

Abstract
The physiological barriers of the brain impair drug delivery for treatment of many neurological disorders. One delivery approach that has not been investigated for their ability to penetrate the brain is RNA-based aptamers. These molecules can impart delivery to peripheral tissues and circulating immune cells, where they act as ligand mimics or can be modified to carry payloads. We developed a library of aptamers and an in vivo evolution protocol to determine whether specific aptamers could be identified that would home to the brain after injection into the peripheral vasculature. Unlike biopanning with recombinant bacteriophage libraries, we found that the aptamer library employed here required more than 15 rounds of in vivo selection for convergence to specific sequences. The aptamer species identified through this approach bound to brain capillary endothelia and penetrated into the parenchyma. The methods described may find general utility for targeting various payloads to the brain.
Keywords: blood–brain barrier, in vivo selection, mouse, RNA
Introduction
The blood–brain barrier (BBB) regulates brain homeostasis and the transport of endogenous and exogenous compounds by controlling their selective and specific uptake, efflux, and metabolism in the brain.1 The BBB consists of an elaborate complex of tight junctions and adherens junctions between adjacent endothelial cells.2 Due to their unique properties, paracellular transport of hydrophilic drugs across BBB endothelia is virtually absent and transcellular transport by passive diffusion is only available to lipophilic molecules or water soluble molecules under 400 Da.2,3 As a result, various targeting strategies are being explored to enhance BBB penetration. Previous attempts encompass increasing the lipophilic nature of the drug, using chimeric protein or antibodies.4 A novel vehicle that has shown promise for targeting peripheral tissues is aptamers.5,6
Aptamers, composed of single-stranded DNA or RNA, are generated from SELEX (Systematic Evolution of Ligands by EXponential enrichment), and gain affinity and specificity to target molecules after rounds of selection. Synthetic aptamers can be modified, for example, by incorporating fluorine or O-methylation at the 2′-OH position to enhance plasma stability and circulating half-life. Previous investigations have shown that aptamers have low toxicity and immunogenicity in vivo compared with other similar agents.7,8 Since the first introduction of SELEX technology in 1990,9,10 it has been applied to the development of moities for clinical diagnosis and in preclinical therapeutic studies.7,11,12 Generally, purified proteins or cultured cells have been utilized for capturing specific nucleic acid aptamers.13,14,15 However, they may not ideally represent the physiological state of the organ or tissue being targeted. Recently, Mi and colleagues identified RNA aptamers that localized to liver-residing colon cancer metastases in mice, suggesting the viability of screening tissue-penetrating aptamers within living animals.16
Previous investigations on aptamer biodistribution after intravenous delivery implies that accessibility to the brain is low. This is likely due to the presence of the BBB.17 Here, we devised an in vivo evolution strategy in wild-type mice, with the goal to identify RNA aptamers with enhanced penetration to brain. After multiple rounds of panning, aptamers which entered brain endothelia and parenchymal cells after peripheral injection emerged.
Results
We first designed a library of aptamers of 40 nucleotides (nt) of random sequence flanked by fixed sequences (Figure 1a and Supplementary Table S1). Because our goal was to identify aptamers that could afford central nervous system delivery, we reasoned that the BBB in situ was the optimal platform for panning compared with artificial BBB culture systems. We administered 2′-fluoropyrimidine–modified RNA (RNase A resistant) random libraries into the mice via tail vein injection (Figure 1a) and subsequently harvested brains for aptamer recovery. Purified RNA was further treated with RNase A and DNase I, amplified, and re-injected into subsequent animals. We sequenced recovered clones via Sanger methods to monitor the progress of SELEX. After 6 and 12 rounds, we found no convergence (data not shown). Therefore, we instituted a negative selection step after round 12. The recovered RNAs were incubated with mouse serum before amplification for subsequent rounds of selection. After round 14, we sequenced the recovered library using an Illumina deep sequencing platform and also performed Sanger sequencing on random selected clones. The data retrieved from Sanger and deep sequencing identified the same sequence (A14) as the top ranked sequence (Table 1).
Figure 1.

In vivo SELEX for aptamers. (a) Schema for SELEX strategy. An RNA library consisting of a 40 nt random region flanked 5′ by a 32 bp left arm containing a T7 transcription start, and 3′ by a 16 bp for PCR amplification was generated, and then RNAs transcribed in the presence of 2′-fluoro nucleosides to enhance nuclease resistance. Purified RNAs were peripherally injected into three C57BL/6 mice and circulated for 1 hour (rounds 1–8) or 3 hours (rounds 9–22). Mice were perfused with DPBS, and RNAs were extracted from whole brain. Recovered RNAs were treated with RNAse A and DNase I, and then converted to double-stranded DNA by RT-PCR. Purified DNAs were transcribed into 2′-F RNA, and full-length RNA was purified for the subsequent round of panning. (b) Predicted secondary structures of aptamers using Mfold (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form). DPBS, Dulbecco's phosphate-buffered saline; nt, nucleotide; RT-PCR, reverse transcription PCR; SELEX, Systematic Evolution of Ligands by EXponential enrichment.
Table 1. Representative sequences from in vivo SELEX.

The library evolved to enrichment after 22 rounds of selection, with three motifs representing almost 90% of sequences (Table 1). Representative sequences identified after SELEX were modeled using Mfold (Figure 1b).18 Strikingly, sequence A14, which was most prominent from round 14 (Table 1) to round 18 (data not shown), was no longer present by round 22, although sequences very similar to A14 emerged. Sequence A15 enriched to 50% in round 22.
Several additional experiments were done to confirm that enrichment was occurring during SELEX. First, we quantified aptamer levels from liver, kidney or brain following tail vein injection of the RNA aptamer libraries amplified after various rounds. Only in brain lysates did we note increased RNA aptamer abundance with successive selections (Figure 2).
Figure 2.
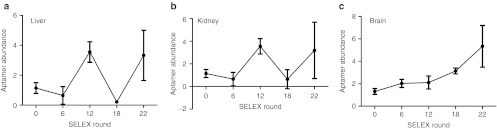
Assessment for aptamer enrichment. RNA (1 nmol) from the starting library (R0), round 6 (R6), round 12 (R12), round 18 (R18), or round 22 (R22) were injected into C57BL/6 mice (three mice/library). Three hours post-injection, the mice were perfused and total RNAs from the (a) liver, (b) kidney or (c) brain were extracted and the relative abundance of RNA library in these organs was quantified by qRT-PCR. Data are normalized to 5S RNA and are presented as mean ± SD. qRT-PCR, quantitative reverse transcription-PCR; SELEX, Systematic Evolution of Ligands by EXponential enrichment.
By sequencing clones from round 22 SELEX, we found 6.67% of clones to be A02, 30% were A09, and 50% were A15 (Table 1 and Figure 3a). We tested the homing potential of these same aptamers using a reverse screening assay.19 A15, A09, A02, and scrambled sequences (SCAP) were transcribed and purified separately, then mixed at equal molar ratios. The mixtures were peripherally injected into mice, and the RNAs were harvested from mouse brain and sequenced. We found that aptamer A15 was the most frequent sequence in brain lysates among the recovered RNAs. Approximately 45% of clones were A15, 30% were A9, and 25% were A2 (Figure 3b). This is similar to what was found from sequencing after SELEX round 22 (Figure 3a), and contrasts the input which was 25% for each aptamer.
Figure 3.
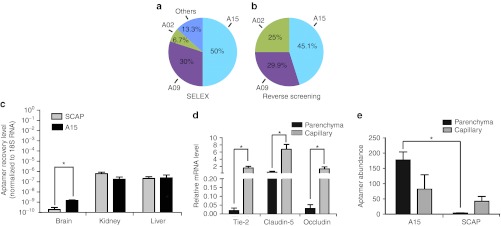
The A15 aptamer shows enhanced specificity to brain. (a) Data represent the percentage of the denoted aptamer present after sequencing of clones from SELEX. (b) Data represents the percentage of the denoted aptamer found in the reverse screening assay. For this, equal amounts of selected RNA motifs (A02, A09 or A15, see Table 1) and scrambled A15 (SCAP) were mixed, and injected into mice. RNA was extracted from the brain, reverse transcribed, cloned, and colonies were sequenced. The frequency of the sequences representing each aptamer is shown. (c) Equal amounts of aptamers (SCAP and A15) were peripherally injected in mice (n = 3–4) and after 4 hours, total RNA was isolated. Aptamer quantitation in brain, kidney, and liver was done using one-step qRT-PCR (5 µg of DNase-treated input RNA) by including 18S RNA as loading control. (d) Quantification of A15 aptamer in capillary versus parenchymal tissues after peripheral injection. Mouse brain capillaries were clarified from brain parenchymal fractions by density centrifugation, and the purity of the capillary fraction was evaluated by assessing the endothelial cell-specific markers Tie-2, Claudin-5, and Occludin. mRNA levels were normalized to GAPDH. Data are expressed as means ± SD (*P < 0.05, Student's t-test; n = 3). (e) qRT-PCR to evaluate aptamer abundance in the indicated fractions. Data are presented as means ± SD using ANOVA and Bonferroni post hoc for significance (*P < 0.05; n = 3 mice per group). ANOVA, analysis of variance; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; qRT-PCR, quantitative reverse transcription-PCR; SELEX, Systematic Evolution of Ligands by EXponential enrichment.
We next asked whether A15 showed increased enrichment in brain over SCAP. Mice were injected with SCAP or A15 and the aptamers were allowed to circulate for several hours. As expected, there was no difference in the levels of aptamers recovered from kidney or liver. However, brain levels did show enrichment (Figure 3c).
In subsequent studies, we tested whether A15 aptamers were retained in capillary cells or brain parenchyma. For this work, we used a capillary depletion protocol which allows separation of the vessels (and endothelia lining them) from the remaining parenchymal cells (e.g., neurons and glia). For this, A15 and control aptamer RNAs were delivered into brain by carotid artery injection, and after circulation blood vessels were separated from the brain parenchyma. The quality of the separation was confirmed by testing the fractions for the vascular markers Tie 2, Claudin-5, and Occludin. In all cases these markers were enriched in the capillary fraction as expected, and conversely, diminished in brain parenchymal fractions (Figure 3d). Next, we assayed for A15 aptamer in the two fractions. To our surprise, quantitative reverse transcription-PCR(qRT-PCR) demonstrated that A15 was most abundant in the blood vessel-depleted brain parenchymal fraction (Figure 3e). These data suggest that the A15 aptamer may penetrate the BBB.
To further test for brain penetration, an in situ hybridization experiment was performed. Aptamers were labeled using DIG-nucleosides. In addition, 2′-O-methyl (2′OMe) residues were incorporated internally to reduce nuclease sensitivity. Sections from animals injected peripherally with A15 or control aptamers were prepared and hybridized with probes specific to the A15 random region. Notably, A15-injected mice revealed positive signal (purple precipitate) in various brain regions including the cortex, hippocampus, cerebellum, and striatum, in contrast to scramble aptamer-injected mice (Figure 4).
Figure 4.

Aptamer biodistribution after peripheral injection. Representative in situ hybridization images from cerebral cortex (Cx), striatum (Str), hippocampus (Hp), and cerebellum (Cb) of mice, collected 3 hours after carotid artery injection with either scrambled (left) or A15 (right) aptamer. The slides were lightly counterstained with methylene green (cell nuclei; light blue). Purple precipitate denotes aptamer hybridization signal (note difference in intensity of aptamer signal between right panels and left panels). Arrows denote regions magnified in insets (lower right, all images) for better visualization of aptamer signal. Sections are representative of those from three different mice/group. Bar = 100 µm and applies to all images. Inset magnification ×5.
The highly impermeable tight junctions between endothelial cells forming the capillaries and venules in the central nervous system of higher vertebrates form the BBB, which impedes the passive diffusion of solutes from the blood into the brain parenchyma. We thus hypothesized that our aptamers must first be internalized into endothelial cells before entry into the brain parenchyma. To test this, we designed a series of in vitro internalization assays to determine whether aptamers with enriched homing to brain were more likely to bind mouse brain-derived endothelial cells than their unselected counterparts.
Aptamer binding (on ice) and internalization (at 37 °C) into endothelial cells were determined by flow cytometry. Cells on ice were treated with biotinylated aptamers (A15 and SCAP), and then subsequently incubated with streptavidin-phycoerythrin (SA-PE) to detect bound aptamer. Fluorescence-activated cell sorting of the cells showed that ~20% more A15 aptamer bound to the endothelial cell surface than control aptamer (Figure 5a, left panel). Assessment of internalized aptamer at 37 °C in permeablized cells showed ~25% more uptake of A15 relative to SCAP (Figure 5a, right panel).
Figure 5.
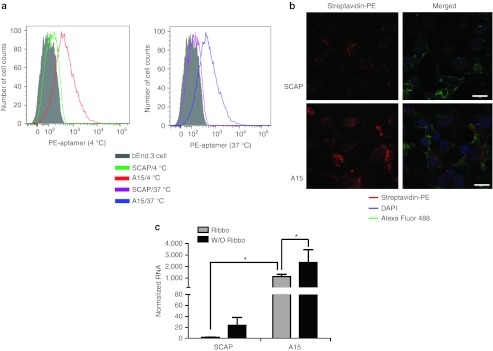
SELEX-derived aptamers show improved binding and uptake into mouse brain endothelial cells in vitro. (a) Flow cytometry to monitor aptamer binding and uptake on bEnd.3 cells. The bEnd.3 cells were treated with the denoted aptamers at 4 or 37 °C. The panels show that A15 binding is enhanced over SCAP (left panel), and that there is extensive uptake at 37 °C relative to SCAP (right panel). (b) Representative confocal images of bEnd.3 cells after incubation with 500 nmol/l SA-PE–conjugated A15 or SCAP, showing cellular uptake with A15, but not SCAP. Green, Alexa-488 labeled wheat germ agglutinin for cell membrane labeling; blue, DAPI to stain nuclei; red, SA-PE–conjugated aptamer. Bar = 20 µm. (c) Quantitation of aptamer internalization into brain endothelia cells. Mouse brain endothelial cells were blocked with 50 µg/ml yeast tRNA and 1 µg/µl poly d(I:C) for 30 minutes, and treated with 1 µmol/l A15 or SCAP for 2 hours at 37 °C. Cells were treated with 0.02 U/µl of Riboshredder to degrade residual RNAs on the cell surface or in the media. Internalized RNA was quantified by qPCR and normalized to GAPDH. The results are presented as means ± SD. Statistical significance was done using ANOVA and Bonferroni post hoc (*P < 0.05; n = 6 per group). ANOVA, analysis of variance; DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; qPCR, quantitative PCR; SA-PE, streptavidin-phycoerythrin; tRNA, transfer RNA.
We also tested for aptamer internalization using microscopy assays. Cells were incubated with SA-PE–labeled aptamers, and then internalized aptamers were visualized using confocal microscopy. We found that A15, but not SCAP, was present in the cytoplasm (Figure 5b). To quantify internalization, mouse brain endothelial cells were incubated with aptamers, followed by treatment with Riboshredder to degrade residual RNAs retained on the cell surface or the culture wells. Resistant, internalized aptamers were quantified by qRT-PCR. Consistent with the above, we observed significantly elevated amounts of internalized A15 aptamer relative to control sequence (Figure 5c). Taken together, these data suggest that the aptamers evolved from in vivo SELEX first target endothelia, followed later by brain penetration.
Discussion
The brain is protected from infectious and toxic agents by a dynamic barrier, the BBB, which also impedes drug transport into the brain via the blood circulation.20 Brain capillary endothelial cells, pericytes, astrocytic foot processes, and nerve endings terminating on the capillary surface constitute the BBB.21 The unique structure of the BBB hinders many therapies directed at brain pathologies. Several noninvasive strategies have been proposed to overcome this problem, such as delivery through the nasal mucosa,22 osmotic opening of the BBB,23 nanoparticle coating,24 transporter vectors,25 and viral vectors.26,27 An untested area of investigation for approaching brain delivery is aptamers. Aptamers have complex secondary and tertiary structure and have been shown to recognize specific targets. Aptamers for specific targets are often identified using SELEX. For conventional SELEX, one bait (i.e., purified protein or tissue culture cells) is traditionally used as input to screen RNAs or DNAs from complex aptamer libraries consisting of pool sizes of 1012–1015.
We reasoned that an unbiased in vivo SELEX protocol performed in situ would be optimal for the identification of aptamers that can reach the brain from the vasculature. Surprisingly, and in contrast to our prior work in phage panning27 and other's work with in vitro SELEX,6 many more rounds of in vivo SELEX was required for enrichment. This may be explained in part by considering the bait complexity and the length of the random sequence. While our 40 nt long random library may offer an advantage for in vitro SELEX, it may increase the complexity of possible secondary and tertiary structures within a given sequence to the point that, when combined with the large repertoire of targets to which the aptamers may bind in vivo, reduce the likelihood for early enrichment. Thus, more rounds are required for eventual selection. Nonetheless, after 14 rounds of panning, some aptamers emerged and further enrichment occurred after 22 rounds.
The timing for our studies may have been critical for evolving aptamers that breached the BBB. For in vivo panning with bacteriophage, circulation times post-delivery range from 5–30 minutes to avoid phage loss.16,27,28 The transcytosis of intact viruses takes longer; only 1% of HIV-1 viral particles cross brain microvascular endothelial cells after 24 hours,29 or 0.1% of adenovirus-5 viral particles after 6 hours.30 For RNA modified for improved stability in blood,8 circulation times of 6–24 hours would be of little benefit as the RNAs would be excreted. Considering the barriers for brain targeting, we let the library circulate for 1–3 hours to balance the time needed for brain endothelia binding with excretion. In other work for liver SELEX, 20-minute circulation times were used.16 It is unclear if we would have found quicker conversion if a shorter circulation time was employed.
In this work, we compared enriched sequences using Illumina deep sequencing and Sanger methods. The sequence distributions from Illumina are consistent with Sanger sequencing, and aptamer A14 was among the top 10 sequences for both platforms (data not shown). To our surprise, the A14 aptamer could be recovered in isolated mouse brain capillary fractions at round 14, but was reduced to undetectable levels (by Sanger sequencing methods) by round 18 (data not shown). In follow-up studies, we could not detect aptamer A14 binding to endothelial cells lines or primary endothelial cell cultures (data not shown). However, we noticed some motifs with consensus sequence similar to A14 after round 22, suggesting the A14 might be competed away by these sequences.
We noticed a high level of RNAs in non-targeted tissues, such as the kidney and liver. As enrichment was not noted upon subsequent rounds of SELEX, we attribute this, at least partially, to rapid clearance of the RNA through the reticuloendothelial system and the kidney due to their low molecular weight. The molecular mass for most aptamers fall in the 5–15 kDa range, and are therefore susceptible to renal filtration even if they are resistant to nuclease-mediated degradation. Similar results have been found in in vivo phage-panning studies.31 Further chemical conjugation to high molecular weight polymers may protect aptamers from renal filtration, and promote persistence in the circulation. Nonetheless, depending on the target use for brain-penetrating aptamers, a fraction of the dose entering the brain may be sufficient for therapeutic benefit, similar to other small molecules.22
It is generally considered that for compounds to cross the BBB, they should have a molecular weight of <400 Da and be lipophilic.2 Given the chemical and physical attributes of our aptamers, it is unlikely that they enter the brain via paracellular aqueous routes or transcellular lipophilic pathways. They may enter via adsorptive-mediated transcytosis, channel and/or receptors for uptake or fluid-phase pinocytosis.32 Recent work suggests that a quadruplex-forming DNA aptamer binds to nucleolin via macropinocytosis.33 Our work in vitro shows that A15 can enter brain endothelia cells under physiological conditions, and in vivo, is evident in brain parenchyma. Whether entry is via fluid-phase pinocytosis or other routes requires further study.
In addition to SELEX, there are many methodologies for selection of random combinatorial libraries such as phage display, ribosome display, and mRNA display. For phage display, phage particles provide the link between genotype (single-stranded DNA) and the selected phenotype (the displayed peptide). For SELEX, the aptamer library contains both the genotype and the phenotype in one molecule. As noted, we did not observe enrichment for over 12 rounds of selection compared with previous phage panning and in vitro SELEX, which suggests that this strategy might be impractical for screening signatures imparted by a disease state; sequences emerge from phage panning within 3–5 rounds making them amenable to screening for disease-specific epitopes.27
In conclusion, we present novel studies demonstrating that aptamers can be panned from the brains of living animals, and that sequences which emerge can enter the brain after peripheral delivery. It will be interesting to investigate whether brain SELEX with a shorter RNA library could accelerate in vivo selection. In addition, to obtain RNA motifs specific to disease phenotypes, a combination of in vitro selection followed by in vivo selection might be preferred.
Materials and Methods
Mouse and cell lines. All animal studies were approved by the University of Iowa Animal Care and Use Committee. We obtained C57BL/6 mice from the Jackson Laboratory (Bar Harbor, ME). Escherichia coli DH5α was grown under aerobic conditions at 37 °C in Luria-Bertani media. Mouse endothelial cell line bEnd.3 was obtained from the American Type Culture Collection (ATCC, Manassas, VA), and cultured in high glucose Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY), supplemented by 10% fetal bovine serum and 1% penicillin-streptomycin, at 37 °C with 5% CO2.
In vivo SELEX. Unless otherwise stated, all oligonucleotides, including DNA libraries were synthesized by Integrated DNA Technologies (Coralville, IA). The single-stranded library sequence was adapted from previous work,14 and contained a central randomized sequence of 40 nt flanked by a 32-nt 5′ and a 16-nt 3′ primer hybridization site. To generate double-stranded DNA, a fill-in reaction with Klenow polymerase (New England Biolabs, Beverly, MA) was performed in the presence of primer SXRP, and incubated at 37 °C for 2 hours. Double-stranded DNAs were excised from 4% agarose gels and purified with Qiaquick gel extraction kits (Qiagen, Valencia, CA), and amplified with primer SXRP and SXFP (12 cycles of 30 seconds at 94 °C, 30 seconds at 60 °C, and 1 minute at 72 °C) in totally 20–50 ml reaction volume. The 88 bp amplicons were purified as above and used as template for in vitro transcription. To produce nuclease-resistant RNA for in vivo injection, 2′-fluoro nucleotide triphosphates were included in the reaction with Durascribe T7 transcription kit (Epicentre, Charlotte, NC) at 37 °C overnight. The transcribed RNA was treated with DNase I (Epicentre), followed by phenol/chloroform extraction. RNA was further concentrated with Amicon YM-10 columns (Millipore, Billerica, MA) and applied to 12% denaturing polyacrylamide gels containing 7 mol/l urea. The expected 71 nt RNA library band was excised and purified as described6 or cleaned using Zymo small RNA PAGE purification kits (Zymo Research, Ervine, CA). Purified RNAs were quantified with a NanoDrop (Thermo Scientific, Wilmington, DE) and formulated in SELEX selection buffer (SSB, 10 mmol/l HEPES, pH 7.4, 50 mmol/l NaCl, 1 mmol/l MgCl2, 1 mmol/l CaCl2, 0.01% bovine serum albumin).
For in vivo selection, 2 nmol of RNA library was prepared in 110 µl SSB, denatured at 94 °C for 3 minutes, and then slowly refolded by cooling to room temperature in the heat block. Renatured RNA was peripherally injected into three 6–8 weeks old C57BL/6 mice, and circulated for 1 or 3 hours. Mice were then perfused with Dulbecco's phosphate-buffered saline (DPBS), brains were removed, rinsed with DPBS, and total brain RNA was extracted using TRIzol Reagent (Life Technologies). Recovered RNAs were treated with RNase A and DNase I at 37 °C for 15 minutes. To generate DNA template for transcription, cDNAs were prepared with OneStep RT-PCR Kit (Qiagen) or Superscript III Reverse Transcriptase (Life Technologies) with primer SXRP, and amplified with Taq PCR Master Mix Kit (Qiagen) for 12–18 cycles. The DNA was purified as above and used to generate 2′-F RNA for the next round of selection. After 6, 12, 14, 18 or 22 rounds of selection, DNA was reverse transcribed from total RNA, cloned into the pGEM-Teasy vector (Promega, Madison, WI), and transformed into Escherichia coli DH5α, and individual colonies from Luria-Bertani plates containing IPTG/X-gal were sequenced by Functional Bioscience (Madison, WI). To generate DNA for deep sequencing, we performed 10 cycles of PCR and submitted the DNA to the Iowa State University DNA Facility to generate libraries for sequencing on the Illumina platform.
qRT-PCR. To assess selection progress, equal amounts of RNA libraries were tail vein injected into three mice. Brain and liver RNAs were extracted by an acid guanidinium-thiocyanate-phenol-chloroform method using TRIzol reagent (Life Technologies) per the vendor's manual. The amount of RNA was quantitated using a Nano-Drop ND-1000 spectrophotometer (Thermo Scientific), and 500 ng DNase I-treated RNA was input for reverse transcription using Superscript III transcriptase (Life Technologies). Relative abundance of RNA species was quantitated using qRT-PCR with specific primers (QFP and QRP) and Power SYBR Green Mster Mix (Life Technologies), and run on the Applied Biosystems Sequence Detection System 7900 (ABI 7900; Life Technologies). Relative RNA levels were normalization to 5S RNA (Primer sets from Life Technologies). To evaluate RNA internalized into endothelial cells, 10,000 bEend.3 cells were plated in 12-well plates, grown to ~90% confluency, washed with cold DPBS and blocked with 100 µg/ml yeast transfer RNA (Sigma-Aldrich, St Louis, MO) and 1 µg/µl poly d(I:C) (Sigma-Aldrich) for 30 minutes. The cells were washed and incubated with 1 µmol/l RNA aptamer in SSB for 2 hours. After removing unbound RNA, the cells were washed with DPBS, treated with 0.02 U Riboshredder (Epicentre) as described,34 rinsed with cold DPBS plus 0.5 mol/l NaCl, and lysed with 500 µl TRIzol reagent (Life Technologies) per well on ice. RNAs were further purified with RNA clean and concentrator (Zymo Research) for quantification. RNA levels were normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) mRNA levels.35 For RNA distribution analysis, 1 nmol of A15 or scrambled aptamer was injected into 3–4 mice, and circulated in the body for 4 hours. The mice were perfused with 20 ml cold PBS, and killed for RNA extraction with Trizol reagent (Life Technologies) followed by purification with Direct-Zol RNA purification kit (Zymo Research). Five microgram of total RNA was input for SuperScript III Platinum SYBR Green One-Step qPCR Kit w/ROX (Life Technologies). All data was analyzed with SDS 2.3 software (Life Technologies) with normalization to 18S RNA.
Cellular uptake and internalization assays. RNAs were purified from denatured polyacrylamide gels and labeled using the Pierce 3′-end biotinylation kit (Thermo Scientific, Rockford, IL) according to the manufacturer's protocol. Biotinylated RNA was purified by phenol/chloroform extraction, precipitated with ethanol, and dissolved in distilled RNase-free water. For conjugation, SA-PE (Prozyme, Hayward, CA) was mixed with biotinylated RNA at a 1:3 molar ratio in SSB at room temperature for 1 hour as before,36 and free dye removed with G-25 columns. Flow cytometry-based internalization assay was as described37 with slight modification. Briefly, bEnd.3 cells were cultured in T150 flasks, the cells were trypsinized and plated on 100 × 20 mm culture dishes 24 hours before the experiment. The cells were washed with DPBS, dissociated from the dish with 0.25% trypsin/EDTA and viable cells counted with trypan staining. SA-PE–labeled aptamer A15 or control aptamer (900 nmol/l) was incubated with 5 × 105 cells in 100 µl SSB at 4 °C for 1 hour. For competition, 5 µmol/l cold unlabeled A15 RNA aptamer or control aptamer was incubated with cells at 4 °C for 30 minutes before adding labeled aptamer. Cells were recovered by low speed centrifugation, and washed three times with DPBS. Cell pellets were resuspended in 500 µl cold DPBS and analyzed using a BD LSR II Violet flow cytometer (Becton Dickinson, San Jose, CA). For each reaction, 30,000 events were collected and analyzed using Flowcyto software version 8.7 (Tree Star, Ashland, OR).
Confocal microscopy was done similar to previous reports.38 The bEnd.3 cells were seeded on 8-well chamber slides to 90% confluence, and washed three times with DPBS. SA-PE–labeled RNA aptamer was prepared as above, denatured at 75 °C for 5 minutes, cooled to room temperature for 15 minutes and 4 µl RNA (500 nmol/l) in 400 µl of binding buffer (with 10% fetal bovine serum) was applied to the cells at 37 °C for 2 hours. The supernatant was aspirated and the slides were washed three times with DPBS. Pre-warmed 4% paraformaldehyde (200 µl) was applied to each well at 37 °C for 15 minutes. Cells were washed once with DPBS, and 1 µg/ml Hoechst 33258 (200 µl) was added to each well. After 5 minutes, 5 µg/ml per well of Wheat Germ Agglutinin, Alexa Fluor 488 (Life Technologies) was added on ice for 15 minutes. Slides were mounted with Vector Shield (Vector Laboratories, Burlingame, CA) and internalized aptamer visualized using a Zeiss 710 confocal microscope (Carol Zeiss, Oberkochen, Germany).
Aptamer enrichment assays. A reverse screening assay19 was performed by injecting a mixture of 100 pmol of individual aptamers into three mice via tail vein. After 1 hour of circulation, hearts were perfused with 10 ml PBS, mouse brain and kidney were collected, and homogenized for RNA extraction as described. After RT-PCR, sequences were cloned into the pGEM-T Easy vector (Promega) and used to transform Escherichia coli; blue/white screening was used to identify colonies containing aptamer inserts, which were sequenced. The frequency of the specific aptamer was determined.
Capillary depletion assay was done as described earlier39,40 using three mice per treatment. Each mouse received 1 nmol of A15 aptamer or one of two control aptamers via tail vein injection. After 1 hour, mice were killed by decapitation, the brain was quickly removed, arachnoid membranes were peeled away, and the choroid plexus was discarded. Mouse brain weight was recorded, and, following an initial mincing brains were homogenized using a glass homogenizer (8–10 strokes) in 2 ml of physiologic buffer (10 mmol/l HEPES; pH 7.4, 141 mmol/l NaCl, 4 mmol/l KCl, 2.8 mmol/l CaCl2, 1 mmol/l MgSO4, 1 mmol/l NaH2PO4, and 10 mmol/l D-glucose). A 26% dextran solution was added (final dextran concentration, 18%) and homogenized again (three strokes). An aliquot of the total homogenate was saved, and the remainder was centrifuged at 5,400g (~9,000 rpm) for 30 minutes at 4 °C in a SW55ti rotor (Beckmean Coulter, Brea, CA). The supernatant (brain parenchyma) and pellet (vasculature) were carefully separated. Light microscopic examination of the pellet and supernatant showed that the pellet consisted of brain vasculature, whereas the supernatant was essentially devoid of vasculature. The purity of the separated fractions was further evaluated by quantifying mRNA levels of endothelial cell markers41 after RNA extraction. RNA from each sample was reverse transcribed with either QRP primer or 5S RNA RT primer from Life Technologies, and amplified with corresponding primers.
In situ hybridization. Probes were designed to the random part of the aptamer. The hybridization was performed as previously published42 with some modification (M. Keiser, unpublished data). Briefly, mice received 1 nmol of A15 or control aptamer via carotid artery injection. Mice were killed and the brain quickly harvested and embedded in optimal cutting temperature for sectioning. The sections were fixed, washed, and dehydrated, and incubated with prehybridization buffer (50% formamide, 5× SSC, 0.5% blocking solution (Roche Applied Science, Brandford, CT), 0.1% Tween-20, 0.1% CHAPS, 50 µg/ml yeast RNA, 5 mmol/l EDTA, 50 µg/ml heparin) for 1 hour in a humid chamber followed by adding 100 nmol/l probe, and hybridized at 42 °C overnight. The next day, the slides were incubated with 50 µg/ml RNase A for 30 minutes at 37 °C and with extensive wash in 2× and 0.1× SSC for 1 hour. The slides were blocked with blocking buffer (20% sheep serum in 50 mmol/l Tris–HCl; pH 7.5, 150 mmol/l sodium chloride, 10 mmol/l potassium chloride, 1% Tween-20) at room temperature. Two hours later, anti-DIG antibody 1:2,000 (Anti-DIG alkaline phosphatase FAB fragments; Roche Applied Science) in blocking buffer was added and incubated at 4 °C in humid chamber for 48 hours. Then the slides were washed with KTBT (50 mmol/l Tris–HCl; pH 7.5, 150 mmol/l sodium chloride, 10 mmol/l potassium chloride, 1% Tween-20) and alkaline phosphatase buffer (100 mmol/l Tris–HCl, 50 mmol/l magnesium chloride, 100 mmol/l sodium chloride, 0.1% Tween-20, pH 9.6), and developed with 1-Step NBT/BCIP (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate) developing solution (Thermo Scientific) in the dark. The slides were washed with water, and counterstained with 1% methylene green (Sigma-Aldrich) at 37 °C for 10 minutes. After washing, the slides were dehydrated, and mounted with Vectorlab mounting buffer (Vector Laboratories), and observed under Zeiss light microscopy equipped with Olympus camera.
Statistical analysis. Results are expressed as mean ± SD. Significance (P < 0.05) was assessed using the unpaired t-test or one-way analysis of variance analysis and the indicated post hoc analysis using GraphPad Prism (Version 5.0) software (GraphPad, La Jolla, CA).
SUPPLEMENTARY MATERIAL Table S1. Synthesized oligonucleotides and primers.
Acknowledgments
This work was supported by the National Institutes of Health grants (NS063246, DK54759) and the Roy J Carver Trust. The authors thank the University of Iowa Cores (Central Microscopy facility, FACS facility, and DNA facility), James McNamara III, and members of the Davidson and McCray lab for critical discussions. M.A.B. and K.A.L. are employed by Integrated DNA Technologies, Inc. (IDT), which offers oligonucleotides for sale similar to some of the compounds described in the manuscript. IDT is however not a publicly traded company and these authors do not own any shares/equity in IDT. The other authors declared no conflict of interest.
Supplementary Material
Synthesized oligonucleotides and primers.
References
- de Boer AG., and, Gaillard PJ. Drug targeting to the brain. Annu Rev Pharmacol Toxicol. 2007;47:323–355. doi: 10.1146/annurev.pharmtox.47.120505.105237. [DOI] [PubMed] [Google Scholar]
- Chen Y., and, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64:640–665. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- Pavan B, Dalpiaz A, Ciliberti N, Biondi C, Manfredini S., and, Vertuani S. Progress in drug delivery to the central nervous system by the prodrug approach. Molecules. 2008;13:1035–1065. doi: 10.3390/molecules13051035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson BL., and, Breakefield XO. Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci. 2003;4:353–364. doi: 10.1038/nrn1104. [DOI] [PubMed] [Google Scholar]
- Zhou J, Swiderski P, Li H, Zhang J, Neff CP, Akkina R.et al. (2009Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells Nucleic Acids Res 373094–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proske D, Blank M, Buhmann R., and, Resch A. Aptamers–basic research, drug development, and clinical applications. Appl Microbiol Biotechnol. 2005;69:367–374. doi: 10.1007/s00253-005-0193-5. [DOI] [PubMed] [Google Scholar]
- Keefe AD, Pai S., and, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C., and, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Ellington AD., and, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Lassalle HP, Marchal S, Guillemin F, Reinhard A., and, Bezdetnaya L. Aptamers as remarkable diagnostic and therapeutic agents in cancer treatment. Curr Drug Metab. 2012;13:1130–1144. doi: 10.2174/138920012802850038. [DOI] [PubMed] [Google Scholar]
- Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z.et al. (2011Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras J Clin Invest 1212401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua P, Kim S., and, Lee DK. Nucleic acid aptamers targeting cell-surface proteins. Methods. 2011;54:215–225. doi: 10.1016/j.ymeth.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Lupold SE, Hicke BJ, Lin Y., and, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P.et al. (2006Aptamers evolved from live cells as effective molecular probes for cancer study Proc Natl Acad Sci USA 10311838–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA.et al. (2010In vivo selection of tumor-targeting RNA motifs Nat Chem Biol 622–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C.et al. (2004Pharmacokinetics and biodistribution of novel aptamer compositions Pharm Res 212234–2246. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daquinag AC, Zhang Y, Amaya-Manzanares F, Simmons PJ., and, Kolonin MG. An isoform of decorin is a resistin receptor on the surface of adipose progenitor cells. Cell Stem Cell. 2011;9:74–86. doi: 10.1016/j.stem.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP.et al. (2007Modelling of the blood-brain barrier in drug discovery and development Nat Rev Drug Discov 6650–661. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Biopharmaceutical drug targeting to the brain. J Drug Target. 2010;18:157–167. doi: 10.3109/10611860903548354. [DOI] [PubMed] [Google Scholar]
- Banks WA. Developing drugs that can cross the blood-brain barrier: applications to Alzheimer's disease. BMC Neurosci. 2008;9 suppl. 3:S2. doi: 10.1186/1471-2202-9-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Brooke CB, Whitmore AC., and, Johnston RE. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol. 2011;85:10682–10690. doi: 10.1128/JVI.05032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Farrar CT, Saidi LJ, William CM, Hyman BT., and, Spires-Jones TL. Nanoparticles enhance brain delivery of blood-brain barrier-impermeable probes for in vivo optical and magnetic resonance imaging. Proc Natl Acad Sci USA. 2011;108:18837–18842. doi: 10.1073/pnas.1111405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DD., and, Lockman PR. The blood-brain barrier choline transporter as a brain drug delivery vector. Life Sci. 2003;73:1609–1615. doi: 10.1016/s0024-3205(03)00504-6. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM., and, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Chang M., and, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat Med. 2009;15:1215–1218. doi: 10.1038/nm.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini R., and, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B.et al. (2002Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway J Virol 766689–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Han T, Everts M, Zhu ZB, Gillespie GY, Curiel DT.et al. (2007Directing adenovirus across the blood-brain barrier via melanotransferrin (P97) transcytosis pathway in an in vitro model Gene Ther 14523–532. [DOI] [PubMed] [Google Scholar]
- Stojanov K, Georgieva JV, Brinkhuis RP, van Hest JC, Rutjes FP, Dierckx RA.et al. (2012In vivo biodistribution of prion- and GM1-targeted polymersomes following intravenous administration in mice Mol Pharm 91620–1627. [DOI] [PubMed] [Google Scholar]
- Hanss B, Leal-Pinto E, Bruggeman LA, Copeland TD., and, Klotman PE. Identification and characterization of a cell membrane nucleic acid channel. Proc Natl Acad Sci USA. 1998;95:1921–1926. doi: 10.1073/pnas.95.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reyes EM, Teng Y., and, Bates PJ. A new paradigm for aptamer therapeutic AS1411 action: uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 2010;70:8617–8629. doi: 10.1158/0008-5472.CAN-10-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães ML, Byrom M, Yan A, Kelly L, Li N, Furtado R.et al. (2012A general RNA motif for cellular transfection Mol Ther 20616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K.et al. (2011A genetically humanized mouse model for hepatitis C virus infection Nature 474208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Larson T, Nguyen HH, Sokolov KV., and, Ellington AD. Directed evolution of gold nanoparticle delivery to cells. Chem Commun (Camb) 2010;46:392–394. doi: 10.1039/b920865h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G, Ahmed MS, Dolf A, Endl E, Knolle PA., and, Famulok M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat Protoc. 2010;5:1993–2004. doi: 10.1038/nprot.2010.163. [DOI] [PubMed] [Google Scholar]
- Shigdar S, Lin J, Yu Y, Pastuovic M, Wei M., and, Duan W. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991–998. doi: 10.1111/j.1349-7006.2011.01897.x. [DOI] [PubMed] [Google Scholar]
- Triguero D, Buciak J., and, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B., and, Starzyk RM. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc Natl Acad Sci USA. 1991;88:4771–4775. doi: 10.1073/pnas.88.11.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa M, Toki H, Tomi M., and, Hosoya K. Gene expression profiles of ATP-binding cassette transporter A and C subfamilies in mouse retinal vascular endothelial cells. Microvasc Res. 2008;75:68–72. doi: 10.1016/j.mvr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- De Block M., and, Debrouwer D. RNA-RNA in situ hybridization using digoxigenin-labeled probes: the use of high-molecular-weight polyvinyl alcohol in the alkaline phosphatase indoxyl-nitroblue tetrazolium reaction. Anal Biochem. 1993;215:86–89. doi: 10.1006/abio.1993.1558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesized oligonucleotides and primers.