform. The sequencing was carried out for the PFC and cerebellar cortex (CBC) of 30 individuals using the paired-end protocol (Data set 1 (DS1)), and in the PFC of 13 individuals using the single-end strand-specific protocol (Data set 2 (DS2)). The read length was 75 nucleotides (nt) for DS1 and 100 nt for DS2. In the DS1, poly-A+ RNA from five individuals, of similar age per sample, were combined to reduce individual variation, resulting in six PFC and six CBC samples (Supplementary Tables S1 and S2, Figure 1A). In DS2, individual samples were sequenced separately.
Figure 1.

Age-related splicing changes in the human brain. (A) Sample age distribution in PFC (orange) and CBC (gray). Each dot represents an individual; the darker shades of color represent older age. In DS1, five individuals of similar age were combined in each sample, resulting in six PFC- and six CBC-pooled samples with the average ages marked on the horizontal axis. (B) Fraction of genes (right) and segments (left) showing significant age-related splicing changes in DS1 (DS1 age), DS2 (DS2 age), and in both data sets (DS1 and DS2), as well as genes showing significantly different age-related changes in brain regions in DS1 (DS1 tissue:age). (C) Correlation of age-related splicing changes between DS1 and DS2. Shown are the distributions of the Pearson correlation coefficients for non-age-related segments (gray, N=22 360); segments significant only in DS2 (yellow, N=4630); segments significant only in DS1 (orange, N=1648); and segments significant in both data sets (red, N=1484). In all cases, only segments with sufficient coverage in both data sets were used. (D) Correlation of inclusion ratio changes with age measured by RT–PCR (horizontal axis) and RNA-seq (vertical axis). (E) Correlation of inclusion ratio estimates measured by RT–PCR (horizontal axis) and RNA-seq (vertical axis). (F) Correlation of age-related splicing changes measured at the transcript (RNA-seq) and protein (mass spectrometry) levels. Shown are the distributions of the Pearson correlation coefficients for the actual observations (orange) and 10 000 random permutations (gray). The difference between the means of observed and permutation distributions was significant (q=0.0004). At the individual segment level, 4 of 24 segments showed marginally significant correlations (Pearson correlation test, BH-corrected FDR<0.1). (G and H) Examples of age-related splicing changes in PFC for PALM (G) and MART (H) gene transcripts. Shown are: (top) RNA coverage (by all samples from DS1) and gene annotation (Ensembl, v57); (bottom) RNA (gray) and protein (blue) coverage and annotation of zoomed gene regions in newborns (nb), young and old individuals (an average of two pooled samples with similar age). The height of the red lines represents the RNA coverage of splice junctions. The PCR results showing relative abundance of the short (S) and long (L) isoforms for the depicted junctions are displayed to the right of the corresponding coverage plot.
In total, we obtained 181 555 729 read pairs in DS1 and 274 927 771 reads in DS2, with the average sample coverage of 18 million reads (Supplementary Table S3). The reads were mapped to the human genome and the splice junctions, including annotated and hypothetical junctions, were constructed by combining exons within a gene based on the Ensembl annotation (Methods). Allowing up to three mismatches, 64% of the reads could be mapped (Supplementary Tables S3 and S4; Supplementary Figure S1), with 93% of the mapped reads located within gene boundaries: 85% within exons, and 25% on the splice junctions (Supplementary Table S3). Allowing no mismatches during the mapping procedure did not influence our results (Supplementary Figures S2–S4). A total of 8365 genes had alternative exons with sufficient coverage in DS1, and 8061, in DS2 (Methods). A total of 30 122 segments from 6755 genes that had sufficient coverage in both data sets were used in the analysis.
We further identified novel splice variants by mapping reads to hypothetical junctions constructed based on the Ensembl exon annotation. In addition to 200 464 annotated splice junctions, we identified 21 644 novel junctions supported by at least two sequence reads. There were no detectable deviations from the uniform junction coverage for either novel or annotated junctions (Supplementary Figure S5). This finding indicates that at least 10% of transcript isoforms expressed in the human brain are not yet annotated.
Age-related splicing variation in the human brain
To identify age-related splicing changes, we defined gene segments as intervals between nearest splice sites, as well as transcriptional and translation start/stop sites, based on the Ensembl annotation (v57). For each segment, two read categories were counted: reads that map to the segment itself, that could be termed inclusion reads (i), and reads that map to junctions between upstream and downstream segments, or exclusion reads (e). Consequently, the inclusion ratio (i.e., the proportion of transcripts that contains a segment) was estimated as
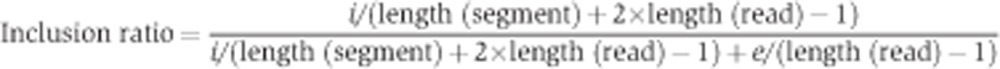 |
The Binomial Generalized Linear Model was applied to test the significance of the inclusion ratio change with age (Methods). Following this approach, 22% of genes detected in DS1 (3132 segments from 1456 genes) and 38% of genes detected in DS2 (6114 segments from 2588 genes) demonstrated significant age-related splicing changes (q<0.05, Figure 1B). A total of 1484 segments from 721 genes were significant in both data sets. Only these segments were used in the further analyses, unless otherwise specified.
The validity of the identified age-related splicing changes was tested using three approaches. First, we compared the identified age-related splicing changes between DS1 and DS2. Even though DS1 and DS2 were generated using alternative sample preparation and sequencing procedures, age-related splicing changes found in each data set overlapped significantly (Fisher’s exact test, P<0.0001) and displayed strong and significant positive correlation for the inclusion ratio changes with age (Figure 1C, Supplementary Table S5).
Second, we measured splicing in all 12 DS1 samples using Affymetrix Exon Arrays. Segments detected on the arrays and showing significant age-related splicing changes in DS1, or in both DS1 and DS2, demonstrated positive and significant correlation for inclusion ratio changes with age between the two methodologies both in PFC and in CBC (Pearson correlation, P<0.0001 in both brain regions) (Supplementary Figure S6).
Third, we tested the inclusion ratio variation of 30 segments showing significant age-related splicing changes in both RNA-seq data sets using PCR. Out of the 30 segments, 24 showed two PCR bands of a size expected for the predicted splicing products. All 24 showed inclusion ratio changes with age that matched the RNA-seq results (ρ=0.93) (Figure 1D and G–H Supplementary Table S6). In addition, not only the inclusion ratio changes with age, but also the inclusion ratio estimates themselves, demonstrated good overall agreement between the PCR and RNA-seq protocols (ρ=0.88) (Figure 1E, Supplementary Table S6). Taken together, these results confirm authenticity and reproducibility of the age-related splicing changes identified by RNA-seq.
Age-related splicing changes at the protein level
Close to 30% of identified age-related splicing changes (26% in DS1 and 33% in DS2) affect segments annotated as protein-coding in all transcript isoforms. Thus, if the corresponding isoforms are translated, these splicing changes should be observed at the protein level. To test this, we took advantage of a data set containing protein measurements from PFC of 12 humans with the same age distribution, collected using label-free mass spectrometry (Somel et al, 2010). Despite the fragmentary nature of mass-spectrometry coverage, sufficient peptide numbers were detected for 24 segments from 19 genes showing significant age-related splicing changes in both data sets (Methods). Importantly, the age-related splicing changes correlated positively and significantly between the mRNA and protein measurements, compared with the correlation coefficient distribution obtained by permutation of the segment labels for RNA and protein measurements among the 24 segments (P=0.0006 for DS1; 0.0001 for DS2; 0.0004 for the integrated DS1/DS2 data set) (Figure 1F). This result further validates the authenticity of identified age-related splicing changes, and indicates the presence of corresponding changes at the protein level.
Splicing changes in development and aging
A previous study showed that gene expression in the human PFC changes at a rapid pace during early postnatal development, but at a much slower rate in adulthood and aging (Somel et al, 2010). Here we show that splicing changes in the human PFC display a similar trend: when classifying changes taking place before 14 years of age as development, and after 25 years as aging, ∼70% of total splicing variation takes place in development: of the 1484 segments showing significant splicing changes with age, 970 showed significant changes in development and 310 in aging, if significance was tested separately for each ontogenetic interval. Notably, splicing changes in development and in aging show strong and opposite directional bias: in development, 75% of segments showed decreased inclusion, while in aging, 77% of segments showed increasing inclusion (Figure 2A). Over lifespan, these two trends comprised a common ‘down–up’ inclusion ratio pattern comprising 62% of segments showing significant age-related changes (Figure 2B). The same bias towards a ‘down–up’ inclusion ratio pattern was observed when we considered only segments showing significant splicing changes in development and/or aging when testing significance separately for each ontogenetic interval (Methods; Supplementary Figures S7 and S8).
Figure 2.

Splicing changes in development and aging. (A) Distribution of the inclusion ratio change index for development (solid line) and aging (dash line) for 1484 age-related segments. The negative index represents the inclusion ratio decrease with age, positive index, the inclusion ratio increase. The colors represent RNA-seq data sets analyzed: yellow—DS1/PFC, gray—DS1/CBC, and red—DS2/PFC. (B) Scatter plot of the inclusion ratio change showing different splice types: violet—skipped exons, yellow—retained introns, gray and white—complex and mixed splice types respectively. Four quadrants of the plot correspond to four patterns of inclusion ratio change with age: down–up, up–up, up–down and down–down. Inclusion ratio change is plotted for the 853 segments showing significant splicing changes with age in both data sets and showing consistent direction of age-related splicing change between data sets in development, as well as in aging. (C) Schematic representation of segment numbers (represented by the size of the pie diagrams) and splicing type fractions (represented by colors) in the four inclusion ratio change patterns shown in panel B. (D) Fractions of NMD segments in the four inclusion ratio change patterns. Numbers on top of the bars show the number of NMD segments and the total number of segments in each pattern. Asterisks indicate significant enrichment of NMD segments in the ‘down–up’ pattern (one-sided Fisher’s exact test, P=0.0002) (E) Distribution of the phastCons scores of segments in the four inclusion ratio change patterns. Asterisks indicate significantly low conservation of segments in the ‘down–up’ pattern (one-sided Wilcoxon test, P<0.0001). (F–K) Gene expression of NMD-related genes, CASC3, UPF2, UPF3B, SMG1, SMG5, and SMG6 in the human PFC. Gene expression was estimated based on published data from (Liu et al, 2012) (green) and (Colantuoni et al, 2011) (violet). Expression intensities for each gene were normalized to mean=0 and s.d.=1. Age scale is log2-transformed, ages are shown in years, MAC stands for ‘months after conception’.
Segments following the ‘down–up’ inclusion ratio pattern displayed a number of distinct features. First, a significantly greater proportion of ‘down–up’ segments, compared with segments following other inclusion patterns, represented retained introns (Fisher’s exact test, P<0.0001; Figure 2C). The prevalence of retained introns in this splicing pattern was not caused by mis-mapping artifacts, as is shown by re-analysis of DS1 and DS2 based on data mapped with no mismatches (Supplementary Figures S3 and S4). Consequently, a significantly greater fraction of the ‘down–up’ segments could be associated with nonsense-mediated decay (NMD) (Fisher’s exact test, P<0.001; Figure 2D). Second, the ‘down–up’ segments were significantly less conserved on the DNA sequence level, compared with other age-related segments (Wilcoxon test, P<0.0001; Figure 2E). This lack of sequence conservation was still present when only retained introns were considered (Wilcoxon test, P<0.0001). Taken together, these features indicate that a fraction of gene segments, preferentially incorporated in early development and late aging, might target these transcripts for NMD.
To test this notion, we assessed the expression of genes involved in the NMD pathway. Of the 21 genes annotated in this pathway (Chang et al, 2007), 15 were expressed in the brain (Colantuoni et al, 2011; Liu et al, 2012) and 6 (UPF2, UPF3b, SMG1, SMG5, SMG6 and CASC3) showed significant age-related expression changes (F-test P<0.01; permutation-based FDR<0.05). Of these six genes, five showed decreased expression in development and increased expression in aging; a profile closely resembling the profile of NMD-associated segments (Figure 2F–K). Finding so strong correlation is unexpected (Fisher’s exact test, P<0.02) and supports the role of the ‘down–up’ segments in triggering NMD.
Age-related splicing changes at rhesus macaque homological segments
To determine the reproducibility of the age-related splicing patterns identified in the human brain, we measured RNA splicing patterns in the PFC of 15 rhesus macaques with ages distributed between 1 day and 28 years, using RNA-seq (Supplementary Table S7). Among the 1484 segments significant in the two human data sets, 496 could be mapped unambiguously to the rhesus macaque genome and had sufficient sequence read coverage. When comparing human and macaque age-related splicing patterns based on the inclusion ratio trajectories of these 496 segments, we observed significantly greater positive correlations than expected by chance (permutation test, P<0.005). Further, if we consider development and aging separately, splicing changes of 321 (of the 496) segments that were significant in development, and 131 segments significant in aging, were consistent between the human and macaque data sets for each of these ontogenetic intervals (Fisher’s exact test, P<0.0001 for development and P<0.005 for aging).
We further tested the consistency of the four main splicing patterns observed in the human PFC, between the human and the macaque data sets. All four main splicing patterns showed significantly greater positive correlations between the human and macaque trajectories of splicing changes with age, independent of the segment selection criteria (mean r between 0.4 and 0.6; permutation test, P<0.005) (Supplementary Figure S9).
Age-related splicing patterns
Gene expression changes found in the human brain over the course of lifespan show discrete patterns (Somel et al, 2010). The age-related splicing changes identified here did not correlate with gene expression changes (Supplementary Figure S10), but displayed discrete splicing patterns. Non-supervised hierarchical clustering of age-related splicing changes in DS1 and DS2 revealed eight main clusters containing 1422 out of 1484 segments (hereafter referred as CL1–8) (Figure 3A, Supplementary Figure S11). Splicing patterns in all eight clusters showed good agreement between the two data sets (Figure 3A).
Figure 3.

Patterns and regulation of age-related splicing changes. (A) Age-related splicing patterns in the PFC (orange) and CBC (gray). The horizontal axis shows the samples’ ages in years. The vertical axis shows the inclusion ratio normalized to mean=0 and s.d.=1 for each segment in each brain region. The points represent the mean inclusion ratios of segments within each pattern in each sample. The lines and the error bars show the cubic spline curves of the mean inclusion ratios and the s.d. of the curves, respectively. The colors represent RNA-seq data sets analyzed: yellow—DS1/PFC, gray—DS1/CBC, and red—DS2/PFC. The numbers above the panels show the ID and the numbers of segments and genes in each pattern. The percentages shown within the panels indicate the percentage of splicing variation found in development and in aging. The vertical dotted line indicates separation between developmental and aging intervals. Numbers of segments separately significant in development or in aging are shown at the bottom of the panels. (B) Top: numbers of SFs with significant enrichment of SFBM in a given pattern (Fisher’s exact test, P<0.05) (gray bars). The striped bars represent the median numbers of SFs expected by chance, calculated by randomly assigning the same numbers of age-related segments to each pattern 1000 times. The symbols above each bar indicate the significance levels of the permutation-based FDR (***—FDR<0.05, **—FDR<0.10, *—FDR<0.20). Bottom: numbers of SFs with significant SFBM enrichment and positive (red) or negative (blue) correlation between the SF expression profile and the inclusion ratio profiles of the segments containing these SFBM within a pattern, compared with the segments from other patterns (one-sided Wilcoxon test, P<0.05). The striped bars represent the median numbers of correlated SFs expected by chance, calculated by randomly assigning the same numbers of age-related segments to each pattern 1000 times. (C–H) Gene expression of SFs, PTBP1, PTBP2, hnRNPA1, hnRNPF, hnRNPH1, and hnRNPH3, in the human PFC. Gene expression was estimated based on published data from (Liu et al, 2012) (green) and (Colantuoni et al, 2011) (violet). Expression intensities for each gene were normalized to mean=0 and s.d.=1. Age scale is log2-transformed, ages are shown in years, MAC stands for ‘months after conception’.
Functional analysis of genes falling into the eight clusters, using GO biological process taxonomy (Ashburner et al, 2000) and Kyoto Encylopedia of Genes and Genomes (KEGG) pathways (Kanehisa et al, 2010), revealed significant functional enrichment in three clusters: CL6, CL7, and CL8. Notably, both CL6 and CL8 were enriched in genes associated with neural functions, including GO terms ‘synaptogenesis’, ‘behavior’ and ‘learning’ in CL6 and ‘regulation of synaptic transmission’ and ‘regulation of neurogenesis’ in CL8 (Supplementary Table S8).
To test whether the identified age-related splicing changes within each cluster could be regulated by SFs, we searched for enrichment in 36 candidate SF-binding motifs (SFBMs) within the 300 bp regions up- and downstream of segment boundaries. We found that five out of eight clusters (CL1, CL3, CL6, CL7, and CL8) demonstrated greater enrichment of SFBMs than expected by chance (permutation-based FDR<0.2) (Figure 3B). Furthermore, in all five clusters, the expression profiles of the corresponding SFs showed significantly better correlation with the splicing profiles of their target segments than expected by chance (permutation-based FDR<0.2) (Figure 3B). Thus, the observed age-related splicing changes could in part be explained by expression changes of the corresponding SFs. It should be noted that, due to the low reliability of computational SFBM identification, our results are likely to underestimate the role of SFs in the regulation of age-related splicing changes (Barash et al, 2010).
Even with the limited annotation available, several notable associations between SFs and age-related splicing patterns could be observed. In the biggest of all clusters, CL1, corresponding to the ‘down–up’ splicing pattern, the expression level of 8 of the 10 SFs with binding site enrichment in this cluster showed a positive correlation with the inclusion ratio pattern in this cluster, with the remaining 2 SFs showing negative correlation (Figure 3B). Finding such numbers of positively and negatively correlated SFs is unexpected (Fisher’s exact test, P<0.001) and indicates a potential regulatory relationship between the expression of these SFs and the predominant ‘down–up’ splicing pattern we observe in the human brain over the course of lifespan. Among SFs that showed significant SFBM enrichment and positive correlation with the inclusion ratio profiles of CL1 segments are the polypyrimidine tract-binding proteins PTBP1 and PTBP2, as well as other heterogeneous nuclear ribonucleoproteins, including hnRNPA1, hnRNPH1, hnRNPH3 and hnRNPHF (Figure 3C–H, Supplementary Table S9). Notably, PTBP2 expression is specific to the brain (Andres et al, 1999) and both PTBP1 and PTBP2 were shown to function in neuronal development (Grabowski, 2011) and glioma proliferation (Cheung et al, 2009). Further, formation of a protein complex containing PTBP1, PTBP2 and HNRNPH1, as well as cooperative RNA binding by PTBP2 and hnRNPH1 has been demonstrated (Markovtsov et al, 2000).
Gene segments in CL6, a cluster associated with the GO terms ‘learning’, ‘behavior’, and ‘synaptogenesis’, showed significant SFBM enrichment, but negative correlation with the expression profiles of hnRNPH1, hnRNPH3, and hnRNPHF. Similarly, gene segments in CL8 associated with the regulation of neurogenesis and synaptic transmission showed significant SFBM enrichment, but negative correlation with the expression of PTBP1 and PTBP2. Previous research has shown that the location of PTBP1/PTBP2-binding sites upstream or downstream of the spliced segment may result in preferential inhibition or activation of the segment inclusion (Llorian et al, 2010). Negative association between the PTBP1/PTBP2 expression and CL8 splicing pattern could be further confirmed in our study using exons experimentally annotated to be repressed or activated by the PTBP1/PTBP2 in HeLa cells (Llorian et al, 2010). The experimentally annotated exons were significantly over-represented in CL8 (one-sided Fisher’s exact test, P=0.002, odds ratio=6.8). Notably, in agreement with the negative association between the PTBP1/PTBP2 expression and the CL8 splicing pattern, all experimentally annotated exons present in CL8 were repressed by PTBP1/PTBP2 in the cell line (one-sided Fisher’s exact test, P=0.001, odds ratio=7.8).
Age-related splicing differences between cortex and cerebellum
For the vast majority of gene segments, age-related splicing changes in one of the brain regions correlated positively and significantly with age-related splicing changes in the other brain region studied (Figure 4A). Still, of the 3132 segments showing age-related splicing changes in DS1, 15% differed significantly in their age-related splicing patterns between the PFC and CBC (q<0.05). Differences in age-related splicing patterns between the two brain regions identified by RNA-seq were further validated using Affymetrix Exon Arrays (Supplementary Figure S12). Some of these differences affect genes associated with neural functions, such as amyloid beta (A4) precursor protein (APP), which is involved in Alzheimer’s disease (Supplementary Figure S13), bridging integrator 1 (BIN1), which is involved in synaptic vesicle endocytosis (Supplementary Figure S14), or protocadherin gamma (PCDHG), shown to be associated with cell adhesion and synapse formation (Frank et al, 2005; Garrett and Weiner, 2009). Notably, all three major isoform exons of PCDHG have distinctively different age-related splicing patterns in the PFC and CBC (Figure 4B and C). Further, splicing changes with age of PCDHG transcript in the human PFC resembled splicing changes reported in the developing mouse brain (Frank et al, 2005), indicating their evolutionary conservation.
Figure 4.

Age-related splicing changes in PFC and CBC. (A) Correlation of age-related splicing changes between the PFC and CBC in DS1. Shown are distributions of the Pearson correlation coefficients for 24 458 segments with sufficient coverage in both data sets but not significant in DS1 (gray), and for 3132 segments showing significant age-related splicing changes in DS1 (red). (B) Age-related splicing changes in the protocadherin gamma transcript in the human PFC and CBC. RNA-seq coverage in all 12 DS1 samples is shown. (C) Inclusion ratio profiles of the protocadherin gamma C3-C5 exons. The colors represent the RNA-seq data sets analyzed: yellow—DS1/PFC, gray—DS1/CBC, and red—DS2/PFC. The points represent the inclusion ratios and the lines represent the cubic spline curves fitted with three degrees of freedom.
Discussion
We have shown that close to 40% of genes expressed in the human brain experience significant splicing changes over postnatal lifespan. As our study is limited to two brain regions and is based on a relatively small number of individuals, it is likely to underestimate the total extent of age-related splicing changes in the human brain. Although functional importance of these changes is not addressed in our study, ∼30% of identified age-related splicing changes affect the protein-coding portion of the transcript. Although the power of our assay was limited due to the relatively low coverage of the protein mass-spectrometry data, for proteins with sufficient peptide coverage, these age-related splicing changes could still be directly observed at the protein sequence level. Notably, many of the genes with splicing changes confirmed at the protein level were previously implicated in neural functions and dysfunctions. These include: MAPT associated with several neurodegenerative disorders including Alzheimer's disease, frontotemporal dementia, and cortico-basal degeneration (Wang and Liu, 2008); NDRG2 implicated in Alzheimer's disease (Mitchelmore et al, 2004) and potentially involved in the stimulation of neurite outgrowth (Takahashi et al, 2005); NPTN, an adhesion protein involved in the long-term potentiation pathway (Smalla et al, 2000; Bernstein et al, 2007); PALM involved in axonal and dendritic outgrowth, dendritic spine maturation and synapse formation (Kutzleb et al, 1998); and PTPRZ1 potentially involved in the regulation of CNS development (Levy et al, 1993).
Close to 70% of age-related splicing changes take place during development (0–14 years) with the remaining 30% occurring in aging (25–98 years). This is similar to results previously obtained at the gene expression level (Somel et al, 2010). In contrast to gene expression changes, which show approximately equal proportions of increased and decreased expression in both development and aging, age-related splicing changes show strong directional asymmetry: 75% of segments showed decreased inclusion during development, while 77% of segments showed increasing inclusion in aging. As the result, the majority of age-related splicing changes detected in the human brain represent a common pattern reflecting preferential inclusion of certain gene segments in infancy and at a very old age. A larger than average proportion of these inclusion events represent intron retention, and are expected to target the transcripts for NMD. It is therefore notable that five of the six NMD pathway members, that show age-related gene expression changes in the human brain, also show elevated expression in infants and old adults. Splicing pattern similarity between infants and elderly is unexpected. Molecular changes occurring at old age are thought to have deleterious effects, as such changes do not impact on the individual’s reproductive success, and could accumulate rapidly within a population (Brian, 1946, 1952). By contrast, any deleterious changes taking place in early postnatal development would have a strong negative effect on the individual’s fitness, and will be eliminated quickly from a population. Therefore, beneficial and deleterious effects of the NMD increase in the infant and aging human brain remain to be investigated.
Mechanistically, similarity of splicing patterns at young and old age could be caused by consistent changes in the expression of the corresponding SFs. Analysis of major splicing patterns indicates that gene segments showing inclusion decrease in development and increase in aging (CL1) are significantly enriched in the candidate-binding sites of 10 SFs: PTBP1, PTBP2, hnRNPA1, hnRNPH1, hnRNPH2, hnRNPH3, hnRNPHF, Tra2β, SC35, and SRp40. Notably, for all 10 SFs, age-related expression changes correlated significantly with the CL1-splicing profile: positively for 8 and negatively for 2 SFs (Supplementary Table S9), while at most 1 would be expected to correlate by chance. Similar enrichment of SFBMs, and correlation between the expression profiles of SFs and their target segments, could be observed in a total of five out of eight splicing clusters. Overall, more than 50% of age-related splicing changes could be linked to expression changes of the corresponding SFs. Given the rather low reliability of the SFBM identification (Barash et al, 2010), the role of SFs in the regulation of age-related splicing changes is likely to be even greater.
The age-related splicing changes detected using RNA-seq measurements was verified by cross-validation of two independent human and one rhesus macaque RNA-seq data sets, reanalysis of the entire DS1 using Exon Arrays, testing selected splicing changes using PCR, and detecting splicing changes at the protein level. Taken together, the results we obtained using these diverse techniques provide strong support for the robustness and authenticity of our results. Verification of age-related splicing changes in the rhesus macaque data set is particularly notable as, in contrast to humans, all rhesus macaque samples were obtained from the same primate facility where animals were raised in the same environment. Thus, this additional data set addresses all possible confounding variables caused by sample heterogeneity in the human data. Furthermore, close correlation between the human and the macaque patterns of splicing changes with age (Supplementary Figure S9) indicates that these changes are evolutionarily conserved, thus implying potential functionality. With respect to possible pitfalls in data analysis, removal of potentially mis-mapped reads using more stringent mapping criteria did not influence our results either for all the age-related splicing changes (Supplementary Figure S2) or, specifically, for age-related changes in intron retention (Supplementary Figures S3 and S4).
Among the remaining confounding variables, we considered the influence of sex, ethnicity, tissue composition, and RNA quality. This study was not designed to estimate sex-related splicing differences in the human brain. Accordingly, DS1 samples contained both male and female individuals in every sample pool. By contrast, DS2 contained 11 male and 2 female samples (age: 19 days and 2 years) (Supplementary Table S2). There was no detectable effect of these two samples on the statistical analysis, and age-related splicing changes showed high positive correlation between DS1 and DS2. Further, multidimensional scaling analysis based on splicing variation in DS1 and DS2 indicated that splicing variation in both data sets was strongly influenced by age, whereas both sex and ethnicity had no detectable effect (Supplementary Figure S15). To further test the influence of the samples’ ethnicity on age-related splicing changes, we estimated the rates of age-related splicing changes in five African-American samples and five Caucasian samples with similar age distributions (see Methods). Despite the small number of samples tested, our results demonstrated a good agreement for splicing changes with age between these two ethnicities (r=0.75, P<0.0001) (Supplementary Figure S16). Thus, splicing differences between the sexes and/or ethnicities in the human brain could not be assessed in our data, and did not have a detectable effect on the identified age-related splicing changes.
Tissue composition in general, as well as tissue composition changes with age, differs greatly between PFC and cerebellum. By contrast, the vast majority of age-related splicing changes are shared between these brain regions (Figure 4A). Furthermore, there was no enrichment of genes preferentially expressed in specific brain cell types (neurons, astrocytes, oligodendrocytes, and oligodendrocyte precursors) (Cahoy et al, 2008) or in the gray or white matter (Erraji-Benchekroun et al, 2005) among any of the eight main splicing patterns (Supplementary Table S10). Thus, the observed age-related splicing changes are unlikely to be caused by changes in tissue composition.
RNA degradation would result in shortening of the captured transcripts and, consequently, greater 3′-coverage bias. We did observe some 3′-coverage bias in both data sets (Supplementary Figure S17). The differences in the coverage bias among samples, however, did not show any significant association with age.
Finally, our experimental design allowed us to effectively remove false positives caused by biological variability; almost all our analysis is based on segments that are significant in two independent data sets. In addition, we used CBC samples from DS1 to focus on age-related splicing changes that were consistent between the two brain regions.
Taken together, these results strongly suggest that the identified age-related splicing changes are real. Although splicing changes in individual genes have been previously shown to have important roles in the brain development (Dredge et al, 2001; Ule et al, 2005; Blencowe, 2006), our study demonstrated that thousands of yet uncharacterized splicing changes take place in the PFC and cerebellum during human brain development, maturation, and aging. Investigating the implications of these changes on the establishment, and deterioration, of human cognitive functions over the course of lifespan will be a formidable, but necessary, task.
Materials and methods
Sample collection
PFC and CBC samples were dissected from postmortem brains of 30 (DS1) and 13 (DS2) humans (aged 0–98 years) (Supplementary Tables S1 and S2). Human samples were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, USA, the Netherlands Brain Bank, Amsterdam, the Netherlands. Informed consent for the use of human tissues for research was obtained in writing from all donors or from their next of kin. All subjects were defined as normal by forensic pathologists at the corresponding brain bank. All subjects suffered sudden death with no prolonged agonal state. Rhesus macaque samples were obtained from the Suzhou Experimental Animal Center, China. Macaques used in this study suffered sudden deaths for reasons other than their participation in this study and without any relation to the tissue used. CBC dissections were made from the lateral CBC. PFC dissections were made from the frontal part of the superior frontal gyrus. All samples contained an ∼2:1 gray matter to white matter volume ratio.
RNA sequencing
For DS1, samples were prepared by pooling equal quantities of total RNA from five individuals of similar age (Supplementary Table S1; Figure 1A). In all cases, PFC and CBC samples were dissected for the same individuals. All samples had a comparable, and good, RNA quality (Supplementary Table S1). RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA). Oligo(dT) selection was performed twice using Oligotex mRNA Midi Kit (Qiagen). After selection, 100 ng mRNA was first fragmented by addition of 5 × fragmentation buffer (200 mM Tris acetate, pH 8.2, 500 mM potassium acetate, and 150 mM magnesium acetate) with heating at 94 °C for 2 min 30 s in a thermocycler. The fragments were then transferred to ice and run over a Sephadex-G50 column (USA Scientific) to remove the fragmentation ions. We used random hexamer primers (Invitrogen, cat. no. 48190-011) for reverse transcription of fragmented mRNA to double-strand cDNA. Sequencing libraries were prepared according to the paired-end non-strand-specific sample preparation protocol ( http://www.illumina.com). Each sample was sequenced in a separate lane in the Illumina Genome Analyzer II system, using the 75-bp paired-end sequencing protocol.
RNA libraries for the human DS2 and for the rhesus macaque data set were constructed according to the Illumina strand-specific RNA-seq library preparation procedure. Briefly, 100 ng fragmented mRNA was first treated with phosphatase, and then consecutively ligated to v1.5 small RNA 3′ and 5′ adapters. After reverse transcription and 15 cycles of amplification, the library was sequenced using Illumina small RNA sequence primers to the length of 100 bp. All sequence data are available at the Sequence Reads Archive, and the study accession is SRP005169.
Defining gene segments
Segments were defined as intervals between nearest segmentation sites, that is, splice sites and transcriptional and translation start/stop sites based on the Ensembl (Flicek et al, 2010) annotation (v57). Segments were further classified according to their location in (a) constitutive exons (segments annotated as exons in all isoforms), (b) constitutive introns (segments annotated as introns in all isoforms), and (c) alternative regions (segments annotated as exons and introns in different isoforms). In total, 366 163 segments were classified as constitutive exonic, 246 080 as constitutive intronic, and 157 177 as alternative.
For the subsequent analysis, we only retained segments from multi-exon, nuclear, and protein-coding genes with a total length of constitutive coding sequence of at least 100 nt (19 383 genes). Only segments overlapping with a gene region annotated as a protein-coding region (CDS) in at least one isoform were retained. We further excluded all segments classified as constitutive intronic. Thus, un-annotated intron retention events could not be detected in our analysis. Instead, alternative segments that had exactly the same coordinates as introns (annotated intron retention events) were considered as retained introns. Only segments with at least 10 inclusion and 10 exclusion reads in each data sets were used in the analysis. Segments with stop codons more than 50 nt upstream of the next splice site in all usable reading frames were considered as NMD segments. Alternative segments surrounded by two constitutive or retained introns were considered to be skipped exons. The remaining alternative segments were classified as complex alternatives.
Construction of the artificial splice junction library
We generated an artificial splice junction library by connecting the two neighboring exons in all annotated isoforms (known junction sequences) and the two non-neighboring exons (novel junction sequences) using the Ensemble annotation (v57). Considering the read to be 75- and 100-nt-long, short junction sequences (<75 nt) were merged to adjacent exons until a length of 75 nt was reached. The remaining short junction sequences that could not be merged were filtered out. In total, the constructed junction library contained 302 179 known and 2 267 677 novel splice junction sequences.
Mapping
Read pairs containing low-quality nucleotides, as defined by the Illumina sequencing procedure, were filtered out. The remaining reads were mapped to the human reference genome (hg19) and the artificial splice junction library using the Bowtie software (Langmead et al, 2009) with the following parameters: ‘−k 20–best –strata’ and allowing a maximum of three mismatches. We required a minimum of a 6-nt overhang for reads that mapped to exon junction sequences.
In DS1, if only one read from a pair could be mapped, the other read from this pair was truncated at 3′-end to the length of 38 nt (one half of the sequence) and mapped again using the same parameters. If the truncated read still could not be mapped, the read pair was discarded. Upon completion of the mapping procedure, all possible combinations of read pair locations were constructed for each pair. Next, we only retained the locations with paired reads that were on different strands of the same chromosome, with a distance between reads not exceeding 1 Mb. Read pairs that had only one valid location were considered as uniquely mapped. Only uniquely mapped reads were used for further analysis.
In DS2, only reads that could be uniquely mapped to the human genome or splice junctions, using the above procedure, were retained.
Novel splice junctions
All exon combinations not included in isoforms listed in the Ensembl annotation (v57), but supported by at least two (DS1) or are least four (DS2) independent reads (reads with the genome coordinates overlapping by less than 100%), that mapped over the splice junction with at least a 6-nt overhang, were classified as novel junctions.
Inclusion ratio
To estimate the splicing extent for each segment, we calculated the segment inclusion ratio as:
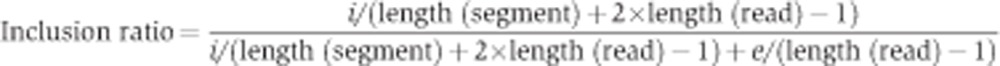 |
where i is the number of inclusion reads (i.e., reads that overlap with the segment), e is the number of exclusion reads (i.e., reads that mapped to the junction that spans the segment), length (segment) is the segment length in nucleotides and length (read) is the read length in nucleotides. Here, the numerator is proportional to the number of mRNA molecules that contained the segment and the denominator is proportional to the number of mRNA molecules that did not contain the segment. For DS2, only reads that mapped to the sense strand were used in the coverage calculation. For exon arrays, the inclusion ratio was calculated as the segment intensity divided by the gene intensity. For protein data, the inclusion ratio was calculated as peptide counts within a segment divided by peptide counts within a gene. For all samples, only segments with i+e≥5 were considered to have sufficient read coverage.
Statistical analysis of splicing based on RNA-seq segment coverage
Let {ij} and {ej} represent the sets containing the total numbers of inclusion and exclusion reads for sample j, respectively (in DS2 only reads that mapped to the sense strand were used). It is possible to consider {ij} and {ej} as results of a binomial trial, where {ij} represents the number of successes and {ej}, the number of failures.
To estimate age-related inclusion ratio changes and differences between the two brain regions in DS1 and age-related inclusion ratio changes in DS2, the following models were fitted using the Generalized Linear Models function in R, with a logit link function:
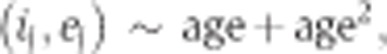 |
where brain−region is categorical variable and age is continuous variable equal to power of 0.25 of donor age.
Significance of each term was estimated by analysis of deviance (log-likelihood test) followed by the Benjamini–Hochberg correction (Benjamini and Hochberg, 1995), with q<0.05. Segments with significant age terms, either linear or quadratic, were considered to be age-related.
The method to analyze splicing described here is converted into a stand-alone java-application and R package and freely available at: http://www.bioinf.fbb.msu.ru/∼mazin.
Additional analysis was performed to find segments where splicing changes significantly during development (before 25 years) and in aging (after 25 years) in DS2. For each period, five samples were chosen to achieve equal power: samples with Brain Bank IDs Maryl_779, Maryl_759, Maryl_1281, 1453 and 1908 were used to test splicing changes in development and samples with Brain Bank IDs 605, 1496, S06/117, S01/118, S00/047 were used to test splicing in aging (Supplementary Table S2). For each ontogenetic period, linear binomial regression to age0.25 was applied, followed by the Benjamini–Hochberg correction. Segments with q<0.05 were considered significant.
To estimate the effect of ethnicity on age-related splicing changes, five African-American samples (Brain Bank IDs: Maryl_779, Maryl_1281, 1453, 1275, 605) and five Caucasian samples (Brain Bank IDs: Maryl_1157, Maryl_759, Maryl_1055, 1908 and 1496) with similar age distributions were selected from DS2. For each segment, a linear fit between inclusion ratio and age0.25 was performed using lm function in R for each ethnicity separately. Calculated linear model slopes were used to assess the agreement between ethnicities.
Inclusion ratio correlation between DS1 and DS2
For each segment, a cubic spline curve was fitted to the inclusion ratios obtained in DS2, with three degrees of freedom. Based on this curve, the inclusion ratio values at points matching ages of samples from DS1 were interpolated. The inclusion ratios obtained from DS1 (PFC) and interpolated in DS2 were used to calculate the Pearson correlation (Figure 1C).
Microarray processing and analysis
RNA isolation, hybridization to microarrays, and data preprocessing were performed as described previously (Somel et al, 2010). Briefly, total RNA was extracted from the same brain samples using Trizol reagent (Invitrogen) and purified with the QIAGEN RNeasy MiniElute kit. RNA quality was assessed with the Agilent 2100 Bioanalyzer system (Supplementary Table S1). For each sample, 2 mg of isolated pooled RNA was used for sample preparation, followed by hybridization to Affymetrix Human Exon 1.0 ST arrays, according to the standard protocol with no modifications ( http://www.affymetrix.com). Before hybridization data analysis, all microarray probes were aligned to the human reference genome (hg19) using the Bowtie software (Langmead et al, 2009). Only probes uniquely matching the human genome perfectly were included in the analysis. Probe intensities were exacted, background corrected, and quantile normalized using the R package ‘affy’ (Gautier et al, 2004). Probes with intensity<100 were excluded from analyses.
To calculate the expression level of gene segments, array probes were mapped to segments. If more than half of the probe length was located within a segment, then the probe was assigned to this segment. In cases where a probe could be mapped to multiple segments, we gave priority to exonic segments. The expression levels of segments were summarized by the median polish function in the R package ‘affy’. The expression level of a gene was calculated as an average intensity of segments located within the coding region of this gene. All microarray data is deposited in the GEO database (accession GSE26586).
To test the significance of age-related splicing changes based on array data, linear regression followed by ANOVA was applied. The following model was used: inclusion ratio∼tissue+age+tissue:age, where tissue denotes either PFC or CBC and age denotes donor age measured at the quadratic root power scale. Because of the low number of samples, no segments were significant at FDR<5%. As array data was not used to identify age-related splicing changes, but only to verify the ones identified using RNA-seq data, we used a relaxed ANOVA significance cutoff (P?0.1) to filter out segments with low signal-to-noise ratio.
To validate the differences in age-related splicing changes between PFC and CBC measured using RNA-seq and Affymetrix Exon Array (Supplementary Figure S11), Pearson correlation coefficients have been calculated between PFC/CBC inclusion ratio differences measured by RNA-seq and Exon Arrays (I(PFC,RNA-seq)−I(CBC,RNA-seq) and I(PFC,Exon Array)−I(CBC,Exon Array)) for each of 601 segments showing significant age-related splicing changes in DS1 and showing trend of change in the Exon Array data ANOVA analysis for the ‘age’ and/or ‘age:tissue’ terms as described above. The I(Tissue,Platform) term denotes the inclusion ratio value of a given tissue and a given platform.
Validation of splicing changes by PCR
First-strand cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen), using the same pooled PFC RNA samples that were used for the RNA-seq DS1 as templates. To check the inclusion ratio change of each selected segment, a primer pair was designed at the two flanks of the segment, and two or three appropriate DS1 samples were used. PCR reactions were performed at 94 °C for 2 min followed by 30 cycles of 95 °C for 20 s, 72 °C for 30 s, and 72 °C for 5 min with rTaq DNA polymerase (TAKARA). Then 5 μl of each PCR product were used for electrophoresis in a 2% (w/v) agarose gel. The size of the fragments was estimated using visual estimation based on DL 2000 DNA marker (TAKARA). After ethidium bromide (EB) staining, 24 out of the total of 30 segments tested by PCR showed two bands with expected product lengths. For the remaining 6 splicing events tested, we did not observe two PCR bands of expected size. These PCR products could not, therefore, be used to calculate changes in inclusion ratio with age. Some of these failed PCRs may indicate errors in our splicing variant predictions, whereas others might result from technical failure. The primer sequences of the 24 valid PCR are listed in Supplementary Table S12. The PCR band intensity quantification was carried out using the ‘Trace Qty’ method in the Quantity One program. In brief, after auto-scaling the image, correcting the background, and detecting the band manually, band intensity was assigned by the ‘Trace Qty’ method. Then, the inclusion ratio was calculated based on the PCR band intensity as Ql/(Ql+Qs), where Ql and Qs are the intensity values of long and short products, respectively. The inclusion ratio changes used to check the correlation between RT–PCR and RNA-seq (Figure 1D) were defined as the RT–PCR or RNA-seq inclusion ratio differences between the two samples tested in RT–PCR. For the segments where three or more samples were tested for RT–PCR, only the two samples with maximum and minimum RNA-seq inclusion ratio were used.
Protein data processing and analysis
In total, 1 301 042 peptide mass spectrum scans from human PFC samples taken from 12 healthy individuals of different ages (0–98 years) were obtained from Somel et al (2010). The peptides were identified by searching against a custom peptide library. This library was created by constructing unique peptides separated by trypsin-sensitive recognition sites among 1484 segments, from 721 genes, with significant age-related splicing change both in DS1 and DS2. The peptides with length <6 amino acids were ignored. The resulting custom library contained 18 029 peptides based on these 341 genes. The decoy library, used to estimate the FDR, was created by randomly shuffling amino acids within each peptide sequence of the custom peptide library but retaining the terminal lysine and arginine. Using the InsPecT (Tsur et al, 2005) peptide identification algorithm, followed by the MS-GF program (Kim et al, 2009), 144 645 peptide counts from 18 014 unique peptides were identified at FDR?0.05.
To ensure sufficient peptide coverage, we required a minimum of 8 peptide counts per protein in each of the 12 samples. In addition, we required that at least one segment showing inclusion ratio changes with respect to age, and at least one constitutive segment, were covered by at least 3 peptide counts each in at least 2 out of 12 samples. As the peptide coverage along the genes is extremely fragmentary, only 24 segments from 19 genes, supported by 5903 peptide counts, corresponding to 168 unique peptides, satisfied these criteria.
To calculate the Pearson correlation between the age-related inclusion ratio changes detected at the protein and RNA levels, the first problem is that ages are not consistent between the RNA-Seq and protein data. Therefore, for DS1, we used the inclusion ratios calculated for six PFC DS1 samples based on the RNA-seq data, and interpolated the inclusion ratios calculated according to the peptide coverage. The peptide inclusion ratios were interpolated at six age points, corresponding to the average ages of six pooled samples, by fitting a quadratic regression curve to the inclusion ratio in the 12 differently aged individual samples. For the integrated data set of DS1 and DS2, the RNA-seq inclusion ratios of total 19 ages were fitted to a quadratic regression curve, then interpolated at 12 age points corresponding to the protein samples. For DS2, 10 samples were analyzed using both RNA-seq and the peptide mass spectrum. These 10 shared samples were used for the correlation calculation. The background distribution was obtained by 10 000 random permutations of segment labels for the 24 segments followed by the correlation test. For each data set, the permutation test P-value was calculated based on the distribution of the mean values of the correlation coefficients obtained in the permutations.
Splicing changes in development and aging
We used the inclusion ratio change index to illustrate the splicing change extent and the direction of the inclusion ratio change in development and aging for each age-related segment. First, we classified 6 ages of DS1 and 13 ages of DS2 as ‘newborn’, ‘young’, and ‘old’. The pools 3 and 4 of DS1 (average age: 18 and 25 years), as well as the samples 8 and 9 from DS2 (ages: 13 and 25 years) were assigned to the ‘young’ category. In DS1, each of the remaining two newborn samples i and two old samples j can be used to calculate  and
and  , where
, where 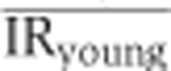 is the mean of inclusion ratios of the two ‘young’ samples. Setting
is the mean of inclusion ratios of the two ‘young’ samples. Setting 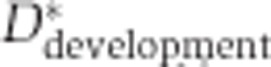 to the maximum of
to the maximum of 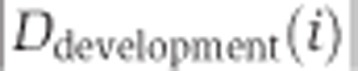 and
and 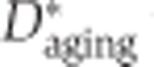 to the maximum of
to the maximum of 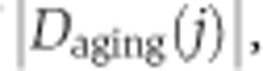 , the inclusion ratio change index of development (
, the inclusion ratio change index of development ( ) and aging (
) and aging (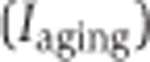 ) can be calculated as
) can be calculated as 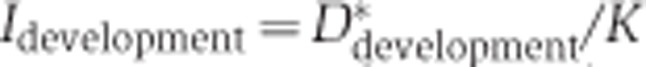 and
and 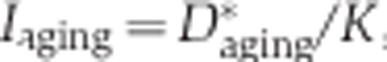 , respectively, where
, respectively, where  . For DS2, the values of seven
. For DS2, the values of seven  and four
and four  terms were calculated, and
terms were calculated, and  and
and 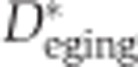 were selected as the second maximum absolute value term. This was done to minimize the influence of outlier inclusion ratio values. The inclusion ratio change indices,
were selected as the second maximum absolute value term. This was done to minimize the influence of outlier inclusion ratio values. The inclusion ratio change indices,  and
and 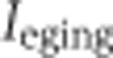 , were calculated as
, were calculated as  , where f(·) denotes the second maximum value. Of the 1484 age-related segments identified based on the whole-lifespan analysis of splicing variation in DS1 and DS2, 853 segments showed splicing changes in both development and aging and had consistent inclusion ratio change direction in the two data sets, i.e.,
, where f(·) denotes the second maximum value. Of the 1484 age-related segments identified based on the whole-lifespan analysis of splicing variation in DS1 and DS2, 853 segments showed splicing changes in both development and aging and had consistent inclusion ratio change direction in the two data sets, i.e.,  and
and 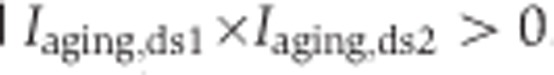 . Only these segments were used in the analyses of developmental and aging splicing changes.
. Only these segments were used in the analyses of developmental and aging splicing changes.
The fraction of splicing changes that take place in development was calculated as  . Among 1484 age-related segments significant in both data sets, the mean values for fraction of splicing changes taking place in development (± 1 s.d.) were 0.676±0.195, 0.640±0.185, and 0.749±0.161 for DS1/PFC, DS1/CBC, and DS2/PFC, respectively.
. Among 1484 age-related segments significant in both data sets, the mean values for fraction of splicing changes taking place in development (± 1 s.d.) were 0.676±0.195, 0.640±0.185, and 0.749±0.161 for DS1/PFC, DS1/CBC, and DS2/PFC, respectively.
‘Down–up’ expression pattern analysis
The list of 12 447 genes expressed in the human PFC, as well as 7071 genes showing significant age-related expression changes in the PFC, was obtained from (Liu et al, 2012). To identify genes following the ‘down–up’ expression pattern, we fit a quadratic curve to the expression data of each of 6690 genes. The age of the samples was counted starting from conception and log2-transformed. Overall, 2247 genes were classified as ‘down–up’ expressed genes.
Conservation analysis
PhastCons scores based on 10 primate species (Siepel et al, 2005) were used to estimate segment conservation. Conservation of a given segment was calculated as an average conservation of nucleotides within this segment.
Rhesus macaque splicing variation analysis
To assess splicing in the rhesus macaque, boundaries of all segments from 721 genes, containing 1484 of the age-related segments identified in the human time series, were mapped to the rhesus genome using the liftOver (Hinrichs et al, 2006) tool. Under the requirement that all the segments in a gene should be mapped to the rhesus macaque genome uniquely, 296 genes containing 538 age-related segments were mapped unambiguously. Next, sequence reads from each of the 15 rhesus samples (Supplementary Table S7) were aligned to the rhesus genome by Tophat, using the junction information of these 296 genes. Inclusion ratios in the rhesus macaque data were calculated as described above. Among the 538 age-related segments, 496 were covered by at least five sequence reads in all rhesus macaque samples, and by at least one read crossing the tested splicing junction in at least one rhesus macaque sample. These segments were used to confirm the splicing pattern found in humans. Further, using the same criteria as for human DS2, 290 of the 496 segments showed significant age-related splicing changes in the rhesus macaque time series data. For comparison between the human and the rhesus macaque time series, the age of rhesus macaque samples was adjusted to the human age based on the maximal lifespan duration difference between the species (120 years for humans and 40 years for rhesus macaques). To calculate the inter-species inclusion ratio correlations, the human inclusion ratios were translated to corresponding rhesus ages by quadratic interpolation under quartic root age axis.
Splicing patterns
To define patterns based on age-related splicing changes, we first constructed dendrograms of segments with significant age-related splicing changes identified in both data sets and with the defined inclusion ratio in all samples (N=1422). This was done by hierarchical clustering, using (1–r) as the distance measure, where r is the Pearson correlation between vectors containing the inclusion ratios of segments from samples in both brain regions and both data sets. We used the ‘complete’ method for hierarchical clustering and cut the tree at a height determined by a manual examination of pattern profiles and by observing pattern functional properties at different levels (h=1.5). This procedure yielded eight patterns with strong within-group correlations.
Functional enrichment analysis
The KEGG pathway annotation (Kanehisa et al, 2010) and the Gene Ontology (GO) annotation (Ashburner et al, 2000) for humans were downloaded from the KEGG ( http://www.genome.jp/kegg/) and Ensembl (version 56) databases, respectively. We used the GO ‘term’ and ‘graph path’ tables, downloaded from the GO database ( http://archive.geneontology.org/latest-termdb/), to associate each GO term with higher level terms. To identify over-represented KEGG pathways or GO terms, we used the Fisher’s exact test and adjusted P-values for multiple testing by the Benjamini–Hochberg correction. Only pathways/terms containing ?3 genes were tested.
Enrichment analysis of SFBMs
Sequences of 29 SFBMs were obtained from http://sfmap.technion.ac.il and another 7 SFBM were collected from previous studies (Faustino and Cooper, 2005; Galarneau and Richard, 2005; Sanford et al, 2009; Bolognani et al, 2010) (Supplementary Table S11). These 36 SFBMs corresponded to 25 SFs. To search for occurrence of these SFBM in the vicinity of splicing sites, the boundary regions for each segment were identified as 300 bp upstream to 300 bp downstream (Barash et al, 2010). Exact matches to each SFBM in each boundary region were searched by a custom Perl script. For each SFBM, we used the Fisher’s exact test to check whether there was an excess instances of this SFBM in a splicing-pattern cluster, compared with all 1422 segments with significant age-related splicing changes in both data sets. The enriched SFBMs were defined as those satisfying the threshold P<0.05.
We further estimated the numbers of significantly enriched SFBM expected by chance, by randomly sampling the same number of age-related segments that were contained within the tested splicing-pattern cluster, and repeating the enrichment test 1000 times. SFs showing significant enrichment of SFBM based on this permutation test were further tested for a correlation between their expression profiles and their target segments’ inclusion ratios. To obtain a better resolution of the temporal expression profiles of SFs, we used published Affymetrix gene array data collected in the PFC and CBC of 23 human individuals (Liu et al, 2012) with an age distribution matching that of DS1. To calculate the correlation between the segments’ inclusion ratio profiles and the SFs’ expression profiles across lifespan, we interpolated the segment inclusion ratio values and the SF expression values from the fitted spline curves at 15 equally distributed time points along the age range. The SF-target segment inclusion ratio correlations were compared with correlations between the same SF and its age-related target segments in the other patterns, using the one-sided Wilcoxon test. The test was conducted in the CBC and PFC separately, and the SFs with significant correlation in both regions were classified as correlated SFs. The expected-by-chance numbers of correlated SFs with enriched SFBMs were calculated by random sampling in the other patterns of the same number of age-related segments that were contained within the tested pattern, and repeating the one-sided Wilcoxon test 1000 times.
Identification of brain region-specific genes
To identify genes expressed differently between the two brain regions, we quantified gene expression levels using counts of reads mapped into constitutive and coding sections of a gene. These numbers and the total numbers of mapped reads were tested using the binomial regression performed as described above (Identification of splicing changes). Genes with a significant (q<0.01) term responsible for brain region differences were considered as brain-region specific. Of these, genes with greater mean expression in PFC than in CBC were classified as PFC-specific and the remaining genes, as CBC-specific.
Multidimensional scaling analysis
Multidimensional Scaling to two dimensions was performed by the isoMDS function from the MASS package in R. Inclusion ratios of each segment in each data set and each brain region were normalized to mean=0 and s.d.=1 before analysis. 1−Pearson correlation coefficient was used as a distance function.
Supplementary Material
Acknowledgments
We thank the NICHD Brain and Tissue Bank for Developmental Disorders, the Netherlands Brain Bank, and Dr HR Zielke in particular for providing the human samples; Suzhou Drug Safety Evaluation and Research Center and C Lian, H Cai and X Zheng in particular for providing the macaque samples; J Dent and J Boyd-Kirkup for editing the manuscript; F Xue for assistance; A Mironov, E Khrameeva, D Malko and all members of the Comparative Biology Group in Shanghai for helpful discussions and suggestions. The study was supported by Chinese Academy of Sciences (grant number KSCX2-EW-R-02-02, KSCX2-EW-J-15-02, and KSCX2-EW-J-15-03); Russian Academy of Sciences programs ‘Molecular and Cellular Biology’ and ‘Basic Science for Medicine’ (state contract 14.740.11.0003); Russian Foundation of Basic Research (grant 09-04-92745); National Natural Science Foundation of China, general program (grant number 31171232); National Natural Science Foundation of China, research fund for international young scientists (grant number 31110258); MS and PK were supported by National Natural Science Foundation of China research grants (grant no. 31010022 & 31050110128); MS was supported by a fellowship from the Chinese Academy of Sciences (2009Y2BS12); PM was partially supported by the ‘Science for Life Extension’ Foundation.
The array data are available at the GEO database, the accession id is GSE26586. All human and macaque RNA-seq data were uploaded to the Short Read Archive. SRA study accession is SRP005169.
Author Contributions: WC, MG, PK conceived and designed the experiments, ZY, XZ, NL, YH, NF, ZN, RZ, HY performed the experiments, PM, JX, XL, ML, LH, YY, YPC analyzed the data, and PM, JX, XL, MS, MG, PK wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G (1999) CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci USA 96: 9873–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y, Calarco JA, Gao WJ, Pan Q, Wang XC, Shai O, Blencowe BJ, Frey BJ (2010) Deciphering the splicing code. Nature 465: 53–59 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate—a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 57: 289–300 [Google Scholar]
- Bernstein HG, Smalla KH, Bogerts B, Gordon-Weeks PR, Beesley PW, Gundelfinger ED, Kreutz MR (2007) The immunolocalization of the synaptic glycoprotein neuroplastin differs substantially between the human and the rodent brain. Brain Res 1134: 107–112 [DOI] [PubMed] [Google Scholar]
- Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y (2010) Sex-specific and lineage-specific alternative splicing in primates. Genome Res 20: 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe BJ (2006) Alternative splicing: new insights from global analyses. Cell 126: 37–47 [DOI] [PubMed] [Google Scholar]
- Bolognani F, Contente-Cuomo T, Perrone-Bizzozero NI (2010) Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res 38: 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian MP (1946) Old age and natural death, Vol. 1: Modern Quarterly.
- Brian MP (1952) An Unsolved Problem Of Biology. . H. K. Lewis: : London,
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33: 95–130 [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28: 264–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco JA, Xing Y, Caceres M, Calarco JP, Xiao X, Pan Q, Lee C, Preuss TM, Blencowe BJ (2007) Global analysis of alternative splicing differences between humans and chimpanzees. Gene Dev 21: 2963–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF (2007) The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74 [DOI] [PubMed] [Google Scholar]
- Cheung HC, Hai T, Zhu W, Baggerly KA, Tsavachidis S, Krahe R, Cote GJ (2009) Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain 132: 2277–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, Kleinman JE (2011) Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Levy N (2006) Altered splicing in prelamin A-associated premature aging phenotypes. Prog Mol Subcell Biol 44: 199–232 [DOI] [PubMed] [Google Scholar]
- Dredge BK, Polydorides AD, Darnell RB (2001) The splice of life: alternative splicing and neurological disease. Nat Rev Neurosci 2: 43–50 [DOI] [PubMed] [Google Scholar]
- Erraji-Benchekroun L, Underwood MD, Arango V, Galfalvy H, Pavlidis P, Smyrniotopoulos P, Mann JJ, Sibille E (2005) Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiat 57: 549–558 [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA (2005) Identification of putative new splicing targets for ETR-3 using sequences identified by systematic evolution of ligands by exponential enrichment. Mol Cell Biol 25: 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kahari A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E et al. (2010) Ensembl 2011. Nucleic Acids Res 39: D800–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Ebert M, Shan WS, Phllips GR, Arndt K, Colman DR, Kemler R (2005) Differential expression of individual gamma-protocadherins during mouse brain development. Mol Cell Neurosci 29: 603–616 [DOI] [PubMed] [Google Scholar]
- Galarneau A, Richard S (2005) Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698 [DOI] [PubMed] [Google Scholar]
- Garrett AM, Weiner JA (2009) Control of CNS synapse development by gamma-protocadherin-mediated astrocyte-neuron contact. J Neurosci 29: 11723–11731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315 [DOI] [PubMed] [Google Scholar]
- Grabowski P (2011) Alternative splicing takes shape during neuronal development. Curr Opin Genet Dev 21: 388–394 [DOI] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, Hillman-Jackson J, Kuhn RM, Pedersen JS, Pohl A, Raney BJ, Rosenbloom KR, Siepel A, Smith KE, Sugnet CW, Sultan-Qurraie A et al. (2006) The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34: D590–D598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan ZY, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38: D355–D360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Gupta N, Bandeira N, Pevzner PA (2009) Spectral dictionaries: integrating de novo peptide sequencing with database search of tandem mass spectra. Mol Cell Proteomics 8: 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzleb C, Sanders G, Yamamoto R, Wang X, Lichte B, Petrasch-Parwez E, Kilimann MW (1998) Paralemmin, a prenyl-palmitoyl-anchored phosphoprotein abundant in neurons and implicated in plasma membrane dynamics and cell process formation. J Cell Biol 143: 795–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JB, Canoll PD, Silvennoinen O, Barnea G, Morse B, Honegger AM, Huang JT, Cannizzaro LA, Park SH, Druck T et al. (1993) The cloning of a receptor-type protein tyrosine phosphatase expressed in the central nervous system. J Biol Chem 268: 10573–10581 [PubMed] [Google Scholar]
- Liu X, Somel M, Tang L, Yan Z, Jiang X, Guo S, Yuan Y, He L, Oleksiak A, Zhang Y, Li N, Hu Y, Chen W, Qiu Z, Paabo S, Khaitovich P (2012) Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res 22: 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, de la Grange P, Ast G, Smith CW (2010) Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 17: 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T (1991) Mechanisms of alternative pre-messenger-RNA splicing. Science 251: 33–34 [DOI] [PubMed] [Google Scholar]
- Markovtsov V, Nikolic JM, Goldman JA, Turck CW, Chou MY, Black DL (2000) Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol 20: 7463–7479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelmore C, Buchmann-Moller S, Rask L, West MJ, Troncoso JC, Jensen NA (2004) NDRG2: a novel Alzheimer's disease associated protein. Neurobiol Dis 16: 48–58 [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey J, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Sanford JR, Wang X, Mort M, VanDuyn N, Cooper DN, Mooney SD, Edenberg HJ, Liu YL (2009) Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res 19: 381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou MM, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla KH, Matthies H, Langnase K, Shabir S, Bockers TM, Wyneken U, Staak S, Krug M, Beesley PW, Gundelfinger ED (2000) The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc Natl Acad Sci USA 97: 4327–4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Guo S, Fu N, Yan Z, Hu HY, Xu Y, Yuan Y, Ning Z, Hu Y, Menzel C, Hu H, Lachmann M, Zeng R, Chen W, Khaitovich P (2010) MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res 20: 1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamada M, Ohata H, Honda K (2005) Ndrg2 promotes neurite outgrowth of NGF-differentiated PC12 cells. Neurosci Lett 388: 157–162 [DOI] [PubMed] [Google Scholar]
- Takeda J, Suzuki Y, Sakate R, Sato Y, Gojobori T, Imanishi T, Sugano S (2010) H-DBAS: human-transcriptome database for alternative splicing: update 2010. Nucleic Acids Res 38: D86–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, Clark TA, Schweitzer AC, Rot G, Curk T, Zupan B, Rogelj B, Shaw CE, Ule J (2011) Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res 21: 1572–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA (2005) Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol 23: 1562–1567 [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB (2005) Nova regulates brain-specific splicing to shape the synapse. Nat Genet 37: 844–852 [DOI] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, Lucier JF, Thibault P, Rancourt C, Tremblay K, Prinos P, Chabot B, Abou Elela S (2009) Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16: 670–U698 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo SJ, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Liu F (2008) Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol 85: 148–175 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.