

Abstract
Side population (SP) cells are previously identified from bone marrow based on their capacity to efflux of the fluorescent dye Hoechst 33342. Recent studies demonstrate that SP cells isolated from various cancer cell lines and primary tumors possess stem-cell-like properties. Thus, targeting tumor SP cells may provide new strategies for treatment in clinic. We previously showed that 1,3,8-trihydroxy-6-methylanthraquinone (emodin), a reactive oxygen species (ROS) generator, enhanced sensitivity of gallbladder cancer SGC-996 cells to cisplatin (CDDP) via generation of ROS and downregulation of multidrug-resistance-associated protein 1 (MRP1). To determine whether emodin also acts effectively on cancer stem cells of gallbladder carcinoma, we use SP cells as a model of cancer stem-cell-like cells. Here, we found that emodin, via ROS-related mechanism and suppressing the function of ATP-binding cassette super-family G member (ABCG2), which is known to be associated with Hoechst dye efflux activity of SP cells, not only reduced the ratio, inhibited clone formation, and eliminated sphere formation of SP cells effectively, but also promoted obviously the intracellular accumulation of doxorubicin, the main substrate of the efflux pump ABCG2. In addition, emodin could sensitize CDDP, via inhibition of expression of ABCG2, to overcome chemoresistance of SP cells. Importantly, similar to the experiment in vitro, emodin/CDDP co-treatment in vivo suppressed the tumor growth derived from SP cells through downregulating ABCG2 expression. Our results suggest that emodin is an effective agent targeting cancer stem-like SP cells of gallbladder carcinoma, either alone or acts as a chemotherapy enhancer.
Introduction
New concepts of “cancer stem cell (CSC) or tumor-initiating cell” have proposed that tumors contain a small subpopulation of cells with self-renew, multiplex differentiation, unlimited proliferation, and high resistance to chemotherapy and radiation, which play important roles in the occurrence, development, and infiltration of tumors [1–4]. In the process of clinical treatments for many malignant tumors, this subset of CSCs can effectively avoid the effects of chemotherapeutic drugs, becoming the roots of tumor recurrence and metastasis. Especially for gallbladder carcinoma, which is the most malignancy of biliary tract tumors with poor prognosis and dismal survival period, chemotherapy is still an important treatment for patients with advanced malignant tumors [5,6]. It is desired to overcome drug resistance of gallbladder cancer cells, particularly of CSCs. Therefore, searching effective chemotherapeutic agents or alternative chemotherapies for gallbladder CSCs may provide new strategies and ideas for treatment in clinic.
Dye exclusion is a valuable technique used in isolating and identifying CSCs, based on the activity of ABC transport, such as ABCG2, yielding a side population (SP) that retains less of the fluorescent DNA-binding dye Hoechst 33342 [7,8]. Originally identified as murine hematopoietic stem cells from bone marrow [9], SP cells have been observed in several mammalian malignant tumor tissues and well-established cancer cell lines [10–16]. Moreover, many studies reveal that SP cells have stem-cell-like characteristics, suggesting that SP sorting can enrich CSCs [11–16].
1,3,8-Trihydroxy-6-methylanthraquinone (emodin) is a kind of natural anthraquinone contained in the traditional Chinese herbal medicines. Our group has previously found that emodin promoted arsenic-trioxide-induced apoptosis in various cancer cell types in a reactive oxygen species (ROS)–dependent manner in vitro and in vivo [17–19]. We have also demonstrated that emodin is recognized as an effective adjunctive to improve efficacy of cisplatin (CDDP) in prostate cancer cells with over-activated hypoxia inducible factor-1 (HIF-1) and potent multiple drug resistance (MDR) [20]. Of more interest is that emodin is found to enhance sensitivity of gallbladder cancer cell line SGC-996 to CDDP via generation of ROS and multidrug-resistance-associated protein 1 (MRP1) downregulation [21]. Based on chemoresistance of cancer stem-like SP cells, we reason that the chemosensitizing effect of emodin may be mediated through targeting gallbladder cancer stem-like SP cells. Thus, we further explore whether emodin, improving intracellular ROS levels, influences stem cell characteristics, including survival ability of gallbladder cancer stem-like SP cells. In addition, we attempt to investigate whether emodin can sensitize CDDP in SP cells of gallbladder carcinoma. In the current study, we found that emodin could effectively affect stem cell characteristics and weaken survival ability of gallbladder cancer stem-like SP cells via a mechanism of enhancement of intracellular ROS levels and inactivation of the pump ABCG2. Further, our results suggested that emodin could sensitize CDDP, via inhibition of expression of ABCG2, to overcome chemoresistance of gallbladder cancer stem-like SP cells.
Materials and Methods
Cell culture
SGC-996 cells were cultured in RPMI-1640 (GibcoBRL) supplemented with 10% newborn calf serum (Biochrom AG). GBC-SD cells were cultured in Dulbecco's modified Eagle's medium (GibcoBRL), containing 10% newborn calf serum. All media were supplemented with 100 U/mL penicillin and 100 mg/L streptomycin. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
SP cell sorting from gallbladder carcinoma cell lines
For SP analysis, cells were trypsinized and resuspended in ice-cold Hank's balanced salt solution (Invitrogen) supplemented with 2% FBS at a concentration of 1×106 cells/mL. Hoechst 33342 (Sigma) was added at a final concentration of 5 μg/mL in the presence or absence of 50 μg/mL verapamil (Sigma-Aldrich), and the samples were incubated at 37°C for 90 min. During the incubation time, cells were gently tapped every 15 min. Afterward, the cells were washed and resuspended at a final concentration of 1×106 cells/mL. Before testing during flow cytometry (FCM; Becton Dickson), propidium iodide (Sigma) was added at a concentration of 1 μg/mL. Hoechst 33342 was excited with an ultraviolet laser at 350 nm, and fluorescence emission was measured with DF424/44 (Hoechst blue) and DF630/22 (Hoechst red) optical filters.
Emodin treatment and SP analysis
For checking the direct effect of emodin on Hoechst dye exclusion assay, cells were given different concentrations (at 20, 40, and 60 μM) of emodin together incubating for 24 h and the effect on SP was analyzed by FCM. To test the sensitivity of SP and non-SP cells to emodin, SP and non-SP cells were seeded at 8×103/mL cells per well in 96-microculture-well plates. After exposing to the agents (at 20, 40, and 60 μM) as indicated for 24 h, cell viability was assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT; Sigma) assay as previously described [20,21]. The control group was without treatment by the agents.
Clone formation analysis
To examine clonogenic ability, SP and non-SP cells were plated at 250 cells per well in 6-well culture dishes. Triplicate wells were performed for each group. After exposing to emodin (40 μM) (Sigma) as indicated for 2 weeks, cells were fixed in methanol for 15 min and then stained with crystal violet (Sigma) for 30 min. Clones with >50 cells were scored, and the clone formation efficiency was calculated according to the formula: (the clone number/the plated cell number)×100%. Cells without emodin treatment were set as control group.
Spheroid formation assay
After sorting by FCM, SP and non-SP cells were seeded at 500 cells per well in 12-well plates in DMEM or RPMI-1640 medium supplemented with 20 ng/mL epidermal growth factor (R&D Systems), 10 ng/mL basic fibroblast growth factor (R&D Systems), and 10 ng/mL leukemia inhibiting factor (R&D Systems) in a humidified atmosphere with 5% CO2 at 37°C. After exposing to emodin (40 μM) as indicated for 10 days, spherical clusters of cells were scored, and the neoplastic sphere formation efficiency was calculated according to the formula: (the clone number/the plated cell number)×100%. The control group was without treatment by emodin.
ROS detection
2,7-Dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma) was used as ROS capture in the cells. It was cleaved intracellularly by nonspecific esterases to form 2,7-dichlorodihydrofluorescein (DCFH), which was further oxidized by ROS and becomes a highly fluorescent compound 2,7-dichlorofluorescein (DCF). Sorted SP and non-SP cells were exposed to 5 μM of DCFH-DA, respectively, at 37°C for 15 min. After washing once with ice-cold PBS, cells were harvested and kept on ice for an immediate detection by flow cytometer FACS Calibur (Becton Dickson). The average intensity of DCF stands for intracellular ROS levels.
Cell viability assay
To test the sensitivity of SP and non-SP cells to emodin alone, cisplatin alone, and emodin plus cisplatin, SP and non-SP cells were seeded at 8×103/mL cells per well in 96-microculture-well plates. After exposing to the agents as indicated for 24 h, cell viability was assayed using MTT assay. The control group was without treatment by the agents.
In vivo tumorigenicity and treatment
All animal experiments were done in accordance with institutional guidelines for animal welfare. SP and non-SP cells were harvested, washed, and resuspended in serum-free optimum medium and then injected subcutaneously into 6-week-old BALB/c-nu/nu mice (n=6 mice per group, purchased from Shanghai Experimental Animal Center). Tumor size was measured every 2 days with a caliper, and tumor volumes were calculated using the formula [22]: π/6×a×b2, where a and b were the long and short diameters, respectively. When tumor size of respective cell transplantation model was ∼100 mm3, mice were sorted into 4 equal groups. The tumor-bearing mice were intraperitoneally administered with physiological saline water as a control, emodin (50 mg/kg), CDDP (2 mg/kg), and emodin/CDDP every 2 days. Tumor size was measured every 2 days with a caliper, and tumor volumes were calculated. After 8 days, experimental measurement was recorded, the mice were sacrificed, and tumor weight was measured.
Doxorubicin fluorescence recording
The fluorescent agent doxorubicin (excitation at 488 nm, emission at 595 nm) was used at a concentration of 1 μg/mL. After being pretreated by emodin for 24 h, SP and non-SP cells were exposed to doxorubicin at 37°C for 30 min. After washing once with ice-cold PBS, cells were harvested and kept on ice. Doxorubicin retention in cells was evaluated by recording of doxorubicin fluorescence in fluorescence microscope by FCM. To investigate the impact of verapamil on the retention of doxorubicin, SGC-996 and GBC-SD cells were exposed to doxorubicin for 30 min after being pretreated by verapamil (50 μg/mL) at 37°C for 20 min.
Reverse transcriptase–polymerase chain reaction
Expression of the ABCG2, MRP1, MRP2, and MDR1 was monitored by reverse transcriptase–polymerase chain reaction (RT-PCR). Sorted SP and non-SP cells were lysed with 1 mL of trizol reagent (Invitrogen), and then the samples were processed according to the manufacturer's protocol to obtain total cellular RNA. One microgram of the isolated total RNA was reverse transcribed using random primers and AMV reverse transcriptase (Promega) for 5 min at 70°C, 5 min on ice, and 60 min at 37°C. The single-stranded cDNA was amplified by PCR using GoTaq DNA polymerase (Promega). PCR of MRP1 and MRP2 genes was performed under the following conditions: 30 s, 94°C; 30 s, 58°C; 30 s, 72°C; 34 cycles. PCR of MDR1 gene was performed under the following conditions: 30 s, 94°C; 30 s, 55°C; 60 s, 72°C; 35 cycles. PCR of ABCG2 gene was performed under the following conditions: 30 s, 94°C; 30 s, 55°C; 60 s, 72°C; 28 cycles. After PCR of ABCG2, MRP1, MRP2, and MDR1 genes was performed, equal amounts of RT-PCR products were loaded on 1.0% agarose gels, respectively. Sorted SP and non-SP cells were seeded in 12-well plates. After cells were treated by drugs, the RT-PCR product of ABCG2 was detected. GAPDH was used as an internal control. The primers for ABCG2 were 5′- TGGCTGTCATGGCTTCAGTA-3′ and 5′- GCCACGTGATTCTTCCACAA-3′. The primers for MRP1 were 5′-TGGTGGGCCTCTCAGTGTCTTA-3′ and 5′-TCGGTAGCGCAGGCAGTAGTTC-3′. The primers for MRP2 were 5′-ATGCTTCCTGGGGATAAT-3′ and 5′-TCAAAGGCACGGATAACT-3′. The primers for MDR1 were 5′-CCCATCATTGCAATAGCAGG-3′ and 5′-GTTCAAACTTCTGCTCCTGA-3′. The primers for GAPDH were 5′-TGGGGAAGGTGAAGGTCGG-3′ and 5′-CTGGAAGATGGTGATGGGA-3′.
ABCG2 siRNA transfection and SP cell resorting
To determine ABCG2 responsible for SP phenotype, ABCG2 siRNA oligonucleotides were transiently transfected, using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. A nonspecific siRNA was transfected as mock. Forty-eight hours later, cells were lysed for RT-PCR to verify the efficiency of silencing. After that, SP cells were resorted by FACS. The sequences of siRNA for ABCG2 were 5′-UAAUGAUGUCCAAGAAGAAGUCUGC-3′ and 3′-GCAGACUUCUUCUUGGACAUCAUUA-5′.
In situ hybridization for ABCG2 in xenograft tumor sections
In situ hybridization was used to localize ABCG2 mRNAs in transplanted tumor sections using digoxigenin-labeled sense (Roche Diagnostics GmbH) and antisense ABCG2 probes. The probes, generated by RT-PCR analysis, were used to hybridize the samples embedded in paraffin. Slides of the tumor tissue were de-paraffined and rehydrated before incubation with Protease K at 37°C for 15 min. Sections were then washed 3 times in 0.1 M Tris-buffered saline (TBS)/diethyl pyrocarbonate for 15 min, incubated with 5× standard sodium citrate (SSC) solution at room temperature for 15 min, and incubated with RNA probe sequentially. After 48 h of hybridization at 37°C, the sections were washed with graded-diluted SSC solutions at 37°C for 15 min. Then, the sections were incubated with antibody against digoxigenin at room temperature for 3 h, and then washed with 0.5 M TBS and 0.01 M TBS (pH 9.5). Hybridization signal was visualized by 5-Bromo-4-Chloro-3′-Indolyphosphate p-Toluidine Salt/Nitro-Blue Tetrazolium Chloride (BCIP/NBT; Sigma). The reaction was stopped by washing in water for 5 min. Slides were then counterstained by nuclear fast red, mounted using an aqueous solution, and photographed.
Statistical analysis
Data were shown as mean value±SD. SPSS17.0 software was used for statistical analysis. Analysis of variance was applied for comparison of the means of 2 or multiple groups. A value of P<0.05 was considered significant.
Results
A small population of SP cells exists in SGC-996 and GBC-SD cells and emodin reduces the ratio of SP cells
The proportions of SP cells from SGC-996 and GBC-SD cells were examined by staining cells with Hoechst 33342 dye to generate a Hoechst Blue–Red profile. As a control, the ABC transporter inhibitor verapamil was added to inhibit the efflux of Hoechst 33342. The proportions of SP cells were 1.0% (Fig. 1B-a) and 0.8% (Fig. 1C-a) from SGC-996 and GBC-SD cells, respectively. In the presence of verapamil (Fig. 1B-b, C-b), no more SP cells were detected with the same threshold. As shown in Fig. 1D, under our experimental conditions with treatment of emodin, a gradual decrease of SP fraction was seen in a dose-dependent fashion. As expected, at the concentrations of 20, 40, and 60 μM of emodin treatment, the proportions of SP detected from SGC-996 were 0.84% (Fig. 1B-c), 0.63% (Fig. 1B-d), and 0.46% (Fig. 1B-e), respectively, while the proportions of SP from GBC-SD were 0.67% (Fig. 1C-c), 0.43% (Fig. 1C-d), and 0.27% (Fig. 1C-e), respectively.
FIG. 1.

Side population (SP) analysis and emodin treatment. (A) Chemical structure of emodin. (B) The proportion of SP cells in human gallbladder carcinoma cell line SGC-996 was 1.0% (B-a), which reduced to 0% in the presence of verapamil (B-b). For checking the direct effect of emodin on Hoechst dye exclusion assay, cells were given at the concentrations of 20 μM (B-c), 40 μM (B-d), and 60 μM (B-e) emodin together incubating for 24 h and the effect on SP was analyzed by flow cytometry. (C) Similar to (B), the proportion of SP cells in GBC-SD was 0.8% (C-a) and, as a control, verapamil was added to inhibit the efflux of Hoechst 33342 (C-b). SP cells were reanalyzed after 20 μM (C-c), 40 μM (C-d), and 60 μM (C-e) emodin treatment, respectively. (D) Under experimental conditions with treatment of emodin, a gradual decrease of SP was seen in the SP fraction dose dependently. The ratio of SP cells was calculated: SP/Total cells. Each experiment was repeated 3 times.
Emodin effectively kills SP cells
To verify this effect on SP and non-SP cells by emodin (at 20, 40, and 60 μM), we measure the cell number by seeding sorted cells in 96-microculture-well plates. After treatment for 24 h, cell viability was assayed using MTT. Our data showed that emodin could effectively kill SP cells in a dose-dependent manner, while there virtually was no obvious effect on non-SP cells (Fig. 2A), suggesting that emodin selectively killed CSCs.
FIG. 2.

Emodin acted effectively on SP cells. (A) Emodin effectively kills SP cells from SGC-996 and GBC-SD. (B) Emodin inhibited clone formation of SP cells. (C) Emodin effectively eliminated sphere formation of SP cells. (a) Stands for SP group and non-SP group from SGC-996; (b) stands for SP group and non-SP group from GBC-SD. Columns, mean of 3 experiments; bars, SD. *P, #P<0.05, experimental group compared with the control group. Each experiment was repeated 3 times.
Emodin inhibits clone formation and effectively eliminates sphere formation of SP cells
Huang et al. previously reported that SP cells had stronger clonogenicity as one of the characteristics of CSCs [23]. To confirm that emodin treatment can inhibit gallbladder carcinoma SP properties, clone formation ability of SP cells was studied in the presence or absence of emodin. As shown in Fig. 2B-a, after culturing the cells for 14 days, an average of 126 clones per well were found following 250 SP cells from SGC-996 cells were initially plated in the untreated group. However, only 76 clones were observed under the culture condition in the treated group (Fig. 2B-a). Similarly, the clone formation ability of SP cells from GBC-SD was also sharply suppressed by emodin (Fig. 2B-b).
The ability to form spheres in nonadherent culture is one of the characteristics of CSCs [24,25]. To test the effects of emodin on sphere formation of SP cells, both cell lines were cultured in our suspension culture system and shaken every day. The sphere-forming efficiency of SP cells from SGC-996 cells was 7%, while there was only 0.2% in the agent treatment group (Fig. 2C-a). Similarly, GBC-SD spheres could be serially subcultured, and the sphere-forming efficiency stayed relatively 5.9%. In sharp contrast, SP cells from GBC-SD cells could not form spheres under the agent treatment (Fig. 2C-b). In Fig. 2B we also found that although non-SP cells had the ability to form clones, its ability was much lower than SP cells and treatment of 40 μM emodin had no effect on non-SP cells. But non-SP cells could not form tumor spheroids (Fig. 2C).
SP cells show lower levels of cellular ROS than non-SP cells and emodin effectively enhances cellular ROS levels
It had recently been shown that similar to normal tissue stem cells, subsets of CSCs in some tumors contain lower ROS levels and stronger ROS defenses compared with their nontumorigenic progeny, which might contribute to tumor radio-resistance [26]. Cellular ROS levels of SP cells were lower than that of non-SP cells from SGC-996 and GBC-SD (Fig. 3A), suggesting that lower ROS levels might be helpful for cancer stem-like SP cells to maintain stem cell properties and protect them from endogenous and exogenous damages.
FIG. 3.

Reactive oxygen species (ROS) detection. (A) Cellular ROS levels of SP cells were lower than that of non-SP cells from SGC-996 and GBC-SD. (B, C) Exposure of SP and non-SP cells to emodin resulted in an immediate elevation of cellular ROS levels in a dose-dependent manner. (a) Stands for SP group; (b) stands for non-SP group. Columns, mean of 3 experiments; bars, SD. *P<0.05, experimental group compared with the control group. Each experiment was repeated 3 times.
Emodin is able to generate ROS in a variety of tumor cells [17,19–21]. We found that exposure of SP and non-SP cells to emodin resulted in the elevation of cellular ROS levels in a dose-dependent manner (Fig. 3B, C). These data indicated that the effect of emodin on enhancement of ROS levels was correlated to the lost of stem cell characteristics and weaken survival ability in SP cells. Elevating ROS levels in CSCs might be a useful method for improving local and systemic oncological therapies.
ABCG2 is responsible for SP phenotype
ABCG2 had been implicated in high Hoechst 33342 dye efflux capacity that marked the SP phenotype [27,28]. We found that ABCG2 expression in SP was higher than that in non-SP cells from the 2 cell lines (Fig. 4A). Meanwhile RT-PCR showed that SP and non-SP cells from SGC-996 cells expressed both MRP1 and MRP2 at the same levels, but the MDR1 expression was low to an undetectable level; there were no differences for MRP1, MRP2, and MDR1 expression between SP and non-SP cells from GBC-SD cells (Fig. 4A). Results showed that after silencing the gene ABCG2, any SP cells from GBC-SD cells could not be got by FACS, meanwhile the proportion of SP cells from SGC-996 cells was only 0.2% left (the original ratio was 1.0%) (Fig. 4B-c). Thus, it can be seen that ABCG2 is the requirement of the phenotype of the SP cells.
FIG. 4.

ABCG2 is responsible for SP phenotype and the effects of emodin on ABCG2. (A) Differential expression of ABCG2 mRNA in SP cell and non-SP cells SGC-996 (A-a) and GBC-SD (A-b). (B-a, B-b) Cells were transfected with nonspecific siRNA (NC) or ABCG2 siRNA. (B-c) After silencing the gene ABCG2, SP cells were sorted. (C-a, C-b) Emodin promotes the intracellular accumulation of doxorubicin of SP cells. (C) Retention of doxorubicin is prolonged by treatment with verapamil in SGC-996 (C-c and GBC-SD C-d). (D) Dose-dependent emodin has no effect on the mRNA expression of ABCG2. (a) Stands for SP group and non-SP group from SGC-996; (b) stands for SP group and non-SP group from GBC-SD. Each experiment was repeated 3 times.
Emodin promotes the intracellular accumulation of doxorubicin, but dose-dependent emodin has no effect on the mRNA expression of ABCG2
The anticancer drug doxorubicin had a natural red fluorescence; thus, its uptake and retention in the cell can be monitored indirectly under FCM. The mean fluorescence intensity of doxorubicin, after being uptaken, is a common readout. In Fig. 4C-a and C-b, we compared the effect of emodin treatment on the retention of doxorubicin between the SP and non-SP cells. We found that dose-dependent emodin could effectively promote the intracellular accumulation of doxorubicin of SP cells, but had no effect on that of non-SP cells. As shown in Fig. 4C-c, the retention of doxorubicin was prolonged by treatment with verapamil.
Due to the above findings that emodin could promote the intracellular accumulation of doxorubicin of SP cells, we then questioned whether the observed effects of emodin could be correlated to regulated expression of ABCG2 gene, which is the main factor for doxorubicin retention [29,30]. To ascertain the action of emodin on ABCG2 expression, RT-PCR for ABCG2 mRNA was performed. Results from RT-PCR showed that there were no differences among dose-dependent emodin treatments in each group (Fig. 4D). These data suggest that emodin could inactive the pump function of ABCG2 responsible for doxorubicin retention, but it could not suppress the expression of ABCG2.
Emodin sensitizes SP cells to the anticancer agent CDDP
To examine the chemoresistance of cancer stem-like SP cells and sensitization of emodin on cell viability, SP and non-SP cells from SGC-996 and GBC-SD cells were treated with CDDP only, emodin only, and co-treated. As demonstrated in the cell viability assay, SP cells were highly resistant to CDDP treatment. SP cells remained 93.72% and 96.58% viable, respectively, from SGC-996 and GBC-SD cells when treated with CDDP at a dosage of 2 μg/mL (Table 1). However, a sharp decrease (27.54% and 29.51%) in cell viability was observed when non-SP cells were exposed to the same dosage (Table 1). These data clearly demonstrated the chemoresistant characteristic of SP cells.
Table 1.
Emodin Sensitizes Side Population Cells to the Anticancer Agent CDDP
| Group | Con (absorbance) | CDDP (absorbance) | Emodin (absorbance) | CDDP+emodin (absorbance) |
|---|---|---|---|---|
| SGC-996 | ||||
| SP | 1.115±0.003 | 1.045±0.064b | 0.873±0.048a,b | 0.616±0.034a |
| Non-SP | 1.117±0.003 | 0.809±0.016a | 1.103±0.012 | 0.687±0.059a |
| GBC-SD | ||||
| SP | 1.181±0.06 | 1.14±0.137b | 0.936±0.11a,b | 0.63±0.097a |
| Non-SP | 1.177±0.051 | 0.83±0.097a | 1.145±0.05 | 0.657±0.054a |
P<0.05, experimental group compared with the control group.
P<0.05, the SP group compared with the non-SP group.
Emodin sensitizes SP cells to the anticancer agent CDDP. Each experiment was repeated 3 times.
CDDP, cisplatin; SP, side population.
More importantly, despite the differences in CDDP sensitivity, both SP and non-SP cells were extremely sensitive to emodin/CDDP co-treatment, as evidenced by the 44.74%, 46.61%, 38.53%, and 44.21% inhibition in cell viability for SP cells from SGC-996, SP cells from GBC-SD, non-SP cells from SGC-996, and non-SP cells from GBC-SD, respectively, suggesting that the enhancement of the toxicity by emodin co-treatment was effective in targeting both SP and non-SP cells. Notably, we observed that emodin effectively sensitized SP cells while no obvious reduction in viable cell number was observed in non-SP cells by emodin alone (Table 1). These findings suggest that emodin may be an effective agent in targeting resistant SP cells.
Emodin/CDDP combination markedly downregulates ABCG2 expression of SP cells
It has been reported that overexpression of ABCG2 is not only a key marker for SP cells and an important factor for maintaining the microenvironment and stem cell characteristics of SP cells, but also often renders cancer cells a constitutive characteristic of multidrug resistance [28,31]. The previous experiments showed that SP cells expressing high level of ABCG2 from SGC-996 and GBC-SD cells could be partially killed by using emodin in combination with chemotherapeutic drug CDDP in vitro and in vivo. To verify whether the effect of emodin/CDDP co-treatment could be correlated to downregulate ABCG2 gene, we measured the expression of ABCG2 of the control, emodin-only, CDDP-only, and emodin/CDDP combination samples. Our results showed that CDDP/emodin co-treatment resulted in a downregulation of ABCG2 in the SP cells from both the cell lines, while there was no change in the non-SP cells (Fig. 5).
FIG. 5.
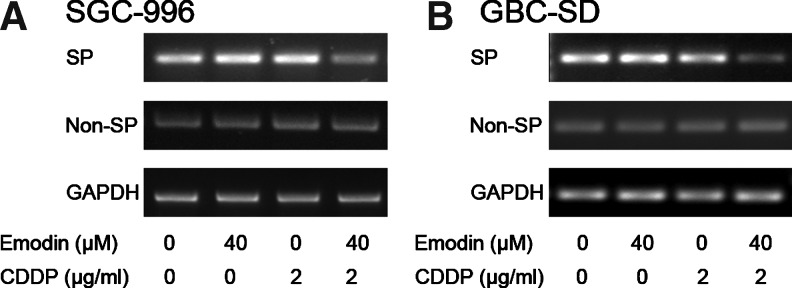
Emodin/cisplatin (CDDP) combination could markedly downregulate ABCG2 expression of SP cells from SGC-996 (A) and GBC-SD (B). Each experiment was repeated 3 times.
Emodin sensitizes SP and non-SP cells to CDDP cytotoxicity in xenograft tumors without displaying obvious toxic effects
When the transplanted tumor volume reached 100 mm3, the mice were synchronously administered with physiological saline, emodin only, CDDP only, and emodin/CDDP for 8 days. Our results showed that mice exposed to emodin/CDDP therapy had significantly smaller tumors than mice in other groups of both SP and non-SP groups (Tables 2 and 3). The tumor volumes of the SP groups under emodin treatment distinctly reduced from both SGC-996 and GBC-SD cells, while those of the non-SP groups had no obvious difference compared with each control group (Tables 2 and 3). Following CDDP treatment, the SP groups showed more resistance than that of the non-SP groups of transplantation from both SGC-996 and GBC-SD cells (Tables 2 and 3). Also, emodin/CDDP treatment could sharply inhibit tumor weight from both SP and non-SP groups (Fig. 6A, B). As shown in Fig. 6C, no notable differences on the body weight loss of mice were observed among these groups, demonstrating that emodin/CDDP co-treatment had no obvious side effects in vivo. These data support the idea that SP-derived tumor cells had robust chemoresistance and emodin administration in combination with the chemotherapeutic drug CDDP could significantly suppress the tumorigenicity in vivo.
Table 2.
In Vivo Tumorigenicity and Treatment (Side Population and Nonside Population Cells of SGC-996)
| Group | Volume D0 (cm3) | Volume D2 (cm3) | Volume D4 (cm3) | Volume D6 (cm3) | Volume D8 (cm3) |
|---|---|---|---|---|---|
| SP (SGC-996) | |||||
| Con | 0.106±0.012 | 0.14±0.052 | 0.199±0.017 | 0.315±0.048 | 0.46±0.04 |
| CDDP | 0.106±0.016 | 0.121±0.023 | 0.178±0.028 | 0.261±0.06 | 0.413±0.029 |
| Emodin | 0.102±0.015 | 0.103±0.014 | 0.133±0.037a | 0.202±0.032a | 0.304±0.018a |
| CDDP+emodin | 0.106±0.012 | 0.098±0.017 | 0.09±0.012a | 0.089±0.011a | 0.083±0.01a |
| Non-SP (SGC-996) | |||||
| Con | 0.102±0.01 | 0.127±0.018 | 0.19±0.037 | 0.288±0.029 | 0.359±0.041 |
| CDDP | 0.102±0.011 | 0.099±0.011a | 0.131±0.013a | 0.198±0.016a | 0.279±0.028a |
| Emodin | 0.102±0.098 | 0.123±0.027 | 0.182±0.019 | 0.277±0.025 | 0.353±0.016 |
| CDDP+emodin | 0.105±0.012 | 0.093±0.013a | 0.085±0.012a | 0.075±0.012a | 0.07±0.013a |
P<0.05, experimental group compared with the control group.
Emodin sensitizes SP and non-SP cells of SGC-996 to CDDP cytotoxicity in xenograft tumors. D, day; (n=6).
Table 3.
In Vivo Tumorigenicity and Treatment (Side Population and Nonside Population Cells of GBC-SD)
| Group | Volume D0 (mm3) | Volume D2 (mm3) | Volume D4 (mm3) | Volume D6 (mm3) | Volume D8 (mm3) |
|---|---|---|---|---|---|
| SP (GBC-SD) | |||||
| Con | 0.101±0.011 | 0.139±0.013 | 0.199±0.022 | 0.277±0.021 | 0.379±0.025 |
| CDDP | 0.101±0.012 | 0.123±0.012 | 0.174±0.021 | 0.212±0.02 | 0.301±0.049 |
| Emodin | 0.102±0.074 | 0.108±0.098 | 0.142±0.015a | 0.183±0.029a | 0.258±0.036a |
| CDDP+emodin | 0.103±0.01 | 0.092±0.011a | 0.084±0.01a | 0.078±0.011a | 0.074±0.009a |
| Non-SP (GBC-SD) | |||||
| Con | 0.102±0.008 | 0.121±0.017 | 0.17±0.025 | 0.232±0.037 | 0.312±0.04 |
| CDDP | 0.102±0.008 | 0.103±0.008a | 0.136±0.028a | 0.181±0.022a | 0.25±0.028a |
| Emodin | 0.102±0.01 | 0.12±0.019 | 0.169±0.014 | 0.221±0.017 | 0.309±0.027 |
| CDDP+emodin | 0.1±0.007 | 0.093±0.002a | 0.084±0.005a | 0.074±0.005a | 0.07±0.005a |
P<0.05, experimental group compared with the control group.
Emodin sensitizes SP and non-SP cells of GBC-SD to CDDP cytotoxicity in xenograft tumors. D, day; (n=6).
FIG. 6.

Emodin administration in combination with the chemotherapeutic drug CDDP in vivo. (A, B) Average weight of transplanted tumors after the mice were exposed to treatments. (a) Stands for SP group; (b) stands for non-SP group. (C) Average body weight of tumor-bearing mice. (a) Stands for SP group and non-SP group from SGC-996; (b) stands for SP group and non-SP group from GBC-SD. Columns, mean; bars, SD. *P<0.05, experimental group compared with the control group (n=6).
Emodin/CDDP combination inhibits the mRNA level of ABCG2 in xenograft tumors of the SP group
In situ hybridization of transplantation tissue for ABCG2 mRNA was used to show the effect of emodin/CDDP combination on ABCG2 expression in vivo. The results of the reporter assay showed that the emodin/CDDP treatment on the SP group from SGC-996 cells was lower than other 3 groups in vivo (Fig. 7A). Consistently, in situ hybridization of the xenograft tissues of SP cells from GBC-SD cells showed the same phenomenon (Fig. 7B). But in the transplantation tissue of non-SP group from both the cell lines, the expression of ABCG2 was not affected by emodin/CDDP combination, emodin-only, or CDDP-only sample (Fig. 7).
FIG. 7.
In situ hybridization for ABCG2 mRNA in xenograft tumor sections. Emodin/CDDP combination inhibited the mRNA expression of ABCG2 in xenograft tumors of the SP group from the cell line SGC-996 (A-a) and GBC-SD (B-a). But in the tumor tissues of non-SP group, ABCG2 could not be downregulated by emodin/CDDP combination, emodin-only, or CDDP-only sample (A-b, B-b). Obviously, in control from each cell line, ABCG2-positive signals of SP group were stronger than that of non-SP group (n=6). The deep black represented positive hybridization signal for ABCG2 mRNA in black and white print. (a) Stands for SP group; (b) stands for non-SP group.
Discussion
Conventional therapies for gallbladder cancer are believed to mainly eliminate the majority of differentiated cancer cells but spare CSCs, which are thought to be associated with recurrence [3,32]. Thus, it is important to develop new therapies targeting CSCs. Putative CSCs in gallbladder cancer were identified with CD44+CD133+ and CD133+, respectively, as sorting markers [33,34]. Yin et al. [35] obtained gallbladder CSC-like cells using suspension cultures of GBC-SD cells in serum-free culture medium containing CDDP. In the present study, we used SP cells as a model of CSC-like cells. On the basis of the ability to efflux the fluorescent DNA-binding dye Hoechst 33342, Goodell et al. [9] first identified SP cells from mouse bone marrow as a small cell population that was highly enriched for hematopoietic stem cells and endowed with long-term repopulating capacity. Ever since their discovery, SP cells have been detected in many tumor tissues, such as esophageal carcinoma [23] and laryngeal [14]. The existence of SP cells has been proven to play an important role in tumor growth and relapse in many solid tumors [10–16,23]. Zhou et al. [36] first proved that ABCG2 was a molecular determinant of the SP phenotype. A number of other studies in a wide variety of organs have also indicated that ABCG2 is responsible for Hoechst 33342 dye efflux pattern and confers the SP cell phenotype [11–15,23]. In our previous work we sorted SP cells from a human gallbladder carcinoma cell line, SGC-996, and found that SP cells possess the characteristics of CSCs, such as asymmetrical cell division, more rapid proliferation, higher clonogenicity, stronger tumorigenicity, more migratory, invasive abilities, more resistant to chemotherapy, and express higher levels of ABCG2 [37]. Zhang et al. [38] recently isolated SP cells from other common gallbladder carcinoma cell line, GBC-SD, and also confirmed that SP cells showed characteristics of CSCs, such as higher clonogenicity and stronger tumorigenicity. These reports provide a strong evidence to use SP cells for CSC study. In this experiment, the proportion of SP cells from the commonly studied human gallbladder carcinoma cell lines SGC-996 and GBC-SD was 1.0% and 0.8%, respectively, both of which reduce to 0% in the presence of verapamil. Moreover, after silencing the gene ABCG2, any SP cells from GBC-SD cells could not be got by FACS, meanwhile the proportion of SP cells from SGC-996 cells was only 0.2% left (the original ratio was 1.0%). In addition, we found that SP cells of SGC-996 and GBC-SD cells showed higher expression of ABCG2 than non-SP cells, meanwhile there were no differences of other main and common ABC transporters MDR1, MRP1, and MRP2 between SP and non-SP cells. Thus, we propose that ABCG2 is the requirement of the phenotype of the SP cells. Widely expressed in stem cells, ABCG2 is also recognized as a universal marker of stem cells [28]. Therefore, SP cells from gallbladder cancer may correspond to CSCs, and at least they enrich more CSCs.
Emodin is one of the main active components enriched in the root and rhizome of many traditional Chinese medicinal herbs, especially from the Rhizoma and Radix families. Emodin has long been studied for anti-inflammatory, antibacterial, diuretic, immunosuppressive, and chemopreventive effects. The anticancer effect of emodin is found to be mediated via induction of apoptosis, inhibition of cancer cell growth, antiproliferation, and antiadhesion [22,39,40]. Recently, we have found that emodin can facilitate cytotoxicity in gallbladder carcinoma in the ROS-dependent manner [21]. Its chemosensitizing effect leads us to hypothesize that emodin may work as an agent targeting cancer stem-like SP cells of gallbladder carcinoma. Here, we showed for the first time that emodin has anti-CSC effects, as indicated by strong inhibition on cell growth in vitro and in vivo.
In the present study we showed that emodin inhibited SP cells of SGC-996 and GBC-SD cells. SP cells are due to the action of the ABC transporter ABCG2, alias breast cancer resistance protein (BCRP) [16,28]. ABCG2 functions as a high-capacity transporter with a wide range of substrates, including various chemotherapy drugs, and it has been shown to participate in the multidrug resistance of tumors and lead to a limitation of chemotherapeutics [8,31,41]. Intriguingly, CSCs are also supposed to be responsible for the acquisition of multidrug chemoresistance and lead to the cancer relapse [4]. SP cells and chemoresistance suggest a close link between ABCG2 and CSCs. Although the molecular mechanisms regulating the expression of ABCG2 remain unclear, many agents have been found to regulate the expression of ABCG2, such as human placental lactogen, human prolactin, and folate [42–44]. Yin et al. found that transforming growth factor-beta decreased ABCG2 gene expression in SP cells from MCF-7 [27]. Katayama et al. [45] showed that dofequidar fumarate sensitizes SP cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Our results showed that emodin in combination with CDDP downregulated ABCG2 expression and facilitated cytotoxicity in SP cells; however, the combination exerted little effect on the expression of ABCG2 in non-SP cells. In difference from other reporters, there is lower ABCG2 expression in non-SP cells, indicating that ABCG2 does not occupy the main role as drug efflux pumps, or protect the organism from a range of xenobiotics as the primary biological role [41]. The expression of ABCG2 in the physiological function may be regulated by multiple pathways in a complex network. Combined with the prior research on inhibition of MRP1 by emodin/CDDP co-treatment in gallbladder cancer [21], these results may explain the fact that drug efflux pumps play important roles in drug resistance of gallbladder cancer and emodin in combination with CDDP can suppress different ABC family resistance proteins in SP and non-SP cells. Different from MDR1 and MRP1, ABCG2 is also a molecular determinant of the SP phenotype [27,28], which suggests that new therapeutic strategies targeting ABCG2 may effectively eliminate CSCs and overcome current chemotherapeutic limitations.
ROS, produced by the metabolism of oxygen, have been implicated in processes as diverse as cancer, cardiovascular disease, and aging. It has recently been shown that central nervous system stem cells, hematopoietic stem cells, human and murine breast CSCs, and early progenitors contain lower levels of ROS than their more mature progenies, which are critical for maintaining stem cell function [26,46–48]. Das et al. [49] consider that hypoxic microenvironments play an important role in the trafficking of normal stem cells and SP cells, and hypoxia enhances tumor stemness by increasing the invasive and tumorigenic SP fraction. Taken together, these findings indicate that stem cells in diverse systems have conserved this lower ROS attribute, which may help to protect their genomes from endogenous and exogenous ROS-mediated damage. In accord with the above-mentioned studies, we found that cellular ROS levels of SP cells were lower than that of non-SP cells from SGC-996 and GBC-SD cells, which suggests that lower ROS levels might be helpful for cancer stem-like SP cells to maintain stem cell properties and protect them from endogenous and exogenous damages [26,45,46]. Emodin was able to generate ROS in a variety of tumor cells [17,19–21]. We found that exposure of SP and non-SP cells to emodin resulted in elevation of cellular ROS levels in a dose-dependent manner. These data indicate that the effect of emodin on enhancement of ROS levels was related to failure stem cell characteristics and weaken survival ability in SP cells [26]. Overcoming low ROS levels in CSCs might be a useful method for improving local and systemic oncological therapies. But for non-SP cells, it is not easy for emodin to damage the cell defensive system, because the intracellular ROS of non-SP cells is less sensitive to hypoxia and hyperoxia regulator than that of SP cells. Besides, we observed that emodin could promote SP cellular accumulation of doxorubicin that is the main substrate of ABCG2 [29,30,50], but it could not suppress the expression of ABCG2. This finding indicates that emodin, via alteration of tumor microenvironment and inactivation of pump function, could affect the drug efflux ABCG2 and ultimately influence doxorubicin retention. Some studies have reported that ABCG2 expression is upregulated by hypoxia and injury via hypoxia inducible transcription factor (HIF) signaling [51,52]. HIF is accompanied with hypoxia, combating overproduction of ROS [20]. But we found that emodin-producing ROS could not change the expression of ABCG2. In summary, emodin, via ROS-related mechanism and inactivating the efflux pump ABCG2/BCRP function, affects survival ability of SP cells and doxorubicin retention. How emodin contributes to inactivation of ABCG2 remains to be clarified in the future.
As emodin/CDDP co-treatment could inhibit ABCG2 in vitro, we tried to treat transplantation tumors with emodin in combination with CDDP in vivo. Our study demonstrates that this combination is effective in killing of SP cells in vivo via inhibition of ABCG2. Interestingly and promisingly, both in vitro and in vivo data show the consistent results, which implies that emodin is a promising chemotherapy-sensitizing agent targeting CSCs. In summary, emodin not only effectively impairs the survival of gallbladder cancer stem-like SP cells via enhancement of intracellular ROS level and inactivation of the pump ABCG2, but also sensitizes CDDP to overcome chemoresistance of SP cells via suppression of the expression of ABCG2. Therefore, emodin may work as an effective therapeutic agent for the treatment of gallbladder carcinoma.
Acknowledgments
The authors thank Prof. Zhao-Yuan Hou and Prof. Wei-Qiang Gao for the very helpful editing of this article. This work was supported by grants from Shanghai Science and Technology Commission, China (09411960800, J. Wang).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Spillane JB. Henderson MA. Cancer stem cells: a review. ANZ J Surg. 2007;77:464–468. doi: 10.1111/j.1445-2197.2007.04096.x. [DOI] [PubMed] [Google Scholar]
- 2.Dalerba P. Cho RW. Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 3.Dean M. Fojo T. Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 4.Reya T. Morrison SJ. Clarke MF. Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Biswas PK. Carcinoma gallbladder. Mymensingh Med J. 2010;19:477–481. [PubMed] [Google Scholar]
- 6.Abahssain H. Afchain P. Melas N. Ismaili N. Rahali R. Rabti HM. Errihani H. [Chemotherapy in gallbladder carcinoma] Presse Med. 2010;39:1238–1245. doi: 10.1016/j.lpm.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Fong D. Yeh A. Naftalovich R. Choi TH. Chan MM. Curcumin inhibits the side population (SP) phenotype of the rat C6 glioma cell line: towards targeting of cancer stem cells with phytochemicals. Cancer Lett. 2010;293:65–72. doi: 10.1016/j.canlet.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathew G. Timm EA., Jr. Sotomayor P. Godoy A. Montecinos VP. Smith GJ. Huss WJ. ABCG2-mediated DyeCycle Violet efflux defined side population in benign and malignant prostate. Cell Cycle. 2009;8:1053–1061. doi: 10.4161/cc.8.7.8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodell MA. Brose K. Paradis G. Conner AS. Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ailles LE. Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Oka M. Toyoda C. Kaneko Y. Nakazawa Y. Aizu-Yokota E. Takehana M. Characterization and localization of side population cells in the lens. Mol Vis. 2010;16:945–953. [PMC free article] [PubMed] [Google Scholar]
- 12.Sung JM. Cho HJ. Yi H. Lee CH. Kim HS. Kim DK. Abd El-Aty AM. Kim JS. Landowski CP. Hediger MA. Shin HC. Characterization of a stem cell population in lung cancer A549 cells. Biochem Biophys Res Commun. 2008;371:163–167. doi: 10.1016/j.bbrc.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 13.Tabor MH. Clay MR. Owen JH. Bradford CR. Carey TE. Wolf GT. Prince ME. Head and neck cancer stem cells: the side population. Laryngoscope. 2011;121:527–533. doi: 10.1002/lary.21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wan G. Zhou L. Xie M. Chen H. Tian J. Characterization of side population cells from laryngeal cancer cell lines. Head Neck. 2010;32:1302–1309. doi: 10.1002/hed.21325. [DOI] [PubMed] [Google Scholar]
- 15.Wang B. Yang H. Huang YZ. Yan RH. Liu FJ. Zhang JN. Biologic characteristics of the side population of human small cell lung cancer cell line H446. Chin J Cancer. 2010;29:254–260. doi: 10.5732/cjc.009.10330. [DOI] [PubMed] [Google Scholar]
- 16.Wu C. Alman BA. Side population cells in human cancers. Cancer Lett. 2008;268:1–9. doi: 10.1016/j.canlet.2008.03.048. [DOI] [PubMed] [Google Scholar]
- 17.Cai J. Niu X. Chen Y. Hu Q. Shi G. Wu H. Wang J. Yi J. Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia. 2008;10:41–51. doi: 10.1593/neo.07754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J. Li H. Chen YY. Wang XJ. Shi GY. Hu QS. Kang XL. Lu Y. Tang XM. Guo QS. Yi J. Anthraquinones sensitize tumor cells to arsenic cytotoxicity in vitro and in vivo via reactive oxygen species-mediated dual regulation of apoptosis. Free Radic Biol Med. 2004;37:2027–2041. doi: 10.1016/j.freeradbiomed.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 19.Yi J. Yang J. He R. Gao F. Sang H. Tang X. Ye RD. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 2004;64:108–116. doi: 10.1158/0008-5472.can-2820-2. [DOI] [PubMed] [Google Scholar]
- 20.Huang XZ. Wang J. Huang C. Chen YY. Shi GY. Hu QS. Yi J. Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells: the mechanisms involve ROS-mediated suppression of multidrug resistance and hypoxia inducible factor-1. Cancer Biol Ther. 2008;7:468–475. doi: 10.4161/cbt.7.3.5457. [DOI] [PubMed] [Google Scholar]
- 21.Wang W. Sun YP. Huang XZ. He M. Chen YY. Shi GY. Li H. Yi J. Wang J. Emodin enhances sensitivity of gallbladder cancer cells to platinum drugs via glutathion depletion and MRP1 downregulation. Biochem Pharmacol. 2010;79:1134–1140. doi: 10.1016/j.bcp.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Cha TL. Qiu L. Chen CT. Wen Y. Hung MC. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Res. 2005;65:2287–2295. doi: 10.1158/0008-5472.CAN-04-3250. [DOI] [PubMed] [Google Scholar]
- 23.Huang D. Gao Q. Guo L. Zhang C. Jiang W. Li H. Wang J. Han X. Shi Y. Lu SH. Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev. 2009;18:465–473. doi: 10.1089/scd.2008.0033. [DOI] [PubMed] [Google Scholar]
- 24.Singh SK. Clarke ID. Terasaki M. Bonn VE. Hawkins C. Squire J. Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 25.Reynolds BA. Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 26.Clarke MF. Diehn M. Cho RW. Lobo NA. Kalisky T. Dorie MJ. Kulp AN. Qian DL. Lam JS, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin L. Castagnino P. Assoian RK. ABCG2 expression and side population abundance regulated by a transforming growth factor beta-directed epithelial-mesenchymal transition. Cancer Res. 2008;68:800–807. doi: 10.1158/0008-5472.CAN-07-2545. [DOI] [PubMed] [Google Scholar]
- 28.Ding XW. Wu JH. Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010;86:631–637. doi: 10.1016/j.lfs.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Lopez JP. Wang-Rodriguez J. Chang C. Chen JS. Pardo FS. Aguilera J. Ongkeko WM. Gefitinib inhibition of drug resistance to doxorubicin by inactivating ABCG2 in thyroid cancer cell lines. Arch Otolaryngol Head Neck Surg. 2007;133:1022–1027. doi: 10.1001/archotol.133.10.1022. [DOI] [PubMed] [Google Scholar]
- 30.Dai CL. Tiwari AK. Wu CP. Su XD. Wang SR. Liu DG. Ashby CR., Jr. Huang Y. Robey RW, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robey RW. To KK. Polgar O. Dohse M. Fetsch P. Dean M. Bates SE. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61:3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donnenberg VS. Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 33.Shi C. Tian R. Wang M. Wang X. Jiang J. Zhang Z. Li X. He Z. Gong W. Qin R. CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol Ther. 2010;10:1182–1190. doi: 10.4161/cbt.10.11.13664. [DOI] [PubMed] [Google Scholar]
- 34.Shi CJ. Gao J. Wang M. Wang X. Tian R. Zhu F. Shen M. Qin RY. CD133(+) gallbladder carcinoma cells exhibit self-renewal ability and tumorigenicity. World J Gastroenterol. 2011;17:2965–2971. doi: 10.3748/wjg.v17.i24.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin BB. Wu SJ. Zong HJ. Ma BJ. Cai D. Preliminary screening and identification of stem cell-like sphere clones in a gallbladder cancer cell line GBC-SD. J Zhejiang Univ Sci B. 2011;12:256–263. doi: 10.1631/jzus.B1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou S. Morris JJ. Barnes Y. Lan L. Schuetz JD. Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99:12339–12344. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li XX. Wang J. Wang HL. Wang W. Yin XB. Li QW. Chen YY. Yi J. Characterization of cancer stem-like cells derived from a side population of a human gallbladder carcinoma cell line, SGC-996. Biochem Biophys Res Commun. 2012;419:728–734. doi: 10.1016/j.bbrc.2012.02.090. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z. Zhu F. Xiao L. Wang M. Tian R. Shi C. Qin R. Side population cells in human gallbladder cancer cell line GBC-SD regulated by TGF-beta-induced epithelial-mesenchymal transition. J Huazhong Univ Sci Technolog Med Sci. 2011;31:749–755. doi: 10.1007/s11596-011-0671-1. [DOI] [PubMed] [Google Scholar]
- 39.Huang Q. Shen HM. Shui G. Wenk MR. Ong CN. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006;66:5807–5815. doi: 10.1158/0008-5472.CAN-06-0077. [DOI] [PubMed] [Google Scholar]
- 40.Su YT. Chang HL. Shyue SK. Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005;70:229–241. doi: 10.1016/j.bcp.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 41.Velamakanni S. Wei SL. Janvilisri T. van Veen HW. ABCG transporters: structure, substrate specificities and physiological roles: a brief overview. J Bioenerg Biomembr. 2007;39:465–471. doi: 10.1007/s10863-007-9122-x. [DOI] [PubMed] [Google Scholar]
- 42.Wang H. Unadkat JD. Mao Q. Hormonal regulation of BCRP expression in human placental BeWo cells. Pharm Res. 2008;25:444–452. doi: 10.1007/s11095-007-9432-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H. Lee EW. Zhou L. Leung PC. Ross DD. Unadkat JD. Mao Q. Progesterone receptor (PR) isoforms PRA and PRB differentially regulate expression of the breast cancer resistance protein in human placental choriocarcinoma BeWo cells. Mol Pharmacol. 2008;73:845–854. doi: 10.1124/mol.107.041087. [DOI] [PubMed] [Google Scholar]
- 44.Ifergan I. Shafran A. Jansen G. Hooijberg JH. Scheffer GL. Assaraf YG. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J Biol Chem. 2004;279:25527–25534. doi: 10.1074/jbc.M401725200. [DOI] [PubMed] [Google Scholar]
- 45.Katayama R. Koike S. Sato S. Sugimoto Y. Tsuruo T. Fujita N. Dofequidar fumarate sensitizes cancer stem-like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Cancer Sci. 2009;100:2060–2068. doi: 10.1111/j.1349-7006.2009.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilliland DG. Tothova Z. Kollipara R. Huntly BJ. Lee BH. Castrillon DH. Cullen DE. McDowell EP. Lazo-Kallanian S, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Noble M. Smith J. Ladi E. Mayer-Proschel M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A. 2000;97:10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito K. Hirao A. Arai F. Matsuoka S. Mak TW. Suda T. Regulation of oxidative stress by ATM is required for the self-renewal of haematopoietic stem cells. Blood. 2004;104:109a. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 49.Das B. Tsuchida R. Malkin D. Koren G. Baruchel S. Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26:1818–1830. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 50.Robey RW. Honjo Y. Morisaki K. Nadjem TA. Runge S. Risbood M. Poruchynsky MS. Bates SE. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–1978. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin CM. Ferdous A. Gallardo T. Humphries C. Sadek H. Caprioli A. Garcia JA. Szweda LI. Garry MG. Garry DJ. Hypoxia-inducible factor-2alpha transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res. 2008;102:1075–1081. doi: 10.1161/CIRCRESAHA.107.161729. [DOI] [PubMed] [Google Scholar]
- 52.Schuetz JD. Krishnamurthy P. Ross DD. Nakanishi T. Bailey-Dell K. Zhou S. Mercer KE. Sarkadi B. Sorrentino BP. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–24225. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]