

Abstract
Most advanced prostate cancers (PCa) will develop into the castration-resistant stage following androgen deprivation therapy, yet the molecular mechanisms remain unclear. In this study, we found PrLZ, a newly identified Prostate Leucine Zipper gene that is highly expressed in PCa could interact with the androgen receptor (AR) directly leading to enhance AR transactivation in the castration-resistant condition. PrLZ might enhance AR transactivation via a change of AR conformation that leads to promotion of AR nuclear translocation and suppression of AR degradation via modulating the proteasome pathway, which resulted in increased prostate-specific antigen expression and promoted PCa growth at the castration-resistant stage. Clinical PCa sample survey from same-patient paired specimens found increased PrLZ expression in castration-resistant PCa following the classical androgen deprivation therapy. Targeting the AR-PrLZ complex via ASC-J9® or PrLZ-siRNA resulted in suppression of PCa growth in various human PCa cells and in vivo mouse PCa models. Together, these data not only strengthen PrLZ roles in the transition from androgen dependence to androgen independence during the castration-resistant stage, but they may also provide a new potential therapy to battle PCa at the castration-resistant stage.
Introduction
Prostate cancer (PCa) is one of most commonly diagnosed malignancies in the USA (1) and the incidence is increasing in the Asian countries (2). Although androgen deprivation therapy (ADT) is the current standard treatment for the advanced PCa, most patients may fail to respond to treatment after an average of 12–18 months and tumors progress into the castration-resistant stage without a clear mechanism (3). Interestingly, immunohistochemical analyses have shown that most tumors at this castration-resistant stage continue to express the androgen receptor (AR) and the AR target gene prostate-specific antigen (PSA), suggesting that the AR signals may be still functional, regardless of ADT treatment (4,5).
Early studies proposed several mechanisms to explain such a puzzle. For example, overexpression/gene amplification of AR (6) or AR mutation to more active forms (7,8) could lead to a response at a relatively lower concentration of androgen after ADT (9,10). AR could also be activated by other steroid hormones and/or antiandrogens after an alteration of the expression ratio of AR to AR coregulators (11–14). Some growth factors and/or kinases may also alter the AR phosphorylation status to activate AR (15–18). However, no effective therapeutic approaches have been developed based on these mechanisms to successfully cure PCa at the castration-resistant stage.
The newly identified Prostate Leucine Zipper gene (PrLZ) (19) belongs to the tumor protein D52 family that has been reported to play important roles in exocytotic secretion and tumor progression (20). PrLZ contains a conserved domain of the D52 family, including a coiled-coil motif, multiple canonical serine/threonine phosphorylation sites and two PEST domains flanking the leucine zipper, which have been linked with regulating protein stability (21). Our previously reported PrLZ is highly expressed in PCa and can protect PCa cells from apoptosis induced by androgen deprivation, indicating its potential linkage to the progression of PCa (19,22,23). The detailed mechanism explaining how PrLZ contributes to PCa progression, however, remains unclear.
In this study, we found that PrLZ could function as an AR activator to transactivate AR target genes and increase AR-mediated PCa cell growth. PrLZ expression was increased after PCa developed into the castration-resistant stage. These new findings might provide us a reasonable explanation why PCa continues to grow and develop into the castration-resistant stage with little sensitivity to ADT treatment. Targeting this AR-PrLZ loop to suppress the PrLZ-mediated AR transactivation and AR-mediated PCa growth may be developed as a potential therapeutic approach to battle PCa at the castration-resistant stage.
Materials and methods
Plasmids, reagents and primers
VP16-AR, PSG5-AR, PGL3-ARE4-Luciferase, HA-tagged Ubiquitin, MDM2, pVP16-AR truncated domains and pCDNA3-Flag-tagged AR full length or functional domain plasmid constructs are available in our laboratory (24). Gal4-PrLZ, PcDNA3-Flag-PrLZ, PBABE-Puro-PrLZ, PGL3-PrLZ-promotor-luciferase and pM-PrLZ truncated domains were constructed according to the standard protocols. The primers for cloning human PrLZ 5kb promoter were described previously (25): forward: 5′-CGA CGC GTT ACA CTG ACA TAT TTG AG-3′, reverse: 5′-GGA CTC GAG TCC AGA CAA ACG CAG-3′. Quantitative PCR primers for PrLZ were as follows: forward: 5′-GAG ATG GAC TTA TAT GAG GAC TAC-3′, reverse: 5′-TTG CTG CTA ACA CTT GAG AC-3′. Small interfering RNA (siRNA) specific to PrLZ was designed and synthesized by Invitrogen. 5-Hydroxy-1,7-bis(3,4-dimethoxyphenyl)-1,4,6-heptatrien-3-one (ASC-J9®), first screened for the disruption of normal AR and its coregulators, was provided by Androscience (26). MG132 and AC-DEVD-CHO were purchased from Calbiochem. 5α-Dihydrotestosterone (DHT), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), collagenase I and the anti-Flag M2 antibody were purchased from Sigma. The anti-AR polyclonal antibodies, G122-77 and G122-25, were from BD Pharmingen™; anti-PSA, anti-AR (C-19) and anti-tubulin antibodies were from Santa Cruz; anti-PrLZ polyclonal antibody was kindly provided by Dr Ruoxiang Wang (Winship Cancer Center, Emory University).
Cell culture, transfections and growth assay
The human Pca, PC3, PC3-AR9, LNCaP and C4-2 cells were maintained in RPMI-1640 medium, and Cos-1 and TRAMP-C1 cells were maintained in Dulbecco’s modified Eagle’s medium. Both media contained 10% fetal calf serum. Cell transfections were performed as described previously by lipofectamine 2000 according to standard procedures (Invitrogen) (22). Cell growth rate was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide proliferation assay.
Western blot and immunoprecipitation and glutathione-S-transferase pull-down assay
Western blot and immunoprecipitation assays were performed as previously reported (22,27). Briefly, AR and PrLZ proteins were immunoprecipitated from RIPA lysates, and the immunoprecipitated proteins were eluted in the presence of 1% Triton X-100 and 0.2% sodium dodecyl sulfate. Western blotting on immunoprecipitates was performed using horseradish peroxidase labeled mAbs and detected using the enhanced chemiluminescent detection system (Amersham, Piscataway, NJ). The glutathione-S-transferase (GST) pull-down assay was described previously (27).
Mammalian two-hybrid assay and luciferase reporter assays
The interaction domain of AR and PrLZ were determined by mammalian two-hybrid assay. Briefly, HEK 293 cells were plated on 24-well dishes with 10% charcoal-stripped fetal bovine serum Dulbecco’s modified Eagle’s medium for 24h. Cells were cotransfected with pG5-Luc reporter plasmid and PM-PrLZ plus pVP16-AR-F (full length), pVP16-AR-N (N-terminal domain), pVP16-AR-D (DNA-binding domain), pVP16-AR-DL (DNA-binding domain plus ligand-binding domain) or pVP16-AR-L (ligand-binding domain). To determine PrLZ truncated domain interaction with full length AR, pG5-luciferase reporter plasmid and pVP16-AR plus different PrLZ truncated domain plasmids were transfected into HEK 293 cell as indicated in the figure legends. pRL-TK luciferase was transfected as the internal control. Luciferase activities were measured using GloMax 20/20 Luminometer (Promega, WI).
The luciferase reporter assay was performed according to our previous study (28). The cells were lysed and the luciferase activity was detected by the dual luciferase assay according to standard procedures (Promega). Briefly, cells were plated on 24-well dishes and transiently transfected with 0.6 μg PCDNA3-AR or/and 0.3 μg ARE4-Luc plus 1ng pRL-TK as internal control using lipofectamine 2000. All the plasmids DNA were normalized by PCDNA3 control vector before transfection. Cells were then harvested for the dual luciferase assay. Firefly luciferase activities were normalized to Renilla luciferase activities. All data are mean ± SE from at least three independent experiments.
Immunofluorescence staining
Immunofluorescence staining was performed as we described previously (22). The primary antibody against AR (C-19) was diluted at 1:250 for staining. Florescent staining signals were examined with a confocal microscope.
In vitro transcription translation and limited trypsinization assay
PrLZ and AR in vitro transcription and translation, in the presence of unlabeled or [35S]-labeled-methionine, were performed according to the manufacturer’s instruction (Promega SP6/T7 TNT kits). Limited trypsinization assays were performed as reported previously (29). In brief, 5 µl of AR labeled translation mixture was incubated at 4°C with 1 µl of PrLZ unlabeled translation mixture for 1h. Limited trypsinization was performed by addition of 5 µl of trypsin solution (20 or 100mg/ml) at 4°C for 15min. The reaction was stopped by adding 2× sodium dodecyl sulfate-sample buffer. The samples were electrophoresed in 13.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and autoradiography was performed overnight.
Human prostate cancer specimens and immunohistochemical analysis
The human androgen naive/sensitive and castration-resistant PCa specimens were originally obtained from PCa patients and stored in the archives of the Department of Urology, Xi’an Jiaotong University, China. The androgen-dependent patients were defined as first diagnosed prostate cancer and the castration-resistant patients were defined as recurrence of tumor following ADT by chemical or surgical approach. Of the 13 sets of cases, 5 out of 13 androgen-dependent patients had localized PCa and had prostatectomy. The rest of the androgen-dependent cases and all postcastration cases are metastatic cancer who underwent ultrasound-guided transrectum prostate biopsy or transurethral prostate resection. Use of the human specimens followed the regulations of the Institutional Review Board Committee of Xi’an Jiaotong University. Representative androgen dependent and refractory samples from the 13 sets of patients before and after castration were subjected to immunohistochemical examination for PrLZ and AR expression level during PCa progression. The primary antibodies of the rabbit anti-PrLZ and the rabbit anti-AR (C-19) were used for immunohistochemical staining (IHC) and the protein expressions were scored as negative (score = 1), weak (score =2), moderate (score= 3) or strong (score =4) using a system that has been validated previously. The positive staining signals were quantitated by Image J software.
Statistics
Results were analyzed by using the statistical software SPSS for windows 10.0 and presented as mean ± SE. Data P < 0.05 was considered the threshold value for statistical significance.
Results
PrLZ promotes PCa cells proliferation and AR transactivation during the the condition of castration-resistance stage
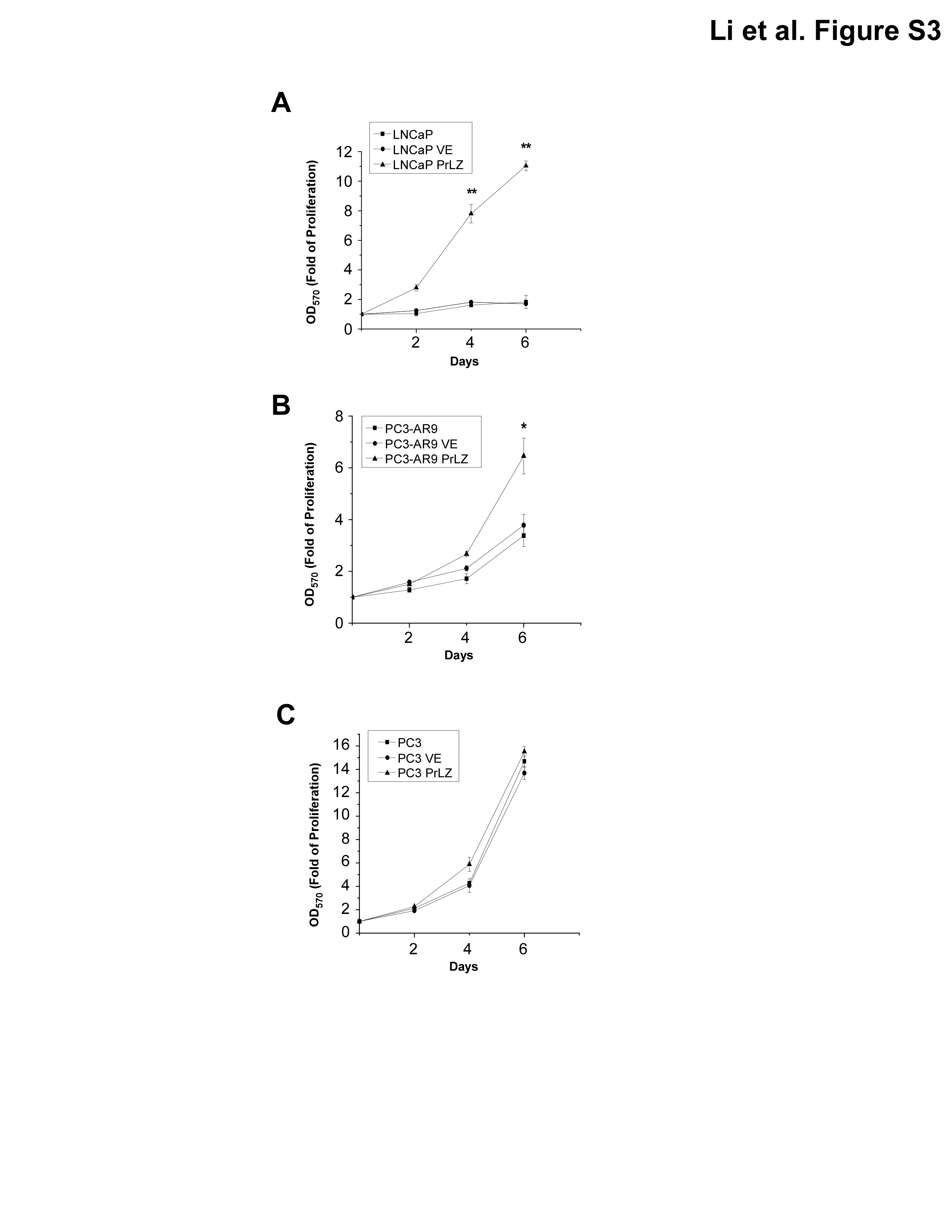
Previous studies found that overexpression of PrLZ might be able to increase PCa LNCaP cells growth, but detailed mechanisms remain unclear (19,22,30). In this study, we hypothesize that PrLZ may function through AR-mediated signals to promote PCa cells growth. We first stably transfected PrLZ into various human PCa cells and found increased PrLZ expression enhanced cell growth in AR-positive LNCaP and PC3-AR9 as compared with vector-transfected control cells in castration-resistant condition (1nM DHT) (Supplementary Figure 1A and B, available at Carcinogenesis Online). In contrast, we found little difference of cell growth in AR-negative PC3 cells after stably transfecting PrLZ (Supplementary Figure 1C, available at Carcinogenesis Online). These results suggested that addition of PrLZ might enhance the PCa cell growth via AR-mediated signals.
To further confirm the above conclusion that PrLZ functions through AR-mediated signals to promote PCa cell growth, we performed AR transactivation via ARE-luciferase reporter assays and found that addition of PrLZ resulted in enhanced AR transactivation in the presence of 1nM DHT in LNCaP and PC3-AR9 cells (Supplementary Figure 1D and E, available at Carcinogenesis Online). Furthermore, adding the PrLZ-siRNA to knockdown endogenous PrLZ resulted in suppression of AR transactivation in PCa C4-2 cells (Supplementary Figure 1F, available at Carcinogenesis Online).
Interestingly, we found enhanced AR transactivation could then lead to increased protein and mRNA expressions of PrLZ in the LNCaP and C4-2 cells (Supplementary Figure 1G and H, available at Carcinogenesis Online), suggesting a positive feed-back-loop regulation mechanism between PrLZ and AR, which was further confirmed at the transcriptional level showing addition of AR (Supplementary Figure 1I, available at Carcinogenesis Online) or PrLZ (Supplementary Figure 1J, available at Carcinogenesis Online) could result in increased luciferase reporter activity linked to the 5′ promoter of PrLZ. Further analysis by chromatin immunoprecipitation assay demonstrated that AR could directly bind to PrLZ promoter in vivo via showing the direct binding of AR with ARE (TGAACTgttTGTACC) located on the-345/-359 region of 5′ promoter of PrLZ (Supplementary Figure 1K, available at Carcinogenesis Online). We also confirmed this important finding that PrLZ could enhance AR binding to target genes using chromatin immunoprecipitation assay in LNCaP and castration-resistant C4-2 cells. Our data showed PrLZ could also bind to the promoter ARE regions of two other AR target genes, PSA and TMPRSS2 (Supplementary Figure 2, available at Carcinogenesis Online). Together, our data showed PrLZ knockdown could suppress the AR binding to PSA and TMPRSS2 promoter ARE region and addition of PrLZ could increase AR binding to PSA and TMPRSS2 promoter ARE regions in the castration-resistant condition (Supplementary Figure 2, available at Carcinogenesis Online).
Together, results from Supplementary Figures 1A–K and 2, available at Carcinogenesis Online, demonstrated that PrLZ could promote PCa cells proliferation via enhanced AR-mediated signals and the enhanced AR-mediated signals could then increase PrLZ expression via transcriptional regulation via a positive feed-back-loop mechanism.
PrLZ functions as AR activator to transactivate AR
To further confirm that PrLZ, and not 1nM DHT that exists in the castration-resistant condition, can transactivate AR, we then examined the above findings in charcoal-stripped serum condition that keep DHT concentration at nearly 0nM in the media. Western blot data showed that the addition of PrLZ enhanced AR transactivation, nearly 2-fold in LNCaP and PC3-AR9 cells (Figure 1A and 1B) and in a dosage-dependent manner in Cos-1 cells (Figure 1C). Consistently, knocking down endogenous PrLZ with PrLZ-siRNA resulted in suppression of AR transactivation in PCa C4-2 cells in charcoal-stripped serum condition (Figure 1D). Results from western blot analysis confirmed the above findings showing increased PrLZ in LNCaP cells enhanced PSA in charcoal-stripped serum condition (Figure 1E), and knockdown of PrLZ with PrLZ-siRNA resulted in reduced expression of AR target gene, PSA, in C4-2 cells (Figure 1F).
Fig. 1.

PrLZ enhances AR transactivation using ARE4-luciferase assay in charcoal-stripped serum medium (A–F). PrLZ enhances AR transactivation near 2.5-fold in stably transfected LNCaP (A) and PC3-AR9 (B), and also dosage-dependently increased AR transactivation in Cos-1 cells (C). PrLZ-siRNA inhibits AR transactivation in stably transfected C4-2 (D) cells. *P < 0.05 and **P < 0.01 versus vector control (VE). Values represent the mean ± SD of at least three independent experiments. PSA and AR expression levels were confirmed in overexpressed of PrLZ LNCaP cells (E) and knocked-down PrLZ C4-2 cells (F).
Together, results from Figure 1A–F suggest that PrLZ, and not 1nM DHT that exists in the castration-resistant condition, is able to transactivate PCa AR.
PrLZ interacts with AR directly
To dissect the potential mechanism(s) why PrLZ could transactivate AR in charcoal-stripped serum condition, we determined whether PrLZ could interact directly with AR. We transfected AR and Flag-tagged PrLZ in various cells, then cell lysate extracts were immunoprecipitated with anti-Flag or anti-AR antibodies, with normal IgG as a negative control. The results showed PrLZ was able to co-immunoprecipitate with AR in both LNCaP and PC3 cells as well as in Cos-1 cells (Figure 2A–C). Importantly, we confirmed that endogenous AR could interact with endogenous PrLZ using co-immunoprecipitation assay in PCa C4-2 cells in charcoal-stripped serum condition, suggesting PrLZ could bind AR in the castration-resistant PCa (Figure 2D).
Fig. 2.

The interaction between AR and PrLZ using immunoprecipitation assay in charcoal-stripped serum condition (A–I). Total lysates from LNCaP (A), PC3 (B) and Cos-1 (C) cells transiently cotransfected with AR and Flag-PrLZ, or from C4-2 cells (D), which have high expressions of endogenous AR and PrLZ, were immunoprecipitated and blotted with PrLZ and AR antibodies. The interaction of AR and PrLZ was confirmed by in vitro GST pull-down assay (E) or by mammalian two-hybrid assay in Cos-1 cells (F). The domains responsible for the interaction between truncated AR [Flag-tagged AR full length (AR-F), AR DNA-binding domain (AR-D), AR DNA-binding domain plus N-terminal (AR-DN), AR ligand-binding domain (AR-L)] and PrLZ were mapped in Cos-1 cells by immunoprecipitation assay. (G). The results were further confirmed by mammalian two-hybrid assay in Cos-1 cells by cotransfection of PrLZ and/or AR different truncated domain as indicated (H and I). **P < 0.01 versus vector control, and values represent the mean ± SD of at least three independent experiments.
We further applied in vitro GST pull-down and mammalian two-hybrid assays to confirm such interaction exists in PCa cells (Figure 2E and 2F). To dissect which domain within AR and PrLZ is responsible for AR-PrLZ interaction, we employed the mammalian two-hybrid interaction and co-immunoprecipitation assays. Our data demonstrated that PrLZ N-terminal PEST domain (aa 1–86) could interact with AR ligand-binding domain (Figure 2G–I).
PrLZ interacts with AR and induces the alteration of AR conformation
What are the consequences of PrLZ interaction with AR? We first asked if PrLZ is similar to other ligands/activators that could change AR conformation upon binding to AR. Using protease digestion assay for conformation change (29), we found that in the absence of ligand the AR is completely degraded by trypsin (Figure 3A, lane 3). Addition of ligand-DHT then induced the AR conformation change that led to a different trypsin-digested pattern (Figure 3A, lanes 8 and 9). Interestingly, addition of PrLZ to bind to AR also led to a new trypsin-digested pattern, suggesting PrLZ could function as an activator to transactivate AR via binding to AR and alter its conformation.
Fig. 3.

PrLZ induces the alteration of AR conformation and promotes nuclear translocation of AR (A–G). AR conformation alteration was analyzed by limited trypsin digestion in the presence of 10nM DHT or activator PrLZ (A). Results showed new different AR digested peptides (labeled as #, PrLZ + AR) or increased intensity of AR digested peptides (labeled as *, 10nM DHT + AR) as compared with control (labeled as $, AR). AR translocation was analyzed by immunofluorescence staining of AR. Cells were serum starved for 24h prior to performing the experiment. Data showed that PrLZ promotes AR translocation from cytosol to nuclei in PrLZ stably transfected LNCaP cells (LNCaP-PrLZ) (B), and in PC3-AR9-PrLZ (D) cells compared with vector control-transfected cells. In contrast, AR nuclear staining was significantly reduced in C4-2 cells transfected with PrLZ-siRNA (F) compared with scramble RNA-transfected control cells. 4′,6-Diamidino-2-phenylindole DNA staining indicates the locations of cell nuclei. Merged images of 4′,6-diamidino-2-phenylindole and AR IHC staining photos showed increased AR nuclear localization. Magnification ×400. The quantitation of PrLZ promoted AR nuclear translocation was further determined by assaying AR protein levels in nuclear versus cytoplasmic fractions in LNCaP (C), PC3-AR9 (E) and C4-2 cells (G).
PrLZ promotes AR translocation from cytoplasm to nuclei
We hypothesized that the changed AR protein structure upon PrLZ binding (Figure 3A) could influence the cytosol–nuclear distribution of AR and consequently induce the AR transactivation. In both LNCaP and PC3-AR9 cells, our data showed that the majority of AR remained in the cytoplasm in charcoal-stripped serum condition, which is consistent with previous reports (29,31). However, addition of PrLZ led to AR translocation from cytoplasm to nuclei in the LNCaP and PC3-AR9 cells (Figure 3B–E). In contrast, using siRNA to knockdown endogenous PrLZ resulted in significant reduction of the AR nuclear localization in C4-2 cells (Figure 3F and 3G). Together, results from Figure 3A–G clearly pointed out that binding of PrLZ to AR led to changed AR conformation, and translocation of AR from cytoplasm to nuclei in the charcoal-stripped serum condition.
PrLZ suppresses AR degradation through affecting proteasome-degradation pathway
PrLZ interaction with AR not only changes AR conformation and promotes AR nuclear translocation, it may also alter AR expression at the protein level. We found that addition of PrLZ in LNCaP and PC3-AR9 cells or PrLZ-siRNA in C4-2 cells resulted in little change in AR mRNA expression in charcoal-stripped serum condition (data not shown). However, as shown by western blot analysis, AR protein expression was altered after addition of PrLZ or PrLZ-siRNA following 30 μg/ml cycloheximide treatment. The increased PrLZ expression significantly suppressed AR degradation in LNCaP and PC3-AR9 cells (Figure 4A and 4B), and knockdown of endogenous PrLZ by PrLZ-siRNA promoted AR degradation in C4-2 cells (Figure 4C).
Fig. 4.

PrLZ suppresses AR degradation through the proteasome-dependent manners (A–F). The AR protein degradation was assayed by western blot in stably transfected PrLZ cDNA or siRNA construct LNCaP (A), PC3-AR9 (B) and C4-2 (C) cells followed by treatment with 30 µg/ml cycloheximide. Quantitative results were analyzed by linear regression and plotted. For assaying AR protein degradation mechanisms (D), C4-2 cells stably transfected with PrLZ siRNA were treated with 10 µM MG132, 10 µM AC-DEVD-CHO or both for 4h, and then harvested for western blot assay. For AR ubiquitination assay, Cos-1 cells were cotransfected with AR plus PrLZ or Mdm2 in the presence or absence of HA-Ubiquitin (HA-Ub) vector, and cells were then harvested for ubiquitination assay (E) and immunoprecipitation assay (F).
Early studies suggested that AR could be degraded through two independent pathways, one involving the proteasome pathway (32) and the other involving caspase 3 signals (33). We first targeted PrLZ via stable transfection with PrLZ-siRNA in C4-2 cells treating with 10 µM MG132, a specific proteasome inhibitor (34), or 10 µM AC-DEVD-CHO, a specific caspase 3/7 inhibitor (35), or with both for 4h under serum-free conditions. Results showed MG132 alone or MG132 plus AC-DEVD-CHO could partially increase AR protein, whereas AC-DEVD-CHO alone failed to alter AR degradation (Figure 4D).
To further dissect the detailed mechanisms of PrLZ-regulated AR degradation mechanisms, we cotransfected AR and/or PrLZ plasmids into Cos-1 cells. Our results clearly showed that PrLZ could bind to AR and stabilize AR through competing for the binding of MDM2 E3 ligase toward AR and consequently reduce the AR ubiquitination (Figure 4E and 4F). This indicates PrLZ can suppress AR degradation partially via a proteasome-mediated pathway.
PrLZ enhances PCa cell growth in charcoal-stripped serum condition
Results from Figures 1–4 clearly demonstrated that PrLZ could function as an activator of AR in charcoal-stripped serum condition to promote AR target gene PSA expression, which includes changed AR transformation, enhanced AR nuclear translocation and suppressed AR degradation. These findings raise an interesting hypothesis that PrLZ may play important roles during transition from androgen-dependent to androgen-independent PCa growth that occurs in most PCa patients during their later castration-resistant stage. We first examined PrLZ effect on PCa growth and found enhanced AR transactivation could consequently increase PCa cell growth in the charcoal-stripped serum condition. As shown in Supplementary Figure 3A and B, available at Carcinogenesis Online, stable transfection of PrLZ into AR-positive LNCaP or PC3-AR9 cells led to increased cell growth as compared with those cells transfected with vector control in charcoal-stripped serum condition. As expected, we found little difference of cell growth in AR-negative PC3 cells after stably transfecting PrLZ (Supplementary Figure 3C, available at Carcinogenesis Online). Using in vitro PCa tumorigenicity assay, we further confirmed the above findings that PrLZ could promote PCa cell growth in charcoal-stripped serum condition (Supplementary Figure 4A–D, available at Carcinogenesis Online).
Increased PrLZ expression in PCa patients’ tumors after ADT treatment
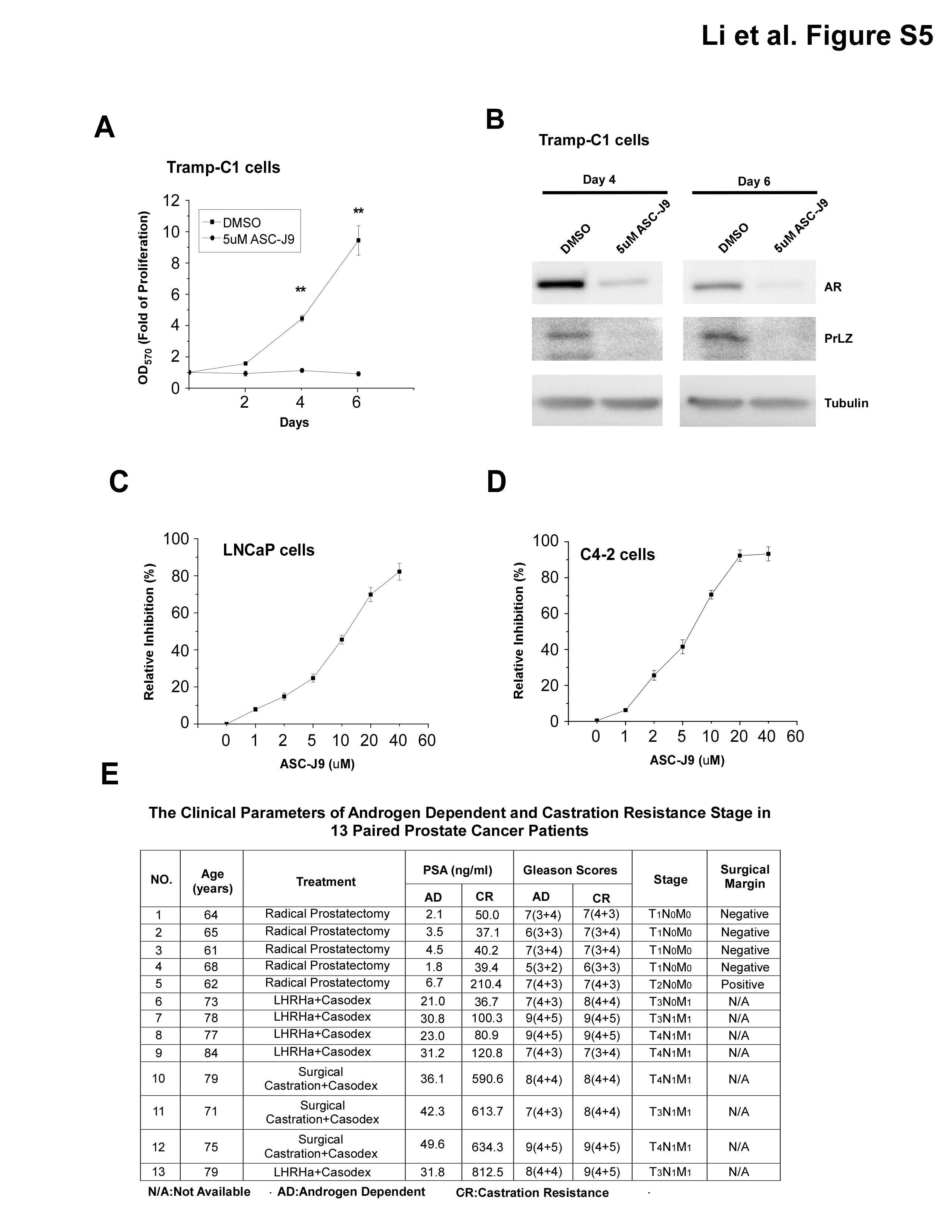
To further support our conclusion generated mainly from various human PCa cell lines, we applied IHC staining to compare the expression of PrLZ in 13 sets of human PCa samples from the same patient before and after ADT treatment. The clinical parameters of those paired patients are as shown in Supplementary Figure 5E, available at Carcinogenesis Online. Results shown in Figure 5A–C showed 13 sets of human PCa samples had increased PrLZ and PSA expression after ADT treatment.
Fig. 5.

Increased PrLZ expression in clinical PCa patients’ tumors and cells (A–D). IHC staining and HE staining shows that PrLZ and AR staining in 13 sets of primary PCa specimens before and after ADT treatment from the same patients. (A): Lanes 1 and 2 show the representative patient’s specimen progressed from androgen dependent (AD) to castration resistant (CR), lane 3 shows PrLZ expression is highly related to the expression of AR at the castration-resistant stage, magnification ×200. Quantitative data of AR and PrLZ (B) expression and serum PSA (C) in the 13 sets of specimens were summarized by analysis of variance analysis. The increased expression of PrLZ were confirmed in AR-positive castration-resistant C4-2 cells compared with those in hormone-dependent LNCaP and LAPC4 cells cultured in serum-free condition for 24h (D).
In addition, we found that PrLZ expression was much higher in AR-positive castration-resistant C4-2 cells, which originated from a LNCaP xenograft tumor in a nude mouse after castration compared with those found in hormone-sensitive LNCaP and LAPC4 cells in charcoal-stripped serum condition (Figure 5D).
Together, results from Figure 5A–C demonstrated that enhanced PrLZ, AR and PSA expression after ADT were shown in castration-resistance PCa patients, suggesting that the increased PrLZ might transactivate AR and promote AR-mediated PCa cell growth even in the Castration Resistance Prostate Cancer (CRPC) stage, which at least partially explained why AR was continuously transactivated in the CRPC patients even though at low level castration androgen levels.
In summary, results from Figures 1–5 provide a reasonable explanation at the molecular level to explain why tumors could continue to grow at castration-resistant stage even when PCa patients continue to receive ADT treatment maintaining androgen at the castration level.
Targeting PrLZ-AR as a new potential therapeutic approach to battle PCa
Results from Figures 1–5 and Supplementary Figures 1–4, available at Carcinogenesis Online, concluded that PrLZ could transactivate, translocate and degradate AR that resulted in the promotion of AR-mediated PCa cell growth. Increased PrLZ expression at the castration-resistant stage further suggested that the PrLZ might promote PCa cell growth during ADT, and could be an important factor contributing to PCa progression into the castration-resistant stage. Because current ADT mainly targets androgens via reduction/prevention of androgens binding to AR (5,36,37), we hypothesize that blocking the PrLZ-AR regulation loop might represent a new potential therapeutic target to battle PCa.
We chose PCa C4-2 cells to test this hypothesis since C4-2 cells represent PCa cells at the castration-resistant stage with cell growth in an androgen-independent manner (38). We first applied PrLZ-siRNA to suppress endogenous PrLZ expression (Figure 1F) and examined its effects on C4-2 cell growth. As shown in Figure 6A–C, reduction of PrLZ via PrLZ-siRNA led to suppression of C4-2 cell growth in charcoal-stripped serum condition, or in the presence of 1nM DHT (castration-resistant condition after ADT), or 10nM DHT (before ADT and castration-resistant condition), suggesting targeting PrLZ to suppress PCa may represent a new potential therapeutic approach to battle PCa at the castration-resistant stage.
Fig. 6.

Targeting PrLZ-AR complex as a new potential therapeutic approach to battle PCa (A–I). Knockdown of endogenous PrLZ by siRNA suppresses C4-2 cell growth in the media containing 10% charcoal-stripped serum in the absence (A) or presence of 1nM (B) or 10nM (C) DHT. Treatment with 5 µM ASC-J9® can inhibit AR-positive cells growth in LNCaP cells (D) and PC3-AR9 cells (E). Moreover, 5 µM ASC-J9® treatment resulted in more suppression effect than PrLZ-siRNA in the C4-2 cells and simultaneous addition of ASC-J9® and PrLZ-siRNA led to better suppression effects (F). *P < 0.05 and **P < 0.01 versus control cells (VE). All values represent the mean ± SE of at least three independent experiments. Western blot results showed ASC-J9® mediated PCa cells growth inhibition was via degradation of AR protein and inhibition of PrLZ expression as a sequential mechanism event in LNCaP (G), PC3-AR9 (H) and C4-2 (I) cells.
We then applied an AR degradation enhancer, ASC-J9®, that could promote AR degradation in selective cells with little side effects (39–42), to examine the ASC-J9®-induced AR degradation effects on PrLZ-AR-mediated PCa cell growth. We found 5 µM ASC-J9® (the IC50 in LNCaP and C4-2 are about 5 µM, Supplementary Figure 5C and D, available at Carcinogenesis Online) could effectively suppress PCa cell growth in LNCaP and PC3-AR9 cells transfected with PrLZ (Figure 6D and 6E). In C4-2 cells that are rich with endogenous PrLZ expression, adding 5 µM ASC-J9® resulted in better suppression effect than PrLZ-siRNA and simultaneous addition of ASC-J9® and PrLZ-siRNA led to the best suppression effects (Figure 6F). Western blot analyses also confirmed that 5 µM ASC-J9® could degrade AR-PrLZ complex in LNCaP, PC3-AR9 and C4-2 cells (Figure 6G–I).
We then extended these in vitro human PCa cell line studies into various in vivo PCa studies. We first applied the TRAMP mouse model, which spontaneously develops prostate tumors to test the drug effect on the AR-PrLZ complex and cancer growth (43). Prior to performing the in vivo PCa study, we tested whether ASC-J9® treatment influenced the cell growth and AR-PrLZ complex formation in the mouse TRAMP-C1 cells isolated from TRAMP PCa. Indeed, our results showed that 5 µM ASC-J9® could effectively suppress cell growth and degrade AR-PrLZ complex in this TRAMP-C1 cells (Supplementary Figure 5A and B, available at Carcinogenesis Online).
We then subcutaneously xenografted TRAMP PCa tumors into SCID mice and injected ASC-J9® intraperitoneal at 100mg/kg body weight every other day for 4 weeks. The results showed ASC-J9® could suppress TRAMP PCa growth in vivo (Supplementary Figure 6A, available at Carcinogenesis Online). We then orthotopically implanted TRAMP PCa tissues into anterior prostates of SCID mice and injected with ASC-J9® intraperitoneal at 100mg/kg body weight every other day for 4 weeks. Sixteen tumors were harvested from each group. We obtained consistent results showing ASC-J9® could suppress the growth of orthotopically implanted TRAMP PCa (Supplementary Figure 6B, available at Carcinogenesis Online).
In addition to in vivo mouse PCa models, we examined the effect of ASC-J9® on AR-PrLZ complex function using the human PCa tumors. Human PCa tumors were established from human PCa patients and grafted under renal capsule in SCID mice. Six weeks after implantation, ASC-J9® was injected intraperitoneal and PSA was used to monitor the tumor growth (data not shown). Twenty tumors were implanted in each test group. After 4 weeks of treatment, our results confirmed ASC-J9® could effectively suppress human PCa tumor growth in vivo (Supplementary Figure 6C, available at Carcinogenesis Online).
We also applied IHC staining and western blot analyses to confirm that 5 µM ASC-J9® could effectively degrade AR-PrLZ complex in harvested PCa tumors (Supplementary Figure 6D–I, available at Carcinogenesis Online).
Together, results from Figure 6A–I and Supplementary Figures 5 and 6, available at Carcinogenesis Online, all demonstrated that we could apply ASC-J9® and/or PrLZ-SiRNA to suppress PCa tumor growth before and after castration-resistant stages.
Discussions
Previous studies showed that increased expression of PrLZ promoted PCa LNCaP cells growth in the normal androgen conditions (19,22). In this report, we defined that PrLZ could also promote PCa cell proliferation in the castration-resistance androgen condition, which indicates that PrLZ may play an important role in different stages of PCa progression, including in CRPC stage.
PrLZ belongs to the tumor protein D52 family, and the C-terminal region of PrLZ is identical to tumor protein D52. In the N-terminal region, however, PrLZ extends with a unique sequence of 46 amino acid residues and a potential PEST domain (49–80 amino acid) (19,44), the latter are involved in protein degradation. Taking advantage of this structural feature, we showed that PrLZ N-terminal unique sequence and PEST domain could interact with AR ligand-binding domain, which could further transactivate and translocate AR in the castration androgen level.
It has been clearly elucidated that upon activation by androgen, AR may then change the conformation to interact with specific DNA sequences, located in the flanking regions of target genes, resulting in modulation of the expression of these genes (45). Androgen binding to AR may change the receptor structure in a manner that alter the sensitivity of proteases to degrade AR (46). Our demonstration that PrLZ binding to AR can alter the trypsin-digested pattern suggested PrLZ could function as an activator to transactivate AR to modulate its downstream gene expression.
Early studies suggested that AR could be degraded by two independent pathways: one involved proteasome pathways and the other involved caspase 3 signals (47). The ubiquitin–proteasome pathway has been studied intensively in recent years and represents a very common degradation pathway for a large variety of proteins (48). Mdm2, a E3 ligase gene, regulates AR stabilization by inducing AR ubiquitylation and subsequent degradation by the proteasome (27). From our Co-IP assay, we have demonstrated that PrLZ PEST domain can bind to the AR and compete for binding with Mdm2, which leads to the decreased ubiquitination of AR and further stabilization of AR. However, the detailed information as to how these three proteins interact may need further investigation.
A previous study was also showed that PrLZ expression increases from primary localized tumors to bone metastasis and also increased from 38.9% in hormone naive metastasis to 52.8% in CRPC PCa tumors through tissue microarray assay (49). We confirmed this conclusion using IHC for comparing the precious the same patients localized PCa before and after ADT therapy, since the genetics background is almost identical. As expected, our present results show the increased PrLZ expression in 13/13 cases. This analysis supported the idea of a progressive enhancement of abnormal PrLZ expression during PCa progression, and that the extent of the abnormal PrLZ expression was positively correlated to the some population clinical progression of PCa patients. We noticed that, in addition to the overexpression of PrLZ, AR was found to high expression and constitutive activation in nucleus at CRPC patients after ADT as compared with before ADT, which is in agreement with previous studies (4,50–52). Interestingly, we found PrLZ expression is highly correlated with AR in PCa tissues after ADT, these results strongly support the explanation of the positive regulation loop between PrLZ and AR. It also explains why PrLZ is increased in CRPC stage after ADT therapy. The limitation of this is that further immunohistochemical analysis of larger same patient specimens before and after castration is needed to characterize the relationship of PrLZ and AR and tumor progression.
A fundamental issue in CRPC is that AR continues to function despite androgen is suppressed to castrate levels (4,5). ADT to suppress/prevent androgens binding to AR may prolong progression on an average of 12–18 months and tumors may again progress into CRPC stage and become incurable (3). Targeting AR instead of blocking androgen binding to AR may become another alternative strategy. In this study, we identified PrLZ may go through AR signaling-mediated PCa progression, suggesting target AR-PrLZ interaction to suppress the PrLZ-enhanced AR-mediated androgen-independent PCa growth may be developed as potential therapeutic approach to battle PCa at CRPC stage. Specific reduction of PrLZ via PrLZ-siRNA led to suppression of androgen-independent C4-2 cell growth in the castration-resistant condition, suggesting targeting PrLZ to suppress PCa may represent a new potential therapeutic approach to battle PCa at CRPC stage. We found applied ASC-J9 for the disruption of normal AR and its AR-ARA55 and/or AR-ARA70 coregulators (26) that resulted in AR degradation in selective cells (39–42), which could dramatically inhibit both AR and/or PrLZ positive PCa cells proliferation instead of AR-negative cells. More importantly, ASC-J9® has few side effects and mice have normal serum testosterone and normal sexual activity. Our results may provide potential ways that apply ASC-J9® and/or PrLZ-siRNA to suppress PCa tumor growth at castration-resistant stage.
In summary, results from these studies clearly demonstrated that PrLZ could interact with AR and induce AR transactivation that leads to increased PCa growth. Targeting the AR-PrLZ loop via ASC-J9® or PrLZ-siRNA could be developed as a new potential therapy to battle PCa before and after the castration-resistant stage.
Supplementary material
Supplementary Figures 1–6 and Materials and Methods can be found at http://carcin.oxfordjournals.org/
Funding
National Natural Science Foundation of China (NSFC 81130041, 81072107); National Program on Key Basic Research Project (973 Program NO.2012CB518305); National Institutes of Health (CA122295); George H. Whipple Professorship Endowment at the University of Rochester; Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) at China Medical University.
Conflict of Interest Statement: None declared.
Supplementary Material
Acknowledgements
We thank Dr See-Tong Pang (Division of Uro-oncology, Department of Surgery, Chang Gung Memorial Hospital, Taoyuan, Taiwan) for providing human PCa specimens.
Glossary
Abbreviations:
- ADT
androgen deprivation therapy
- AR
androgen receptor
- DHT
dihydrotestosterone
- IHC
immunohistochemical
- PCa
prostate cancer
- PrLZ
Prostate Leucine Zipper
- PSA
prostate-specific antigen
- siRNA
small interfering RNA
References
- 1. Cutler J.A., et al. (2006). Leading causes of death in the United States. JAMA, 295, 383–4; author reply 384 . [DOI] [PubMed] [Google Scholar]
- 2. Sim H.G., et al. (2005). Changing demography of prostate cancer in Asia. Eur. J. Cancer, 41, 834–845 . [DOI] [PubMed] [Google Scholar]
- 3. Miyamoto H., et al. (2004). Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate, 61, 332–353 . [DOI] [PubMed] [Google Scholar]
- 4. Chen C.D., et al. (2004). Molecular determinants of resistance to antiandrogen therapy. Nat. Med., 10, 33–39 . [DOI] [PubMed] [Google Scholar]
- 5. Niu Y., et al. (2010). Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene, 29, 3593–3604 . [DOI] [PubMed] [Google Scholar]
- 6. Visakorpi T., et al. (1995). In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet., 9, 401–406 . [DOI] [PubMed] [Google Scholar]
- 7. Marcelli M., et al. (2000). Androgen receptor mutations in prostate cancer. Cancer Res., 60, 944–949 . [PubMed] [Google Scholar]
- 8. Bergerat J.P., et al. (2009). Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Hum. Mutat., 30, 145–157 . [DOI] [PubMed] [Google Scholar]
- 9. Gregory C.W., et al. (2001). Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res., 61, 2892–2898 . [PubMed] [Google Scholar]
- 10. Mohler J.L. (2008). Castration-recurrent prostate cancer is not androgen-independent. Adv. Exp. Med. Biol., 617, 223–234 . [DOI] [PubMed] [Google Scholar]
- 11. Chmelar R., et al. (2007). Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer, 120, 719–733 . [DOI] [PubMed] [Google Scholar]
- 12. Heinlein C.A., et al. (2002). Androgen receptor (AR) coregulators: an overview. Endocr. Rev., 23, 175–200 . [DOI] [PubMed] [Google Scholar]
- 13. Yeh S., et al. (1998). From estrogen to androgen receptor: a new pathway for sex hormones in prostate. Proc. Natl. Acad. Sci. U.S.A., 95, 5527–5532 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyamoto H., et al. (1998). Promotion of agonist activity of antiandrogens by the androgen receptor coactivator, ARA70, in human prostate cancer DU145 cells. Proc. Natl. Acad. Sci. U.S.A., 95, 7379–7384 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craft N., et al. (1999). A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med., 5, 280–285 . [DOI] [PubMed] [Google Scholar]
- 16. Papatsoris A.G., et al. (2007). The power and promise of “rewiring” the mitogen-activated protein kinase network in prostate cancer therapeutics. Mol. Cancer Ther., 6, 811–819 . [DOI] [PubMed] [Google Scholar]
- 17. Guo Z., et al. (2006). Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell, 10, 309–319 . [DOI] [PubMed] [Google Scholar]
- 18. Qiu Y., et al. (1998). Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature, 393, 83–85 . [DOI] [PubMed] [Google Scholar]
- 19. Wang R., et al. (2004). PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Res., 64, 1589–1594 . [DOI] [PubMed] [Google Scholar]
- 20. Boutros R., et al. (2004). The tumor protein D52 family: many pieces, many puzzles. Biochem. Biophys. Res. Commun., 325, 1115–1121 . [DOI] [PubMed] [Google Scholar]
- 21. Byrne J.A., et al. (1996). Definition of the tumor protein D52 (TPD52) gene family through cloning of D52 homologues in human (hD53) and mouse (mD52). Genomics, 35, 523–532 . [DOI] [PubMed] [Google Scholar]
- 22. Li L., et al. (2009). PrLZ expression is associated with the progression of prostate cancer LNCaP cells. Mol. Carcinog., 48, 432–440 . [DOI] [PubMed] [Google Scholar]
- 23. Zhang D., et al. (2011). PrLZ protects prostate cancer cells from apoptosis induced by androgen deprivation via the activation of Stat3/Bcl-2 pathway. Cancer Res., 71, 2193–2202 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yeh S., et al. (1999). Differential induction of androgen receptor transactivation by different androgen receptor coactivators in human prostate cancer DU145 cells. Endocrine, 11, 195–202 . [DOI] [PubMed] [Google Scholar]
- 25. Wang J., et al. (2007). Identification and characterization of the novel human prostate cancer-specific PC-1 gene promoter. Biochem. Biophys. Res. Commun., 357, 8–13 . [DOI] [PubMed] [Google Scholar]
- 26. Ohtsu H., et al. (2002). Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J. Med. Chem., 45, 5037–5042 . [DOI] [PubMed] [Google Scholar]
- 27. Lin H.K., et al. (2002). Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J., 21, 4037–4048 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niu Y., et al. (2008). Tissue prostate-specific antigen facilitates refractory prostate tumor progression via enhancing ARA70-regulated androgen receptor transactivation. Cancer Res., 68, 7110–7119 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuil C.W., et al. (1995). Ligand-induced conformational alterations of the androgen receptor analyzed by limited trypsinization. Studies on the mechanism of antiandrogen action. J. Biol. Chem., 270, 27569–27576 . [DOI] [PubMed] [Google Scholar]
- 30. Zhang H., et al. (2007). PC-1/PrLZ contributes to malignant progression in prostate cancer. Cancer Res., 67, 8906–8913 . [DOI] [PubMed] [Google Scholar]
- 31. Tyagi R.K., et al. (2000). Dynamics of intracellular movement and nucleocytoplasmic recycling of the ligand-activated androgen receptor in living cells. Mol. Endocrinol., 14, 1162–1174 . [DOI] [PubMed] [Google Scholar]
- 32. Sheflin L., et al. (2000). Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem. Biophys. Res. Commun., 276, 144–150 . [DOI] [PubMed] [Google Scholar]
- 33. Lin H.K., et al. (2004). Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol. Endocrinol., 18, 2409–2423 . [DOI] [PubMed] [Google Scholar]
- 34. Tsubuki S., et al. (1996). Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem., 119, 572–576 . [DOI] [PubMed] [Google Scholar]
- 35. Nicholson D.W., et al. (1995). Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature, 376, 37–43 . [DOI] [PubMed] [Google Scholar]
- 36. Miyamoto H., et al. (2005). Inhibition of the Akt, cyclooxygenase-2, and matrix metalloproteinase-9 pathways in combination with androgen deprivation therapy: potential therapeutic approaches for prostate cancer. Mol. Carcinog., 44, 1–10 . [DOI] [PubMed] [Google Scholar]
- 37. Miyamoto H., et al. (2005). Does androgen deprivation improve treatment outcomes in patients with low-risk and intermediate-risk prostate cancer? Nat. Clin. Pract. Oncol., 2, 236–237 . [DOI] [PubMed] [Google Scholar]
- 38. Wu H.C., et al. (1994). Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int. J. Cancer, 57, 406–412 . [DOI] [PubMed] [Google Scholar]
- 39. Miyamoto H., et al. (2007). Promotion of bladder cancer development and progression by androgen receptor signals. J. Natl. Cancer Inst., 99, 558–568 . [DOI] [PubMed] [Google Scholar]
- 40. Yang Z., et al. (2007). ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat. Med., 13, 348–353 . [DOI] [PubMed] [Google Scholar]
- 41. Lai J.J., et al. (2009). Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J. Clin. Invest., 119, 3739–3751 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu M.H., et al. (2010). Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci. Transl. Med., 2, 32ra35 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gingrich J.R., et al. (1996). A transgenic mouse prostate cancer model. Toxicol. Pathol., 24, 502–504 . [DOI] [PubMed] [Google Scholar]
- 44. Wang R., et al. (2009). Transcription variants of the prostate-specific PrLZ gene and their interaction with 14-3-3 proteins. Biochem. Biophys. Res. Commun., 389, 455–460 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans R.M. (1988). The steroid and thyroid hormone receptor superfamily. Science, 240, 889–895 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kallio P.J., et al. (1994). Agonists, but not antagonists, alter the conformation of the hormone-binding domain of androgen receptor. Endocrinology, 134, 998–1001 . [DOI] [PubMed] [Google Scholar]
- 47. Lin H.K., et al. (2002). Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J. Biol. Chem., 277, 36570–36576 . [DOI] [PubMed] [Google Scholar]
- 48. Pickart C.M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem., 70, 503–533 . [DOI] [PubMed] [Google Scholar]
- 49. Wang R., et al. (2007). PrLZ is expressed in normal prostate development and in human prostate cancer progression. Clin. Cancer Res., 13, 6040–6048 . [DOI] [PubMed] [Google Scholar]
- 50. van der Kwast T.H., et al. (1991). Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer, 48, 189–193 . [DOI] [PubMed] [Google Scholar]
- 51. Ruizeveld de Winter J.A., et al. (1991). Androgen receptor expression in human tissues: an immunohistochemical study. J. Histochem. Cytochem., 39, 927–936 . [DOI] [PubMed] [Google Scholar]
- 52. Ruizeveld de Winter J.A., et al. (1994). Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol., 144, 735–746 . [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}