

Abstract
Obesity has various deleterious effects on health largely associated with metabolic abnormalities including abnormal glucose and lipid homeostasis that are associated with vascular injury and known cardiac, renal, and cerebrovascular complications. Advanced age is also associated with increased adiposity, decreased lean mass, and increased risk for obesity-related diseases. Although many of these obesity- and age-related disease processes have long been subsumed to be secondary to metabolic or vascular dysfunction, increasing evidence indicates that obesity also modulates nonvascular diseases such as Alzheimer's disease (AD) dementia. The link between peripheral obesity and neurode-generation will be explored, using adipokines and AD as a template. After an introduction to the neuropathology of AD, the relationship between body weight, obesity, and dementia will be reviewed. Then, population-based and experimental studies that address whether leptin modulates brain health and mitigates AD pathways will be explored. These studies will serve as a framework for understanding the role of adipokines in brain health.
Keywords: adiponectin, Alzheimer, amyloid, leptin
Introduction
Understanding how obesity adversely affects the aging brain is hindered by the number of metabolic and hormonal pathways that are altered due to both obesity and aging. These changes include alterations in energy expenditure, reduced respiratory quotient, hyperlipidemia, hyperinsulinemia, glucose intolerance, low-grade inflammation, and changes in adipokine levels. Dissecting the relative role of each of these factors remains a challenge. Of the various changes associated with obesity, altered adipokine signaling may be one mechanism whereby obesity affects brain health. Adipokines are known to affect brain physiology and function, and both population and experimental studies suggest that changes in adipokine function may mitigate the pathogenesis of Alzheimer's disease (AD). The interaction between obesity and AD has been explored in several reviews, usually with an emphasis on insulin signaling pathways.1–4 This review will emphasize the potential role of adipose tissue and adipokines in the pathogenesis of AD with a particular focus on leptin.
Neuropathology of Alzheimer's disease
AD is characterized by an insidious loss of memory and cognition leading to death within 10 years.5–8 AD brains show neuronal loss and reactive gliosis involving limbic regions (including amygdala and hippocampus) and cerebral cortex with relative sparing of the basal ganglia, cerebellum, brainstem, and spinal cord.9 Affected brain regions show an abundance of intracellular and extracellular aggregates (Fig. 1). Intraneuronal aggregates called neurofibrillary tangles consist of hyperphosphorylated tau protein in the form of insoluble paired helical filaments.10 Extracellular deposits called amyloid plaques consist of Aβ peptides in the form of insoluble amyloid fibrils. Many amyloid plaques also contain swollen dystrophic tau-positive neurites and are thus called neuritic plaques. Both plaques and tangles stain avidly using dyes, such as thioflavin S or congo red, that bind to β-pleated sheet structures. Widespread involvement and a high density of plaques and tangles are diagnostic of AD.
Figure 1.

Neuropathology of AD. Images from hematoxylin- and eosin-stained histologic sections (400× magnification) from affected human AD brain show: (A) granulovacuolar degeneration (arrowheads), (B) intracellular neurofibrillary tangles, and (C) waxy extracellular amyloid plaques. As neurofibrillary tangles and amyloid plaques are relatively subtle using routine stains, histologic diagnosis of AD is aided by using dyes that bind to amyloid structures or with antibodies specific for tau or Aβ(D) Thioflavin S staining (fluorescent green color, 100× magnification) of human AD necortex shows abundant extracellular amyloid plaques (arrows) and intraneuronal neurofibrillary tangles (arrowheads). Higher-power images (400× magnification) show (E) a neurofibrillary tangle and (F) an amyloid plaque. (G) Neurofibrillary tangles (arrows, 100×) are stained with antibodies that recognize phosphorylated tau protein epitopes. Note also the presence of clusters of phospho-tau immunoreactivity (circles) that label dystrophic neurites in association with amyloid plaques. (H) Amyloid plaques form in transgenic mouse overexpressing mutant APP that can be labeled with antibodies against Aβ (400× magnification).
Aβ peptides are generated by sequential proteolysis of the amyloid precursor protein (APP) by a series of endoproteolytic proteases historically called “secretases” prior to their cloning.9 β-secretase, now known as β-site APP cleaving enzyme (BACE), cleaves at the N-terminus of the Aβ sequence. An alternative cleavage pathway involving α-secretase results in proteolysis within the Aβ domain, precluding the generation of Aβ peptide. After α-or β-cleavage, the remaining C-terminal APP fragments are then cleaved by the γ-secretase enzymatic complex that consists of four proteins (presenilin, Aph1, Pen2, and nicastrin). Importantly, several mutations within the APP or presenilin genes result in autosomal dominant cerebral Aβ amyloidosis. These autosomal dominant amyloidoses usually result in clinical dementia and AD-like neuropathology, although other clinical syndromes and pathologies are observed, such as cerebral amyloid angiopathy, leading to recurrent hemorrhagic strokes. Finally, the vast majority of these mutations result in increased amyloidogenic Aβ peptide generation. Thus, Aβ is central to the pathogenesis of AD dementia.
Tau protein normally functions to bind and stabilize microtubules.10 Tau is prone to aggregation intracellularly where the protein is ubiquitinated and hyperphosphporylated at multiple sites. Although tau mutations have not been linked to AD dementia, several tau mutations are causative for other related neurodegenerative dementias indicating that tau dysfunction alone is sufficient for neuronal degeneration. Understanding the processes that influence amyloid plaque or neurofibrillary tangle formation may prove invaluable in mitigating AD and related neurodegenerative diseases.
Recent large-scale studies have been examining the relationship between clinical symptomology, various biomarkers of AD dementia, and brain pathology (Fig. 2).5–8,11 Biomarkers currently under investigation include cerebrospinal fluid (CSF) Aβ, CSF tau, structural and functional brain imaging using MRI or PET, and synthetic amyloid imaging compounds that detect cerebral amyloid. The clinical spectrum of disease has been expanded in recognition of the fact that AD is likely a protracted and insidious disease. A predementia stage called “mild cognitive impairment” (MCI) can be defined using careful psychometric testing, sometimes coincident with a subjective decline in memory, reasoning, or visual perception that does not reach the diagnostic threshold of dementia.7 Individuals with MCI are at risk for subsequent dementia. However, even before clinical signs can be detected, abnormal biomarker values can be detected in cognitively normal individuals, leading to the concept of preclinical disease characterized by early biological changes including the onset of CNS pathology.8
Figure 2.
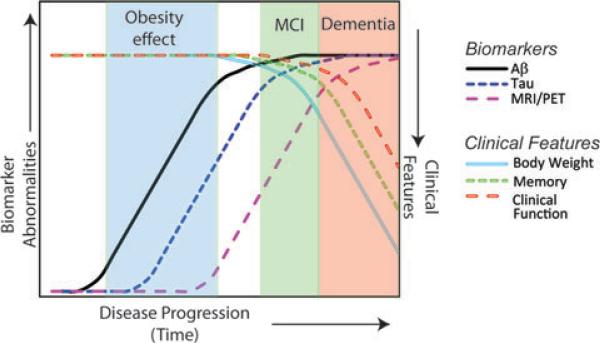
Model of the clinical and biomarker progression of AD. Current efforts are underway to define the relationship between progression of AD (x-axis), various AD biomarkers (left y-axis), and clinical parameters associated with AD (right y-axis). Abnormal biomarker values related to the Aβ peptide (CSF Aβ measurements and/or amyloid dye imaging techniques, black line) indicate that abnormalities related to Aβ can be detected in presymptomatic individuals. Consequently, tau abnormalities and pathology may then develop (blue dotted line), followed by changes in brain structure and function (MRI and PET scans, purple dashed line). The onset of memory decline (empty green dotted line) heralds the clinical designation of “mild cognitive impairment” (MCI), which is typically followed by an overt decline in clinical function (empty red dashed line) and dementia, most commonly diagnosed as AD. Also reflected in this model is the association between obesity and dementia that is thought to occur in the presymptomatic phase of the disease as well as the association between weight loss (empty light blue line) and dementia that typically occurs close to and during the later stages of AD. Adapted and modified from work by Jack et al.11
Body weight, obesity, and dementia
With the aging of the Baby Boomers, the number of Americans 65 and older will more than double from 2000 to 2050, and those 85 and older will increase by fivefold.12–14 Advanced age is the strongest risk factor for AD, and AD is the most common aging-related neurodegenerative disease, with a projected 13 million or more afflicted by 2050 and with a cost of over $1 trillion unless effective interventions are implemented soon.15–18 Obesity rates have also dramatically increased in the last 25 years such that greater than 1 in 3 adults are obese.19 Of all the demographic age groups, Baby Boomers have the highest rates of obesity at 40%.19 These demographics raise the public health concern of increases in aging-and obesity-related diseases.
The relationship between body weight and dementia is complex in that body weight has an age-dependent relationship with dementia (Fig. 2 and Table 1). Individuals with AD have lower body weight,20 and it is plausible that dementia leads to a negative energy balance secondary to malnutrition. However, reductions in body weight is a harbinger of AD even before clinical symptoms of dementia are detected,21,22 suggesting that loss of body weight may be a manifestation of early brain dysfunction. Furthermore, lower body mass indexes (BMIs) are associated with abnormal CSF Aβ and tau levels23 and with increased CNS pathology at autopsy including neurofibrillary tangles and amyloid plaques.24 Thus, weight loss is a consistent feature of AD dementia and correlates with the presence of abnormal biomarkers and increased brain pathology. However, weight loss associated with AD should be interpreted with caution since dual-energy x-ray absorptiometry studies suggest that weight loss is primarily due to sarcopenia and not loss of fat.25 Thus total body weight and BMI may not be valid surrogate measures of obesity in the elderly. Given this propensity for weight loss, studies of elderly cohorts examining the relationship between obesity and AD are mixed,21,22,26–32 perhaps reflecting the difficulties in defining obesity in elderly cohorts based on anthropomorphic measurements such as BMI. For example, using the waist-to-hip ratio as a measure of central obesity reveals that obesity is associated with a significant hazards ratio of 2.5 for the development of AD while BMI in the same elderly cohort was not significantly associated with an increased risk for AD.31
Table 1.
Summary of human studies on the relationships between obesity, AD, leptin, and brain function
| Reference | Study cohort | Adipokine changes |
|---|---|---|
| Stewart et al.21 | Honolulu–Asia Aging Study (32-year prospective longitudinal study); 1,890 Japanese-American men, of which 112 developed AD | Dementia is associated with weight loss. Weight loss precedes the onset of clinical dementia. Weight loss accelerates by the time of diagnosis. |
| Ewers et al.23 | Multicenter cross-sectional study (308 AD, 296 MCI, 147 controls) | Abnormal CSF biomarker signature (tau and β-amyloid) is associated with lower BMI. |
| Buchman et al.24 | Religious Order Study (clinical-pathologic study of Catholic clergy); 298 deceased subjects | AD pathology (using a global pathology measure) is associated with lower BMI. Other pathology (infarcts, Lewy bodies) did not correlate with BMI. |
| Burns et al.25 | University of Kansas Alzheimer and Memory Program cross-sectional study; of 70 early-stage AD, 70 controls | Lean mass is reduced in early AD individuals compared to non-demented controls. Lean mass is associated with whole-brain volume and white matter volume, and global cognitive performance. |
| Gustafson et al.26 | Swedish adults (longitudinal study, 70–88 years old); 392 non-demented individuals, of which 93 developed dementia | Women who developed dementia had higher BMI at age 70, 75, and 79 compared to women who did not develop dementia. No association was observed in men. |
| Gustafson et al.27 | Prospective Population Study of Women in Sweden (32-year-longitudinal study); 1,462 women, of which 161 developed dementia | Elevated midlife waist-to-hip ratio increases risk for dementia. |
| Buchman et al.28 | Religious Order Study (prospective longitudinal clinical-pathologic study of Catholic clergy, 5.5 years); 918 non-demented individuals, of which 151 developed AD | Declining BMI is associated with increased AD risk. |
| Hayden et al.29 | Cache County Study (community-based study in Utah); 3,264 individuals | Obesity (elevated BMI) increases risk of AD in females but not males. Hypertension increases risk of vascular dementia. Diabetes increases risk of vascular dementia in females but not males. |
| Fitzpatrick et al.30 | Multisite community-dwelling cohort (prospective longitudinal study, 5.4 years); 2,798 non-demented adults, of which 480 developed dementia (254 AD without vascular dementia and 213 vascular dementia +/− AD) | Midlife obesity (elevated BMI) increases risk for dementia. In late life, low BMI increases risk for dementia and obesity decreases risk for dementia. |
| Luchsinger et al.31 | Washington Heights–Inwood Columbia Aging Project (longitudinal study of randomly recruited community-dwelling cohort in New York City); 1,459 elderly (65+), of which 145 developed AD (5,734 person-years) | Waist-to-hip ratio (but not BMI) is associated with increased AD risk. |
| Vanhanen et al.32 | Random population study of cardiovascular risk factors and diabetes in Finland (longitudinal study, 3.5 years); 959 subjects (65–74 years old), of which 45 developed probable or possible AD | Metabolic syndrome is associated with AD. |
| Whitmer et al.33 | Kaiser Permanente Northern California Medical Group (longitudinal study, 27 years) 10,276 individuals of which 713 developed dementia | Individuals who were obese or overweight (elevated BMI) at midlife are at increased risk for dementia. |
| Whitmer et al.34 | Kaiser Permanente Northern California Medical Group (longitudinal study, 36 years); 10,136 individuals, of which 477 developed AD and 132 developed vascular dementia | Individuals who were obese or overweight (elevated BMI) at midlife are at increased risk for AD and vascular dementia. |
| Whitmer et al.35 | Kaiser Permanente Northern California Medical Group (longitudinal study, 36 years); 6,583 individuals, of which 1,049 developed dementia | Sagittal abdominal diameter as a measure of central obesity is associated with increased risk for dementia. |
| Kivipelto et al.36 | Cardiovascular Risk Factors, Aging and Dementia (CAIDE) study (random population-based longitudinal study, 21 years); 1,449 individuals, of which 61 developed dementia and 48 developed AD | Midlife obesity (elevated BMI) is associated with risk of dementia and risk of AD. Additional vascular factors (hypertension, hypercholesterolemia) also increase risk for dementia. |
| Chiang et al.37 | Nested case–control study of 157 demented and 628 control subjects | Being either underweight or overweight (by BMI) increases risk for dementia, AD, and vascular dementia. |
| Lieb et al.57 | Framingham study (longitudinal study, 8.3 years); 5,209 individuals, of which 111 developed dementia and 89 developed AD | Higher leptin (single measurement in asymptomatic individuals) is associated with lower risk of dementia and AD. Higher leptin is associated with higher cerebral brain volume by MRI. |
| Nartia et al.78 | Cross-sectional study of 34 elderly individuals without dementia or metabolic syndrome | Higher leptin is associated with increased gray matter volume in the hippocampus and cerebellum. |
| Holden et al.79 | Health, Aging and Body Composition (Health ABC) study (prospective longitudinal cohort study, 5 years) of well-functioning community-dwelling elders in Memphis, TN or Pittsburgh, PA | Higher leptin is associated with less cognitive decline as measured by the Modified Mini-Mental State Exam. |
| Matochik et al.80 | Three human adults with congenital leptin-deficiency treated with leptin; MRI study | Leptin increases gray matter in the anterior cingulate gyrus, inferior parietal lobule, and cerebellum. |
| Paz-Filho et al.81 | A five-year-old boy with congenital leptin deficiency treated with leptin | Baseline tests showed slow neurocognitive development. After two years of leptin treatment, most neurocognitive domains showed substantial improvement, with some areas exceeding expectations for age. |
| Baicy et al.81 | Three human adults with congenital leptin deficiency treated with leptin; fMRI study | Leptin reduces brain activation in insular, parietal, and temporal cortex (linked to hunger) and enhances brain activation in prefrontal cortex (linked to inhibition/satiety). |
| Farooqi et al.83 | Two human individuals with congenital leptin deficiency; fMRI study | Leptin modulates brain activation in ventral striatum and posterolateral ventral striatum (linked to reward/satiety). |
| Rosenbaum et al.84 | Six obese subjects examined at baseline and after 10% weight loss, then treated with leptin or placebo; fMRI study | Weight loss (a state of relative hypoleptinemia) alters neuronal activity in multiple brain regions. These changes in brain activation could be reversed by leptin treatment. |
Analysis of multiple biomarkers of AD has yielded a model in which AD is a protracted and insidious disease, with pathogenic processes occurring years if not decades prior to clinical symptoms.11 Keeping this temporal distinction in mind, it is intriguing that mid-life obesity has been found to increase the risk of developing late-life AD (hazards or odds ratios ranging from ~ 1.4 to 3.6; see Table 1).27,30,33–37 These studies define obesity with different metrics, including BMI, waist circumference, sagittal abdominal diameter, and waist-to-hip ratios. Diabetes mellitus and vascular disease are also associated with dementia.1 However, in as much as such factors can be treated as separate variables, the risk due to diabetes mellitus and vascular disease appears to be independent of the risk due to obesity. One important caveat to these studies is that most cohorts are clinically defined as having AD dementia without autopsy confirmation. Given the ubiquity of multiple brain pathologies in the elderly including a high prevalence of vascular disease, validation of these findings in autopsy-confirmed cohorts is desirable to define the relative contribution of vascular versus nonvascular pathology.38,39 Nonetheless, the fact that obesity confers increased risk for AD during the preclinical stage of AD suggests that obesity may modulate biological pathways early in the pathogenesis of AD. Identifying factors that influence brain pathology before the onset of overt neurodegeneration provides an avenue for possible preventative intervention.
Diet and obesity in experimental AD models
Multiple AD mouse models have been generated, most of which overexpress mutant forms of APP and/or tau.40 In general, expressing APP harboring mutations linked to human disease results in age-dependent accumulation of amyloid plaques and deficits in learning and memory behaviors (Fig. 1H).41,42 Mutant tau transgenic models develop intracellular tau aggregates that can lead to age-dependent neuronal degeneration.43 Although these models overexpress high levels of mutant protein and are therefore not entirely physiologic, these mice have proven invaluable in understanding the factors that influence the neuropathology of AD. APP transgenic mice have been fed various diets to determine whether diet can influence cerebral amyloid deposition. Several studies have documented increases in body weight or adiposity in APP transgenic mice in response to high-calorie diets. Diets range from high-fat diets to high-fat and high-cholesterol diets to high-sucrose diets. Remarkably, despite differences in dietary nutrient content, diet-induced obesity is consistently associated with an increase in cerebral amyloid pathology.44–50 The only exception is a single study in which a Western diet was administered for only four weeks to examine relatively acute changes at an age (one to two months) before amyloid plaque pathology is found.51 Likewise, several dietary regimens associated with weight loss have been tested, including ketogenic diets and caloric-restriction diets. Again, studies that document weight loss uniformly show a correlation between weight reduction and decreased cerebral amyloid pathology.52–56 These studies, performed on multiple different APP transgenic strains using a variety of diets from several independent laboratories, provide strong evidence that diet, body weight, and obesity modulate cerebral amyloid pathology.
Leptin and AD dementia
There are many possible factors and mechanisms that may account for this modulation of AD pathology. Indeed, several studies suggest that adipokines, and in particular leptin, may influence the pathogenesis of AD. A prospective study of 785 participants has indicated that higher circulating levels of leptin are associated with a reduction in AD incidence.57 Higher leptin was associated with a lower incidence of dementia including AD dementia (hazard ratio for AD ~ 0.6), even when correcting for the waist-to-hip~ratio, BMI, and vascular risk factors in multivariate statistical models. Higher leptin levels were also associated with higher total cerebral brain volume in the subset of participants who underwent MRI imaging. Since leptin levels are known to fluctuate over time, it is remarkable that a significant association between leptin and AD could be detected using a single leptin measurement. The major determinant of circulating leptin levels is adipose tissue mass, and typically hyperleptinemia is associated with obesity and central leptin resistance. Thus, it is interesting that there was no association between leptin and AD incidence in the top quartile of participants based on waist-to-hip measurements or in individuals with a BMI > 30. Finally, the mean follow-up time of 8.3 years suggests that many leptin measurements were performed in the preclinical or MCI stages of the disease. This study suggests that leptin during the presymptomatic phase of AD, at least in nonobese individuals, may be neuroprotective and mitigate the progression of AD.
Secreted by adipose tissue, leptin regulates body weight by modulating LR-expressing neurons in the CNS, particularly within the hypothalamus and brainstem.58–60 However, leptin has pleiotropic metabolic effects, regulating energy expenditure, feeding behavior, locomotor activity, bone mass, growth, thermogenesis, fertility, life span, adrenal function, and thyroid function. Thus, mice with congenital absence of leptin (ob/ob mice) or leptin's signaling receptor (db/db mice) exhibit a complex phenotype with abnormalities in virtually every organ system. These diverse effects are most congruous with the absence of leptin acting as a signal of starvation, triggering numerous compensatory changes that secondarily lead to obesity.61 Thus, leptin-deficient models are a complex hybrid of the starvation response and the numerous secondary effects of obesity.
Leptin acts through the longest isoform of leptin receptors (LRb), the only isoform containing the cytoplasmic signaling domain.58–60 Leptin binding triggers phosphorylation of cytoplasmic tyrosine residues that initiate various signaling pathways including JAK2-STAT3, Erk1/2, and PI3K pathways (Fig. 3). Other signaling molecules may be regulated by leptin, such as AMP kinase (AMPK) and mammalian target of rapamycin complex 1 (mTOR1).62–65 However, the signaling mechanisms by which leptin affects these molecules are not entirely known, and it is not known whether these pathways result from a direct effect of leptin on LRb.
Figure 3.
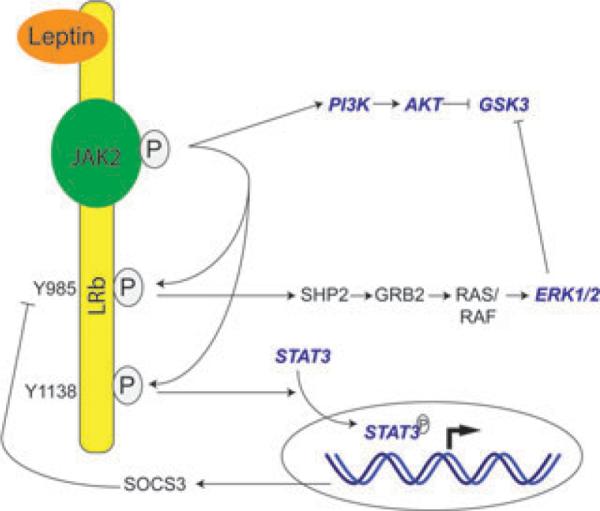
Leptin receptor signaling. LRb is constitutively associated with Janus family of tyrosine kinase 2 (JAK2). Leptin binding to LRb triggers autophosporylation of JAK2155–157 and subsequent phosphorylation of cytoplasmic LRb tyrosine residues, including Y1138 and Y985. Phosphorylation of Y1138 recruits STAT3, which is then phosphorylated and translocated into the nucleus to alter gene expression, including upregulation of suppressor of cytokine signaling 3 (SOCS3) expression.142,155 SOCS3 binds to Y985 as a negative feedback regulator of LRb signaling.141 Y985 serves dual roles in that its phosphorylation promotes binding of the tyrosine phosphatase SHP-2, leading to downstream Erk1/2 phosphorylation/activation.155,158 In cultured cells, LR activation also triggers phosphoinositide 3-kinase (PI3K)-Akt signaling,86,89,93,94 although it is unclear whether Akt phosphorylation occurs in vivo.98,159–161 Both Erk1/2 (via p90RSK) and Akt phosphorylation can lead to downstream GSK3 phsophorylation/inhibition.86 Signaling molecules that are known to affect APP processing are highlighted in blue italics.
LRs are present in both hypothalamic and extrahypothalamic neurons, including neurons of the hippocampus and cerebral cortex.66–74 The major metabolic effects are predominantly due to leptin action on hypothalamic and hindbrain neurons. However, several lines of evidence suggest that leptin has nonmetabolic CNS effects as well. For example, leptin does not exert any metabolic effects in mice prior to weaning despite a large postnatal surge, indicative of a function distinct from its role in metabolism.75,76 The brains of ob/ob mice are smaller with reduced levels of synaptic proteins, abnormalities that are partially reversed by leptin treatment.77 In normal elderly, circulating leptin levels correlate with gray matter volume in various brain regions including the hippocampus,57,78 and inversely correlates with cognitive decline.79 Leptin also reverses neurocognitive deficits and structural abnormalities in multiple brain regions in humans with congenital leptin deficiency.80,81 In leptin-deficient individuals and people with recent weight loss (representing a state of relative leptin deficiency), exogenous leptin alters brain activation in response to food cues.82–84 These findings indicate that leptin has strong effects on brain structure and function outside the hypothalamus.
A growing literature indicates that leptin is neurotrophic. Leptin promotes dendritic growth cones/filipodia outgrowth in hippocampal and cortical neurons in vitro85,86 and exhibits trophic activity on neurons that regulate feeding behavior in vivo.87,88 Leptin increases adult hippocampal neurogenesis89 and enhances hippocampal long-term potentiation by enhancing NMDA receptor function in part through MAPK.90 Indeed, db/db mice exhibit defects in hippocampal neuronal morphology and hippocampus-dependent learning and memory behaviors.91,92 In addition to its neurotrophic role, leptin appears to be neuroprotective,77 as seen in various models of neuronal injury including injury related to stroke, seizure, neurotrophin withdrawal, excitotoxicity, oxidative damage, apoptosis, 6-hyrdoxydopamine, and tumor necrosis factor-α.93–99 The pleotropic effects of leptin on neuronal integrity and function makes it possible that leptin may have beneficial effects on the CNS independent of the pathogenic mechanisms of AD.
More relevant to the pathogenesis of AD, leptin reduces Aβ secretion in cultured neuronal cells or organotypic slices,45,100,101 and chronic leptin treatment with supraphysiologic doses reduces Aβ levels in brain and serum of APP Tg mice.45,102 The reduction of Aβ appears to reduce β-secretase expression and/or activity, and the reduction can be blocked by an inhibitor of the AMPK in cultured cells.45,100,101 Leptin also reduces tau phosphorylation in cultured neuronal cells and organotypic cultures, and this effect was blocked by AMPK and glycogen synthase kinase-3 (GSK3) inhibitors.100,101,103,104 Several issues remain with regards to the mechanisms whereby leptin may modulate AD pathology. Although activating leptin pathways may influence Aβ or tau pathways, it remains unclear whether altered leptin signaling is physiologically relevant in terms of AD pathogenesis. Toward that end, crossing APP transgenic mice onto a leptin-deficient ob/ob background results in worsening of cognitive function, enhancement of cerebrovascular inflammation, and increased cerebral amyloid angiopathy.105 A fundamental issue with understanding the mechanisms of leptin action in vivo is the vast number of metabolic and physiologic parameters that are regulated by leptin. Thus ob/ob mice represent an extreme phenotype compared to diet-induced obesity models. Another question that deserves attention reflects the inherent complexity of CNS circuitry, and thus it is unknown whether leptin acts directly on cortical or hippocampal neurons to inhibit amyloid plaque deposition as opposed to an indirect effect via hypothalamic relay neurons. Finally, most cultured cell lines do not express the long LRb isoform of the leptin receptor, and thus it is important to determine whether leptin's effects are mediated through LRb and the known downstream LRb signaling pathways. For example, although it is proposed that leptin modulates Aβ and tau through AMPK, it is unclear how leptin activates AMPK. In skeletal muscle, much of AMPK activation in vivo is through autonomic innervation and not a direct effect of leptin binding to LRb.64 Leptin actually decreases AMPK activity in the hypothalamus and knockout of AMPK in POMC and AgRP expressing hypothalamic neurons has no effect on their leptin responsivity.63,106
Many of the other receptor-signaling molecules downstream of LRb are known to affect secretase activity or APP trafficking (Fig. 3, highlighted in blue). First, STAT3 increases BACE expression by binding to its promoter and is also thought to mediate Aβ toxicity.107–110 Second, Erk1/2 activation increases α-secretase activity, and may inhibit β- and γ-secretase activity.111–120 PI3K activation increases APP trafficking and the secretion of APP metabolites,121–124 and constitutively active Akt inhibits APP trafficking by feedback inhibition of PI3K.125 Finally, inhibition of GSK3 results in an inhibition of γ-secretase.126,127 Therefore, leptin signaling can potentially intersect with APP processing pathways at multiple levels, and it remains to be determined which pathway mediates leptin's antiamyloidogenic effects in vivo.
One additional consideration is the relationship between obesity, aging, and leptin. Circulating leptin levels are high in obese humans and rodents in correlation with adiposity.60 Aging also results in increased leptin levels and adiposity.128–130 The maintenance of increased fat despite hyperleptinemia indicates that obesity and aging are both states of leptin resistance. The exact mechanisms of leptin resistance are not completely understood but are in part due to defective leptin transport across the blood–brain barrier and in part are intrinsic to leptin-responsive neurons.129–140 Intrinsic neuronal leptin resistance has been linked to increased feedback inhibition by SOCS3 and increased protein tyrosine phosphatase 1B (PTP1B) activity, both of which dampen downstream LR signaling pathways.141–151 This interrelationship between peripheral leptin, central leptin resistance, obesity, and aging should be considered in future clinical and experimental studies on leptin and AD.
Conclusions
Advances in the fields of endocrinology and neuropathology are beginning to reveal a complex relationship between peripheral metabolic factors and brain health. Although obesity increases the risk for AD in humans and diet-induced obesity modulates AD pathology in transgenic mice, the mechanisms that account for these phenomena are not entirely known. In terms of leptin, increased peripheral leptin is associated with reduced incidence of AD in the nonobese elderly, and treating AD transgenic mice with leptin ameliorates AD pathology. It remains to be determined whether central leptin resistance associated with aging and obesity enhances amyloid plaque deposition. Also, the effects of other adipokines, in particular the inflammatory adipokines (e.g., adiponectin, cytokines, and complement), have received little attention with regards to their contribution to AD. Adiponectin is thought to be complementary to the actions of leptin and exhibits anti-inflammatory properties. However, only two small studies have been reported with conflicting results as to whether changes in plasma adiponectin are related to MCI or AD.152,153 Thus, further studies of the relationship between adipokines and AD are warranted. Finally, despite the emphasis on leptin in this review, several genes associated with lipid homeostasis are thought to influence AD risk including genes encoding apolipoprotein E, apolipoprotein J (clusterin), and sortilin-related receptor.154 Undoubtedly, multiple metabolic and hormonal changes associated with obesity and aging will prove to influence the brain in health and disease. Together, this burgeoning field is proving that the brain is under the influence of peripheral metabolism and that an integrated physiologic approach to understanding brain health and disease may reveal novel mechanisms that may be amenable to therapeutic intervention.
Acknowledgments
EBL is supported by NIH/NIA K08-AG039510.
Footnotes
Conflicts of interest
The author declares no conflicts of interest.
References
- 1.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch. Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luchsinger JA, Mayeux R. Adiposity and Alzheimer's disease. Curr. Alzheimer Res. 2007;4:127–134. doi: 10.2174/156720507780362100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitmer RA. The epidemiology of adiposity and dementia. Curr. Alzheimer Res. 2007;4:117–122. doi: 10.2174/156720507780362065. [DOI] [PubMed] [Google Scholar]
- 4.Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer's disease. J. Alzheimers Dis. 2009;16:693–704. doi: 10.3233/JAD-2009-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jack CR, Jr., Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EB, Lee VM. Biology and molecular neuropathology of beta-amyloid protein. In: Dawbarn D, Allen SJ, editors. Neurobiology of Alzheimer's Disease. 3rd Ed. Oxford University Press; Oxford: 2007. pp. 81–110. [Google Scholar]
- 10.Lee VM, Goedert M, Trojanowski JQ. Neurode-generative tauopathies. Annu.Rev.Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 11.Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hetzel L, Smith A. Census Bureau Brief, C2KBR/01-10. U.S. Census Bureau; Washington, DC: 2001. The 65 Years and Over Population: 2000. [Google Scholar]
- 13.Meyer J. Census Bureau Brief C2KBR/01-12. U.S. Census Bureau; Washington, DC: 2001. Age: 2000. [Google Scholar]
- 14.Federal Interagency Forum on Aging-Related Statistics . Older Americans 2008: key indicators of well-being. U.S. Government Printing Office; Washington, DC: 2008. [Google Scholar]
- 15.The Lewin Group Saving lives, saving money: Dividends for Americans investing in Alzheimer research. Alzheimer's Association Report. 2006 2006. [Google Scholar]
- 16.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cogan J, Mitchell O. Perspectives from the President's Commission on Social Security Reform. J. Econom. Perspect. 2003;7:149–172. [Google Scholar]
- 18.Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch. Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 19.Ogden CL, Carroll MD, McDowell MA, Flegal KM. NCHS Data Brief No 1. National Center for Health Statistics; Hyattsville, MD: 2007. Obesity among adults in the United States—no statistically significant change since 2003–2004. [PubMed] [Google Scholar]
- 20.Gillette Guyonnet S, Abellan Van Kan G, Alix E, et al. IANA (International Academy on Nutrition and Aging) Expert group: weight loss and Alzheimer's disease. J. Nutr. Health Aging. 2007;11:38–48. [PubMed] [Google Scholar]
- 21.Stewart R, Masaki K, Xue QL, et al. A 32-year prospective study of change in body weight and incident dementia: the Honoluluy–Asia aging study. Arch. Neurol. 2005;62:55–60. doi: 10.1001/archneur.62.1.55. [DOI] [PubMed] [Google Scholar]
- 22.Barrett-Connor E, Edelstein S, et al. Weight loss precedes dementia in community-dwelling older adults. J. Nutr. Health Aging. 1998;2:113–114. [PubMed] [Google Scholar]
- 23.Ewers M, Schmitz S, Hansson O, et al. Body mass index is associated with biological CSF markers of core brain pathology of Alzheimer's disease. Neurobiol. Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.05.005. [Epub ahead of print]. doi:10.1016/j.neurbiolaging.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchman AS, Schneider JA, Wilson RS, et al. Body mass index in older persons is associated with Alzheimer disease pathology. Neurology. 2006;67:1949–1954. doi: 10.1212/01.wnl.0000247046.90574.0f. [DOI] [PubMed] [Google Scholar]
- 25.Burns JM, Johnson DK, Watts A, et al. Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol. 2010;67:428–433. doi: 10.1001/archneurol.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gustafson D, Rothenberg E, Blennow K, et al. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 2003;163:1524–1528. doi: 10.1001/archinte.163.13.1524. [DOI] [PubMed] [Google Scholar]
- 27.Gustafson DR, Backman K, Waern M, et al. Adiposity indicators and dementia over 32 years in Sweden. Neurology. 2009;73:1559–1566. doi: 10.1212/WNL.0b013e3181c0d4b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchman AS, Wilson RS, Bienias JL, et al. Change in body mass index and risk of incident Alzheimer disease. Neurology. 2005;65:892–897. doi: 10.1212/01.wnl.0000176061.33817.90. [DOI] [PubMed] [Google Scholar]
- 29.Hayden KM, Zandi PP, Lyketsos CG, et al. Vascular risk factors for incident Alzheimer disease and vascular dementia: the Cache County study. Alzheimer Dis. Assoc. Disord. 2006;20:93–100. doi: 10.1097/01.wad.0000213814.43047.86. [DOI] [PubMed] [Google Scholar]
- 30.Fitzpatrick AL, Kuller LH, Lopez OL, et al. Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch. Neurol. 2009;66:336–342. doi: 10.1001/archneurol.2008.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luchsinger JA, Cheng D, Tang MX, et al. Central obesity in the elderly is related to late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011 doi: 10.1097/WAD.0b013e318222f0d4. doi:10.1097/WAD.0b013e318222f0d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanhanen M, Koivisto K, Moilanen L, et al. Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology. 2006;67:843–847. doi: 10.1212/01.wnl.0000234037.91185.99. [DOI] [PubMed] [Google Scholar]
- 33.Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005;330:1360–1362. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitmer RA, Gunderson EP, Quesenberry CP, Jr., et al. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 2007;4:103–109. doi: 10.2174/156720507780362047. [DOI] [PubMed] [Google Scholar]
- 35.Whitmer RA, Gustafson DR, Barrett-Connor E, et al. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- 36.Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005;62:1556–1560. doi: 10.1001/archneur.62.10.1556. [DOI] [PubMed] [Google Scholar]
- 37.Chiang CJ, Yip PK, Wu SC, et al. Midlife risk factors for subtypes of dementia: a nested case-control study in Taiwan. Am. J. Geriatr. Psychiatr. 2007;15:762–771. doi: 10.1097/JGP.0b013e318050c98f. [DOI] [PubMed] [Google Scholar]
- 38.Nelson PT, Smith CD, Abner EA, et al. Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim. Biophys. Acta. 2009;1792:454–469. doi: 10.1016/j.bbadis.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnen JA, Larson EB, Brickell K, et al. Different patterns of cerebral injury in dementia with or without diabetes. Arch. Neurol. 2009;66:315–322. doi: 10.1001/archneurol.2008.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duff K, Suleman F. Transgenic mouse models of Alzheimer's disease: how useful have they been for therapeutic development? Brief Funct. Genomic Proteomic. 2004;3:47–59. doi: 10.1093/bfgp/3.1.47. [DOI] [PubMed] [Google Scholar]
- 41.Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 42.Games D, Adams D, Alessandrini R, et al. Alzheimer-type neuropathology in transgenic mice over-expressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 43.Ishihara T, Hong M, Zhang B, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 44.Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- 45.Fewlass DC, Noboa K, Pi-Sunyer FX, et al. Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. 2004;18:1870–1878. doi: 10.1096/fj.04-2572com. [DOI] [PubMed] [Google Scholar]
- 46.Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 47.Julien C, Tremblay C, Phivilay A, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 48.Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J. Nutr. Health Aging. 2002;6:315–319. [PubMed] [Google Scholar]
- 49.Pedrini S, Thomas C, Brautigam H, et al. Dietary composition modulates brain mass and solubilizable Abeta levels in a mouse model of aggressive Alzheimer's amyloid pathology. Mol. Neurodegener. 2009;4:40. doi: 10.1186/1750-1326-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Z, Ho L, Wang J, et al. Connective tissue growth factor (CTGF) expression in the brain is a downstream effector of insulin resistance- associated promotion of Alzheimer's disease beta-amyloid neuropathology. FASEB J. 2005;19:2081–2082. doi: 10.1096/fj.05-4359fje. [DOI] [PubMed] [Google Scholar]
- 51.Studzinski CM, Li F, Bruce-Keller AJ, et al. Effects of short-term Western diet on cerebral oxidative stress and diabetes related factors in APP x PS1 knock-in mice. J. Neurochem. 2009;108:860–866. doi: 10.1111/j.1471-4159.2008.05798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halagappa VK, Guo Z, Pearson M, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 53.Mouton PR, Chachich ME, Quigley C, et al. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci. Lett. 2009;464:184–187. doi: 10.1016/j.neulet.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel NV, Gordon MN, Connor KE, et al. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 55.Van der Auwera I, Wera S, et al. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr. Metab. 2005;2:28. doi: 10.1186/1743-7075-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Ho L, Qin W, et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- 57.Lieb W, Beiser AS, Vasan RS, et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA. 2009;302:2565–2572. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahima RS, Flier JS. Leptin. Annu. Rev. Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 59.Ahima RS, Osei SY. Leptin signaling. Physiol. Behav. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 60.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 61.Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 62.Andersson U, Filipsson K, Abbott CR, et al. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004;279:12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 63.Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 64.Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 65.Villanueva EC, Munzberg H, Cota D, et al. Complex regulation of mammalian target of rapamycin complex 1 in the basomedial hypothalamus by leptin and nutritional status. Endocrinology. 2009;150:4541–4551. doi: 10.1210/en.2009-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baskin DG, Schwartz MW, Seeley RJ, et al. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J. Histochem. Cytochem. 1999;47:353–362. doi: 10.1177/002215549904700309. [DOI] [PubMed] [Google Scholar]
- 67.Burguera B, Couce ME, Long J, et al. The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology. 2000;71:187–195. doi: 10.1159/000054536. [DOI] [PubMed] [Google Scholar]
- 68.Elmquist JK, Bjorbaek C, Ahima RS, et al. Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- 69.Guan XM, Hess JF, Yu H, et al. Differential expression of mRNA for leptin receptor isoforms in the rat brain. Mol. Cell. Endocrinol. 1997;133:1–7. doi: 10.1016/s0303-7207(97)00138-x. [DOI] [PubMed] [Google Scholar]
- 70.Hakansson ML, Hulting AL, Meister B. Expression of leptin receptor mRNA in the hypothalamic arcuate nucleus—relationship with NPY neurones. Neuroreport. 1996;7:3087–3092. doi: 10.1097/00001756-199611250-00059. [DOI] [PubMed] [Google Scholar]
- 71.Huang XF, Koutcherov I, Lin S, et al. Localization of leptin receptor mRNA expression in mouse brain. Neuroreport. 1996;7:2635–2638. doi: 10.1097/00001756-199611040-00045. [DOI] [PubMed] [Google Scholar]
- 72.Mercer JG, Moar KM, Hoggard N. Localization of leptin receptor (Ob-R) messenger ribonucleic acid in the rodent hindbrain. Endocrinology. 1998;139:29–34. doi: 10.1210/endo.139.1.5685. [DOI] [PubMed] [Google Scholar]
- 73.Savioz A, Charnay Y, Huguenin C, et al. Expression of leptin receptor mRNA (long form splice variant) in the human cerebellum. Neuroreport. 1997;8:3123–3126. doi: 10.1097/00001756-199709290-00023. [DOI] [PubMed] [Google Scholar]
- 74.Scott MM, Lachey JL, Sternson SM, et al. Leptin targets in the mouse brain. J. Comp. Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ahima RS, Hileman SM. Postnatal regulation of hypothalamic neuropeptide expression by leptin: implications for energy balance and body weight regulation. Regul. Pept. 2000;92:1–7. doi: 10.1016/s0167-0115(00)00142-7. [DOI] [PubMed] [Google Scholar]
- 76.Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J. Clin. Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahima RS, Bjorbaek C, Osei S, Flier JS. Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology. 1999;140:2755–2762. doi: 10.1210/endo.140.6.6774. [DOI] [PubMed] [Google Scholar]
- 78.Narita K, Kosaka H, Okazawa H, et al. Relationship between plasma leptin level and brain structure in elderly: a voxel-based morphometric study. Biol. Psychiatr. 2009;65:992–994. doi: 10.1016/j.biopsych.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 79.Holden KF, Lindquist K, Tylavsky FA, et al. Serum leptin level and cognition in the elderly: findings from the Health ABC Study. Neurobiol. Aging. 2009;30:1483–1489. doi: 10.1016/j.neurobiolaging.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matochik JA, London ED, Yildiz BO, et al. Effect of leptin replacement on brain structure in genetically leptin-deficient adults. J. Clin. Endocrinol. Metab. 2005;90:2851–2854. doi: 10.1210/jc.2004-1979. [DOI] [PubMed] [Google Scholar]
- 81.Paz-Filho GJ, Babikian T, Asarnow R, et al. Leptin replacement improves cognitive development. PLoS One. 2008;3:e3098. doi: 10.1371/journal.pone.0003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baicy K, London ED, Monterosso J, et al. Leptin replacement alters brain response to food cues in genetically leptin-deficient adults. Proc. Natl. Acad. Sci. USA. 2007;104:18276–18279. doi: 10.1073/pnas.0706481104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farooqi IS, Bullmore E, Keogh J, et al. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rosenbaum M, Sy M, Pavlovich K, et al. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J. Clin. Invest. 2008;118:2583–2591. doi: 10.1172/JCI35055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O'Malley D, MacDonald N, Mizielinska S, et al. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol. Cell. Neurosci. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Valerio A, Ghisi V, Dossena M, et al. Leptin increases axonal growth cone size in developing mouse cortical neurons by convergent signals inactivating glycogen synthase kinase-3beta. J. Biol. Chem. 2006;281:12950–12958. doi: 10.1074/jbc.M508691200. [DOI] [PubMed] [Google Scholar]
- 87.Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 88.Bouret SG, Gorski JN, Patterson CM, et al. Hypothalamic neural projections are permanently disrupted in diet-induced obese rats. Cell Metab. 2008;7:179–185. doi: 10.1016/j.cmet.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garza JC, Guo M, Zhang W, Lu XY. Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J. Biol. Chem. 2008;283:18238–18247. doi: 10.1074/jbc.M800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stranahan AM, Arumugam TV, Cutler RG, et al. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stranahan AM, Lee K, Martin B, et al. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doherty GH, Oldreive C, Harvey J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience. 2008;154:1297–1307. doi: 10.1016/j.neuroscience.2008.04.052. [DOI] [PubMed] [Google Scholar]
- 94.Guo Z, Jiang H, Xu X, et al. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008;283:1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- 95.Shanley LJ, O'Malley D, Irving AJ, et al. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J. Physiol. 2002;545:933–944. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valerio A, Dossena M, Bertolotti P, et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke. 2009;40:610–617. doi: 10.1161/STROKEAHA.108.528588. [DOI] [PubMed] [Google Scholar]
- 97.Weng Z, Signore AP, Gao Y, et al. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007;282:34479–34491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- 98.Zhang F, Chen J. Leptin protects hippocampal CA1 neurons against ischemic injury. J. Neurochem. 2008;107:578–587. doi: 10.1111/j.1471-4159.2008.05645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang F, Wang S, Signore AP, Chen J. Neuro-protective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38:2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]
- 100.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marwarha G, Dasari B, Prasanthi JR, et al. Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J. Alzheimers Dis. 2010;19:1007–1019. doi: 10.3233/JAD-2010-1298. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Greco SJ, Bryan KJ, Sarkar S, et al. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer's disease. J. Alzheimers Dis. 2010;19:1155–1167. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Greco SJ, Sarkar S, Casadesus G, et al. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci. Lett. 2009;455:191–194. doi: 10.1016/j.neulet.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Greco SJ, Sarkar S, Johnston JM, et al. Leptin reduces Alzheimer's disease-related tau phosphorylation in neuronal cells. Biochem. Biophys. Res. Commun. 2008;376:536–541. doi: 10.1016/j.bbrc.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Claret M, Smith MA, Batterham RL, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wan J, Fu AK, Ip FC, et al. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer's disease. J. Neurosci. 2010;30:6873–6881. doi: 10.1523/JNEUROSCI.0519-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sambamurti K, Kinsey R, Maloney B, et al. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- 109.Tamagno E, Parola M, Bardini P, et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 110.Wen Y, Yu WH, Maloney B, et al. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mills J, Laurent Charest D, Lam F, et al. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J. Neurosci. 1997;17:9415–9422. doi: 10.1523/JNEUROSCI.17-24-09415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Avramovich Y, Amit T, Youdim MB. Nonsteroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J. Biol. Chem. 2002;277:31466–31473. doi: 10.1074/jbc.M201308200. [DOI] [PubMed] [Google Scholar]
- 113.Liu F, Su Y, Li B, Ni B. Regulation of amyloid precursor protein expression and secretion via activation of ERK1/2 by hepatocyte growth factor in HEK293 cells transfected with APP751. Exp. Cell Res. 2003;287:387–396. doi: 10.1016/s0014-4827(03)00152-6. [DOI] [PubMed] [Google Scholar]
- 114.Manthey D, Heck S, Engert S, Behl C. Estrogen induces a rapid secretion of amyloid beta precursor protein via the mitogen-activated protein kinase pathway. Eur. J. Biochem. 2001;268:4285–4291. doi: 10.1046/j.1432-1327.2001.02346.x. [DOI] [PubMed] [Google Scholar]
- 115.Canet-Aviles RM, Anderton M, Hooper NM, et al. Muscarine enhances soluble amyloid precursor protein secretion in human neuroblastoma SH-SY5Y by a pathway dependent on protein kinase C(alpha), src-tyrosine kinase and extracellular signal-regulated kinase but not phospholipase C. Brain Res. Mol. Brain Res. 2002;102:62–72. doi: 10.1016/s0169-328x(02)00184-5. [DOI] [PubMed] [Google Scholar]
- 116.Tamagno E, Guglielmotto M, Giliberto L, et al. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol. Aging. 2009;30:1563–1573. doi: 10.1016/j.neurobiolaging.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 117.Kim SK, Park HJ, Hong HS, et al. ERK1/2 is an endogenous negative regulator of the gamma-secretase activity. FASEB J. 2006;20:157–159. doi: 10.1096/fj.05-4055fje. [DOI] [PubMed] [Google Scholar]
- 118.Kojro E, Postina R, Buro C, et al. The neuropeptide PACAP promotes the alpha-secretase pathway for processing the Alzheimer amyloid precursor protein. FASEB J. 2006;20:512–514. doi: 10.1096/fj.05-4812fje. [DOI] [PubMed] [Google Scholar]
- 119.Bandyopadhyay S, Hartley DM, Cahill CM, et al. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006;84:106–118. doi: 10.1002/jnr.20864. [DOI] [PubMed] [Google Scholar]
- 120.Zhu X, Lee HG, Raina AK, et al. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 121.Adlerz L, Holback S, Multhaup G, Iverfeldt K. IGF-1-induced processing of the amyloid precursor protein family is mediated by different signaling pathways. J. Biol. Chem. 2007;282:10203–10209. doi: 10.1074/jbc.M611183200. [DOI] [PubMed] [Google Scholar]
- 122.Gasparini L, Gouras GK, Wang R, et al. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001;21:2561–2570. doi: 10.1523/JNEUROSCI.21-08-02561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Petanceska SS, Gandy S. The phosphatidylinositol 3-kinase inhibitor wortmannin alters the metabolism of the Alzheimer's amyloid precursor protein. J. Neurochem. 1999;73:2316–2320. doi: 10.1046/j.1471-4159.1999.0732316.x. [DOI] [PubMed] [Google Scholar]
- 124.Solano DC, Sironi M, Bonfini C, et al. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14:1015–1022. doi: 10.1096/fasebj.14.7.1015. [DOI] [PubMed] [Google Scholar]
- 125.Shineman DW, Dain AS, Kim ML, Lee VM. Constitutively active Akt inhibits trafficking of amyloid precursor protein and amyloid precursor protein metabolites through feedback inhibition of phosphoinositide 3-kinase. Biochemistry. 2009;48:3787–3794. doi: 10.1021/bi802070j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 127.Su Y, Ryder J, Li B, et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 128.Ahren B, Mansson S, Gingerich RL, Havel PJ. Regulation of plasma leptin in mice: influence of age, high-fat diet, and fasting. Am.J.Physiol. 1997;273:R113–120. doi: 10.1152/ajpregu.1997.273.1.R113. [DOI] [PubMed] [Google Scholar]
- 129.Li H, Matheny M, Nicolson M, et al. Leptin gene expression increases with age independent of increasing adiposity in rats. Diabetes. 1997;46:2035–2039. doi: 10.2337/diab.46.12.2035. [DOI] [PubMed] [Google Scholar]
- 130.Scarpace PJ, Matheny M, Moore RL, Tumer N. Impaired leptin responsiveness in aged rats. Diabetes. 2000;49:431–435. doi: 10.2337/diabetes.49.3.431. [DOI] [PubMed] [Google Scholar]
- 131.Carrascosa JM, Ros M, Andres A, et al. Changes in the neuroendocrine control of energy homeostasis by adiposity signals during aging. Exp. Gerontol. 2009;44:20–25. doi: 10.1016/j.exger.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 132.Gabriely I, Ma XH, Yang XM, et al. Leptin resistance during aging is independent of fat mass. Diabetes. 2002;51:1016–1021. doi: 10.2337/diabetes.51.4.1016. [DOI] [PubMed] [Google Scholar]
- 133.Ma XH, Muzumdar R, Yang XM, et al. Aging is associated with resistance to effects of leptin on fat distribution and insulin action. J. Gerontol. A Biol. Sci. Med. Sci. 2002;57:B225–231. doi: 10.1093/gerona/57.6.b225. [DOI] [PubMed] [Google Scholar]
- 134.Muzumdar RH, Ma X, Yang X, et al. Central resistance to the inhibitory effects of leptin on stimulated insulin secretion with aging. Neurobiol. Aging. 2006;27:1308–1314. doi: 10.1016/j.neurobiolaging.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 135.Qian H, Azain MJ, Hartzell DL, Baile CA. Increased leptin resistance as rats grow to maturity. Proc. Soc. Exp. Biol. Med. 1998;219:160–165. doi: 10.3181/00379727-219-44330. [DOI] [PubMed] [Google Scholar]
- 136.Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104::1111–1117. doi: 10.1016/s0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 137.Shek EW, Scarpace PJ. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regul. Pept. 2000;92:65–71. doi: 10.1016/s0167-0115(00)00151-8. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Y, Matheny M, Tumer N, Scarpace PJ. Aged-obese rats exhibit robust responses to a melanocortin agonist and antagonist despite leptin resistance. Neurobiol. Aging. 2004;25:1349–1360. doi: 10.1016/j.neurobiolaging.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 139.El-Haschimi K, Pierroz DD, Hileman SM, et al. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandez-Galaz C, Fernandez-Agullo T, Campoy F, et al. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. J. Endocrinol. 2001;171:23–32. doi: 10.1677/joe.0.1710023. [DOI] [PubMed] [Google Scholar]
- 141.Bjorbak C, Lavery HJ, Bates SH, et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 142.Dunn SL, Bjornholm M, Bates SH, et al. Feedback inhibition of leptin receptor/Jak2 signaling via Tyr1138 of the leptin receptor and suppressor of cytokine signaling 3. Mol. Endocrinol. 2005;19:925–938. doi: 10.1210/me.2004-0353. [DOI] [PubMed] [Google Scholar]
- 143.Bjorbaek C, Elmquist JK, Frantz JD, et al. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 144.Peralta S, Carrascosa JM, Gallardo N, et al. Ageing increases SOCS-3 expression in rat hypothalamus: effects of food restriction. Biochem. Biophys. Res. Commun. 2002;296:425–428. doi: 10.1016/s0006-291x(02)00906-3. [DOI] [PubMed] [Google Scholar]
- 145.Wang ZW, Pan WT, Lee Y, et al. The role of leptin resistance in the lipid abnormalities of aging. FASEB J. 2001;15:108–114. doi: 10.1096/fj.00-0310com. [DOI] [PubMed] [Google Scholar]
- 146.Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 147.Cheng A, Uetani N, Simoncic PD, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 148.Morrison CD, White CL, Wang Z, et al. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148:433–440. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.White CL, Whittington A, Barnes MJ, et al. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am. J. Physiol. Endocrinol. Metab. 2009;296:E291–E299. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 151.Zabolotny JM, Kim YB, Welsh LA, et al. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Une K, Takei YA, Tomita N, et al. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer's disease. Eur. J. Neurol. 2011;18:1006–1009. doi: 10.1111/j.1468-1331.2010.03194.x. [DOI] [PubMed] [Google Scholar]
- 153.Bigalke B, Schreitmuller B, Sopova K, et al. Adipocytokines and CD34 progenitor cells in Alzheimer's disease. PLoS One. 2011;6:e20286. doi: 10.1371/journal.pone.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 155.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 156.Kloek C, Haq AK, Dunn SL, et al. Regulation of Jak kinases by intracellular leptin receptor sequences. J. Biol. Chem. 2002;277:41547–41555. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 157.Li C, Friedman JM. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc. Natl. Acad.Sci.USA. 1999;96:9677–9682. doi: 10.1073/pnas.96.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bjorbaek C, Buchholz RM, Davis SM, et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J. Biol. Chem. 2001;276:4747–4755. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- 159.Carvalheira JB, Torsoni MA, Ueno M, et al. Crosstalk between the insulin and leptin signaling systems in rat hypothalamus. Obes. Res. 2005;13:48–57. doi: 10.1038/oby.2005.7. [DOI] [PubMed] [Google Scholar]
- 160.Morton GJ, Gelling RW, Niswender KD, et al. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 161.Zhao AZ, Huan JN, Gupta S, et al. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat. Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]