

Abstract
Background
Data regarding the patterns and the mechanisms of deregulation of the insulin growth factor (IGF) pathway in adult and pediatric gastrointestinal stromal tumors (GISTs) are limited.
Methods
We investigated the expression profiling of the genes encoding the main components of the IGF signaling pathway in 131 GISTs (106 adult, 21 pediatric and 4 young adult) and 25 other soft-tissue sarcomas (STS) using an Affymetrix U133A platform. IGF2 was investigated for loss of imprinting (LOI) whereas IGF1R was analyzed for copy number aberration and mutation.
Results
IGF2 was the most highly overexpressed gene of the IGF pathway in GIST. IGF2 expression was also significantly higher than in other STS. IGF2 expression was correlated to the age onset and mutational status of GIST. Indeed, IGF2 expression was significantly higher in the “adult” group than in the “pediatric” and “young adult” groups. Among adult GIST, IGF2 expression was higher in tumors lacking KIT or PDGFRA mutations in comparison with mutated cases. A trend for a higher expression of IGF2 in resistant GIST in comparison to responsive GIST was also found. Overexpression of IGF2 was not related to LOI. Conversely, the expression of the IGF1R gene was significantly higher in the pediatric group than in the adult group. No copy number gains or mutations of IGF1R were observed.
Conclusion
The IGF pathway is deregulated in GIST with distinct patterns according to age onset and mutational status. The IGF pathway may represent a therapeutic target in patients with primary or secondary resistance to imatinib.
Keywords: GIST, gastrointestinal stromal tumor, IGF2, IGF1R
INTRODUCTION
The majority of gastrointestinal stromal tumors (GIST) are characterized by gain-of-function mutations of the KIT or the PDGFRA genes resulting in the activation of the downstream pathways Ras/MAPK, JAK/STAT3, and PI3K/AKT, playing a crucial role in tumorigenesis (1-2). The discovery of KIT as the biologic driver of GIST favored the rapid development of targeted therapies in the adjuvant and metastatic settings. Currently, imatinib is the only drug approved for first-line therapy of metastatic GIST. Disease control is achieved in approximately 85% of patients with a median progression-free survival (PFS) of 20 to 24 months (3). However, 10-15% of patients exhibit primary resistance, and acquired secondary resistance is ultimately observed in the majority of patients who initially experienced clinical benefit from imatinib (3). Although secondary gene mutation in the KIT kinase domain may represent the most common mechanism of secondary resistance to imatinib in GIST, activation of alternative signaling pathways that bypass the requirement for KIT kinase activity may also play an important role (4). Recent studies have suggested that besides KIT or PDFGRA mutation, aberrations of the insulin-like growth factor signaling pathway may play an important role in GIST tumorigenesis (5-11). This pathway consists of insulin-like growth factor (IGF1 and IGF2), IGF receptors 1 (IGF1R) and 2 (IGF2R), and IGF-binding proteins (12). Similar to KIT, IGF1R induces signaling through the MAP kinase and PI3 kinase cascades, whereas IGF2R has no signal transduction capacity and acts as a scavenger receptor, down-regulating IGF2 (12). The ligand IGF1 functions primarily by activating the IGF1R, whereas IGF2 can act through either the IGF1R or through the insulin receptor A isoform. Particularly high expression of IGF2 was reported in GIST (5,7,10) and was correlated with a high rate of metastatic relapse in patients treated for a localized disease (7). This overexpression is considered as the primary mechanism of the paraneoplastic hypoglycemia reported in several cases of GIST (13-18), but also in several other tumor types including solitary fibrous tumors (Doege-Potter syndrome) (19-20). The IGF2 gene is imprinted and active only on the paternally inherited allele. We and others have reported that loss of imprinting (LOI) was the main mechanism responsible for IGF2 overexpression in several subtypes of carcinomas and mesenchymal tumor (21-23). However, there are no data about the mechanism of overexpression of IGF2 in GIST. We hypothesized that this overexpression was also related to LOI. Moreover, since there is a major overlap between KIT and IGF1R downstream cascades, we hypothesized that IGF2 overexpression was associated with resistance to imatinib. We performed this study in order to address these two hypotheses.
Patients and Methods
Patients
One hundred thirty one patients with a diagnosis of GIST, identified from the Memorial Sloan-Kettering Cancer Center (New York, NY, USA) sarcoma database, were included in this study on the basis of available material for molecular experiments. Patient characteristics are described in Table 1. The proportion of small bowel GIST was slightly higher than that of GIST originating from the stomach. This reflects the poorer prognosis of small bowel GIST patients which are more likely to relapse and to receive imatinib. Pediatric and young adult cases were defined by an age at diagnosis of ≤18 years and >18 but ≤30 years, respectively. In all cases, the pathologic diagnosis was confirmed using standard H&E staining and immunoreactivity for CD117 (DAKO Corp., Carpinteria, California; 1:500) on formalin-fixed paraffin-embedded tissue. KIT (exons 9, 11, 13, 14, and 17) and PDGFRA (exon 12, 14, 18) genotyping was performed as previously described (24). Response to imatinib was assessed according to CHOI criteria (25). “Responsive” samples were obtained by surgical resection of the tumor before any radiological progression of the disease with imatinib treatment. “Resistant” samples were obtained by surgical resection of a lesion with evidence of radiological progression under imatinib treatment. The study was approved by the Institutional Review Board (IRB-protocol 02-060).
TABLE 1.
Patients characteristics (N=131)
| No. of Patients | % | |
|---|---|---|
| Age onset | ||
| Adult (≥ 30 years) | 106 | 81.0 |
| Young adult (≥ 18 and < 30 years) | 4 | 16.0 |
| Children (<18 years) | 21 | 4.0 |
| Sex | ||
| Male | 70 | 53.5 |
| Female | 61 | 46.5 |
| Tumor site | ||
| Small bowel | 57 | 76.5 |
| Stomach | 42 | 15.0 |
| Other | 32 | 8.5 |
| Mutational status | ||
| KIT Exon 11 | 66 | 50.5 |
| KIT Exon 9 | 23 | 17.5 |
| KIT Exon 13 | 1 | 0.5 |
| PDGFRA | 9 | 7.0 |
| Wild-type | 32 | 24.5 |
| Treatment status | ||
| Imatinib-naïve | 78 | 59.5 |
| Imatinib-responsive | 10 | 7.5 |
| Imatinib-resistant | 43 | 33.0 |
Microarray analysis
Total RNA was extracted from frozen tumor tissues with RNeasy Mini Kit (Qiagen, Valencia, CA) and followed by RNase-free DNase treatment (Qiagen, Valencia, CA) to remove DNA contamination shortly before RT. The quality of each RNA sample was tested by using a Bioanalyzer 2100 (Agilent, Santa Clara, CA). Microarray analysis was performed by using the Human Genome U133A expression array (Affymetrix, Inc., Santa Clara, CA; containing 22,000 transcripts) as previously described (26). One hundred thirty one GIST tumors and a control group of 25 sarcomas (five leiomyosarcomas, six fibrosarcomas, four clear-cell sarcomas, four undifferentiated round cell sarcomas, four synovial sarcomas and two dedifferentiated liposarcomas) were analyzed. The gene expression data on the soft tissue sarcoma control group has been previously published, which is publically available at: http://cbio.mskcc.org/Public/SFT (22). Affymetrix probeset used for expression analysis were 202410_x_at and 210881_s_at for IGF2, 203627_at, 203628_at and 208441_at for IGF1R, 203851_at for IGFBP6 and 211972_x_at, 201033_x_at, 208856_x_at, 211720_x_at for RPLP0. For each gene, a mean for the probesets of interest was calculated. Results are presented as ratios: (data relevant gene/average data RPLPO) ×100 to compare expression between samples.
Bisulfite sequencing analysis
Bisulfite treatment of genomic DNA was performed by using an Epitect Bisulfite Kit (Qiagen, Valencia, CA). The IGF2 differentially methylated region (DMR) is located in the 5’ flanking region of the H19 gene and 90kb downstream of IGF2. Nested PCR of bisulfite-converted genomic DNA was performed using the following primers: 1st Sense: 5’-gggaatgtttatttatgtatgaag-3’, 1st Antisense: 5’-taaaaacctcctccacctcc-3’, 2nd Sense: 5’-taatttatttagggtggtgtt-3’, 2nd Antisense: 5’-tccaaacacccccaccttaa-3’. The protocols are available on request. Direct sequencing was performed and the methylation status was determined. For all cases examined, the bisulfite sequences could be divided into two patterns of IGF2 DMR methylation: (i) hypomethylation, whereby all 3 CpG loci were converted to thymidines, indicating that all 3 CpG loci of both alleles were demethylated; and (ii) normal methylation, in which the overlapping of both thymidine and cytosine peaks on a sequencing chromatogram was detected for all 3 CpG loci, indicating that both methylated and demethylated IGF2 DMR alleles are present.
Determination of informative cases and of LOI status
The presence of a polymorphic pattern was examined in each case using ApaI polymorphism for IGF2, as previously described. This restriction enzyme site is found in both genomic DNA and at the cDNA level, and was previously associated with IGF2 loss of imprinting (26). Genomic DNA and cDNA were amplified by polymerase chain reaction (PCR) and the PCR product was digested with 10u ApaI endonuclease for 6 hours at 37°C, then separated on a 3% agarose gel containing ethidium bromide. Genomic DNA samples with two bands of appropriate size (a: 292bp, b: 231bp) were defined as informative. Fragment ‘a’ was the undigested fragment (292 bp, lack of polymorphic sites), and fragments ‘b’ were digested fragments (231 bp and 61 bp, with polymorphic sites). The presence of both fragment ‘a’ and ‘b’ of genomic DNA defined a case as informative. The presence of both fragments ‘a’ and ‘b’ of cDNA were identified as biallelic expression or LOI; while the presence of only fragment ‘a’ or ‘b’ was identified as monoallelic expression or imprinting. To confirm the results, direct sequencing of the amplified genomic DNA and cDNA was performed.
Fluorescence in Situ Hybridization (FISH) for IGF1R copy number analysis
FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking IGF1R: SP.G-RP11-147O9, SP.G-RP11-802L22, SP.R-RP11-1087J7 and SP.R-RP11-320L10, as well as a centromeric chromosome 15 probe, SP.O-15a. BAC clones were chosen according to USCS genome browser (http://genome.uscs.edu). The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described (28). Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems).
Full-Length Sequencing of IGF1R and mutation detection
Genomic DNA extracted from 15 tumors with available frozen tissues was used to amplify putative exonic regions of IGF1R, which were broken into target regions of 500 bp or less, and specific primers were designed using Primer 3. Purified PCR reactions were sequenced bidirectionally with M13 primer and Big Dye Terminator Kit v.3.1 (Applied Biosystems). Bidirectional reads and mapping tables were subjected to a QC filter which excludes reads that have an average Phred quality score of < 10 for bases 100–200. Passing reads were assembled against the PTPRD reference sequence using command line Consed 16.0 (29). Assemblies were passed on to Polyphred 6.02b (30) and Polyscan 3.0 (31).
Real-Time Quantitative Reverse Transcription-PCR
One μg of total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) at 25°C for 10 min, 37°C for 120 min, 85°C for five min and hold at 4°C. Twenty ng/μl of resultant cDNA was used in a quantitative-PCR using an 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA) and pre-designed TaqMan ABI Gene expression Assays (Hs00171254_m1* for IGF2, Hs00609566_m1* for IGF1R, Hs00181853_m1* for IGFBP6). Amplification was carried out at 95°C for 10 min, and 40 cycles (95°C for 15 sec, 60°C for one min). To calculate the efficiency of the PCR and to assess the sensitivity of each assay, we also performed a five point standard curve (80, 26.67, 8.88, 2.96, and 0.98 ng/ul). Triplicates CT values were averaged and amounts of target were interpolated from the standard curves and normalized to GAPDH (reference gene).
RESULTS
GIST show overexpression of IGF2 compared to other soft tissue sarcomas
We analyzed the expression of 12 genes encoding for the main components of the IGF signaling pathway, which were available on the U133A chip: the two ligands (IGF1, IGF2), the two IGF receptors (IGF1R and IGF2R), the seven binding proteins (IGFBP1-7) and the acid-labile subunit IGFALS. In GIST, IGF2 was highly overexpressed by 11 to 375 orders of magnitude above the other signaling molecules of the IGF pathway. IGF2 expression was also significantly higher than in other sarcomas subtypes (fold-change: 10.4, p< 0.0001).
Expression of IGF2 correlates with the age at diagnosis and is inversely associated with IGF1R expression
The pattern of expression of IGF2 was correlated to the age at onset and mutational status of GIST. Indeed, IGF2 expression was significantly higher in the “adult” group than in the pediatric and the young adult groups (fold change: 2.7, p=0.004) (Figure 1). This result was also confirmed by quantitative real time RT-PCR (fold change: 13). Among adult GIST, IGF2 expression was higher in tumors lacking KIT or PDGFRA mutations in comparison with mutated cases (fold change: 1.9, p=0.04) (Figure 1). Conversely, the expression of the IGF1R gene was significantly higher in the pediatric group than in the adult group (fold change: 3.2, p<0.0001). This result was confirmed by quantitative real time RT-PCR (fold change: 9). Since IGF1R overexpression in pediatric GIST has been previously correlated with an increase in the IGF1R gene copy number (Tarn et al., 2008), we analyzed IGF1R status by FISH in 18 cases (12 pediatric GIST and six adult wild-type GIST) selected on the basis of high IGF1R expression. However, no copy number alterations were detected in any of the samples. Additionally, we performed full-length sequencing of the IGF1R cDNA in 15 cases (12 pediatric, 2 wild-type young adults and one young adult with a KIT exon 9 mutation) and did not detect any mutations. Besides IGF1R, IGF2 expression is also negatively correlated with IGFBP6 expression (p<0.0001).
Figure 1.
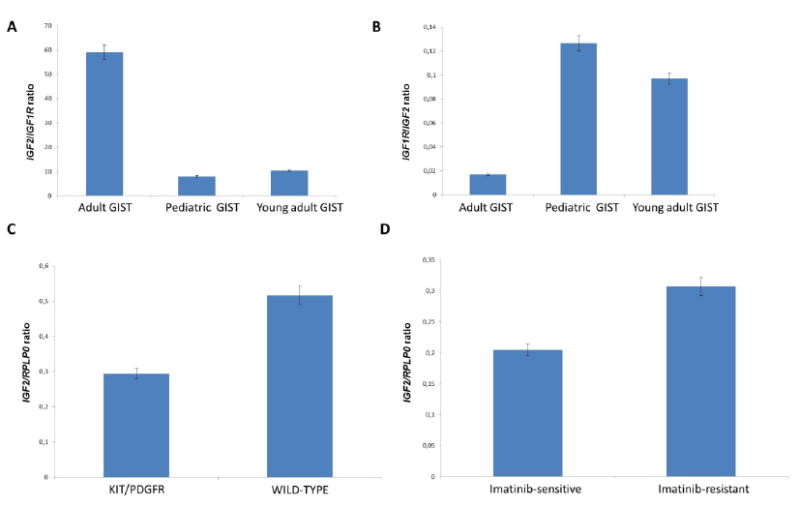
IGF2 and IGF1R mRNA expression in GIST. A: IGF2/IGF1R ratio according to age onset; B: IGF1R/IGF2 ratio according to age onset; C: IGF2/RPLP0 ratio according to mutational status; D: IGF2/RPLP0 according to sensitivity to imatinib.
Acquired imatinib-resistant GIST show marginal IGF2 overexpression
GIST tumor cells can replace the lack of signal due to KIT/PDFGRA inhibition by activating alternative pathways. We hypothesized that IGF2 overexpression and the resulting activation of IGF signaling may represent a possible mechanism of secondary resistance to imatinib in advanced GIST. We compared IGF2 expression in tumor groups according to imatinib status (responsive or resistant). We found a 1.5 fold IGF2 overexpression in resistant GIST compared to responsive GIST (Figure 1), however, it did not reach statistical significance (p=0.15), results being also confirmed by qRT-PCR (fold change: 2.2).
IGF2 overexpression in GIST is associated with IGF2 DMR hypomethylation, but not loss of imprinting
To investigate whether IGF2 overexpression in a subset of GIST is the result of the methylation status of the IGF2 DMR, we investigated this event in 86 of the 131 GIST cases. Hypomethylation of the three CpG loci of the IGF2 DMR was detected in 20 cases (23%). IGF2 mRNA expression was significantly higher in hypomethylated cases than in normal cases (fold-change: 1.6; p=0.005) (Figure 2). Methylation changes of the DMR have been associated with loss of imprinting in several solid tumors. We decided to investigate if this mechanism was involved in GIST. We identified eight informative cases (heterozygous for the ApaI polymorphism in exon 9 of IGF2) among the 21 cases with available RNA. All eight cases had monoallelic expression of IGF2, including five cases with IGF2 DMR hypomethylation.
Figure 2.
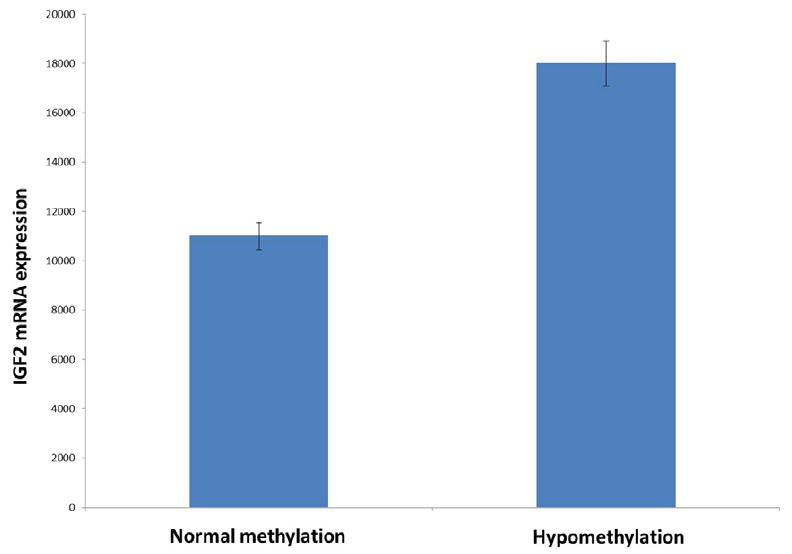
IGF2 mRNA expression according to the methylation status of the IGF2 differentially-methylated region.
DISCUSSION
There is a growing interest in IGF signaling for the development of anticancer therapies due to the crucial role of this pathway in tumorigenesis. We report here the first study assessing the general patterns of deregulation of the IGF pathway in adult and pediatric GISTs. Previous studies have demonstrated that IGF2 was highly overexpressed in GISTs and that this overexpression was associated with poor prognostic features. Our results confirm the overexpression of IGF2 in GIST, and provide additional insights regarding this abnormality. Previous series did not include pediatric and young adult GIST (7, 10). We showed here that the IGF pathway is deregulated in GIST with distinct patterns according to the age onset of the disease. Indeed, we observed that pediatric GISTs are associated with very low levels of IGF2 and high levels of IGF1R. Conversely, adult GISTs express high levels of IGF2 and low level of IGF1R. Among adult GIST, we found that the level of expression of IGF2 is higher in GIST without detectable KIT or PDGFRA mutations, than in mutated tumors. Moreover, by analyzing the expression of the 12 genes encoding the main components of the IGF signaling pathway, we also found that IGF2 expression in GIST is also negatively correlated with IGFBP6 expression (p<0.0001). Although IGFBPs-1-5 binds IGF1 and IGF2 with a similar affinity, IGFBP-6 has a 20 to 100-fold higher affinity to IGF2 than to IGF1 (32-33). IGFBP-6, whose expression is regulated by P53, sequesters IGF2 and thus acts as a tumor suppressor gene in a large variety of cancers. It inhibits growth in lung cancer (34), colorectal cancer (35), prostate cancer (36), neuroblastoma (37) and rhabdomyosarcoma (38), whereas its expression is lower in malignant or metastatic tumors than in benign or non-metastatic tumors (39). Our finding showing a coordinated overexpression of IGF2 and downregulation of its specific inhibitor IGFBP-6 in GIST, strongly suggests that IGF2 signaling plays an important role in GIST tumorigenesis.
Altogether, our results suggest that at least a subset of GIST are characterized by a paracrine or an autocrine loop, which may, through the overexpression of IGF2 (particularly wild-type adult GIST) or IGFR1 (pediatric and young adult GIST), induce an auto-enhancing activation of MAPK and PI3K pathways and therefore tumor cell proliferation. The activation of the IGF pathway in wild-type GIST and its ability to bypass KIT signaling may explain at least in part the lesser sensitivity of these tumors to imatinib. However, further functional studies are needed to confirm these hypotheses.
The mechanisms by which IGF2 and IGFR1 become deregulated in GIST remain to be identified. Genomic imprinting is a mechanism by which there is preferential expression of a gene based on the parental origin of the allele. IGF2 and H19 (which does not encode any protein) are reciprocally imprinted genes, located on human chromosome 11p15.5 (1). Although H19 dos not encode any protein, its expression is associated with silencing of IGF2 (2). IGF2 is exclusively expressed from the paternal allele, whereas H19 is only expressed from the maternal allele. The transcription of the IGF2 gene is regulated by a differentially methylated region (DMR) located in the 5’ flanking region of the H19 gene and 90kb downstream of IGF2. The DMR on the maternal allele is unmethylated, while the DMR on the paternal allele is methylated (40-44). This methylation of the paternal allele ICR blocks the transcription factor CCCTC-binding factor (CTCF) from binding and creating a physical barrier that stops downstream enhancers from augmenting IGF2 promoters. This effectively silences the maternal allele (45-47). Hence, to assess whether the increased expression of IGF2 in GIST was due to LOI, we selected samples heterozygous for ApaI polymorphism for evaluating biallelic expression of IGF2. Strikingly, all the informative cases had monoallelic expression of IGF2 including five cases with IGF2 DMR hypomethylation. Obviously, we cannot exclude that LOI occurs in a minority of cases, however, our results suggest that enhanced paternal allele transcription might be responsible for the increased IGF2 expression in the majority of GIST. Our results are in line with previous studies showing that changes in DNA methylation of the DMR are not associated with biallelic expression in some tumor models (48). However, further studies are needed to investigate the mechanisms involved in the regulation of DNA methylation in GIST. In regards to IGF1R overexpression, our results confirmed more recent reports showing no increase in copy number of the IGF1R (11), as it was firstly suggested in a small series indicating low level amplification in size of wild-type GIST (8). Hence, it appears that that IGF1R amplification is not a common event in GISTs, and that IGF1R overexpression is caused by other mechanisms than copy number abnormalities or activating mutations.
As IGF2 overexpression was previously shown to correlate with poor prognostic features in GIST, in this study we chose to focus on its potential role in resistance to imatinib in patients with advanced disease. Preliminary in vitro data suggested that the IGF pathway may mediate imatinib response or resistance through IGFBP-3 (49). In our series, we found no difference in terms of IGBP3 expression between responsive, resistance or naive GIST (data not shown). However, we found a marginal downregulation of IGF2 in responsive lesions and a statistically significant higher expression of IGF2 in wild-type adult GIST which are known to be less response to imatinib. The most promising future perspectives of our results are first to study the biological role of IGF2 in GIST using in vitro and in vivo models. If preclinical functional studies demonstrate the pathogenetic role of IGF2 in GIST and its involvement in imatinib resistance, therapies targeting the IGF pathway may be beneficial in patients with primary or secondary resistance to imatinib.
Acknowledgments
We thank Pippa McKelvie-Sebileau and Milagros Soto for medical editorial services.
Supported by: PO1 CA047179-15A2 (CRA, SS), P50 CA 140146-01 (CRA,SS), R01-CA102774 (PB), R01 CA102613 (RPD), Life Raft Group (CRA), Starr Cancer Consortium (CRA), GIST Cancer Research Fund (CRA, RPD), Geoffrey Beene Cancer Research Center (RPD), Mr. J.H.L. Pit and Mrs. Pit-van Karnebeek and the Dutch GIST Foundation (RPD), and Swim Across America (RPD), French National Cancer Institute: Soutien pour la formation à la recherche translationnelle en cancérologie 2010 (AI).
Footnotes
Conflict of interest statement
None declared
References
- 1.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 3.GSTM-AG. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–1253. doi: 10.1200/JCO.2009.24.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liegl B, Kepten I, Le C, Zhu M, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernando E, Charytonowicz E, Dudas ME, et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. 2007;13:748–753. doi: 10.1038/nm1560. [DOI] [PubMed] [Google Scholar]
- 6.Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–3215. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braconi C, Bracci R, Bearzi I, et al. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293–1298. doi: 10.1093/annonc/mdn040. [DOI] [PubMed] [Google Scholar]
- 8.Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008;105:8387–8392. doi: 10.1073/pnas.0803383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pantaleo MA, Astolfi A, Di Battista M, et al. Insulin-like growth factor 1 receptor expression in wild-type GISTs: a potential novel therapeutic target. Int J Cancer. 2009;125:2991–2994. doi: 10.1002/ijc.24595. [DOI] [PubMed] [Google Scholar]
- 10.Steigen SE, Schaeffer DF, West RB, Nielsen TO. Expression of insulin-like growth factor 2 in mesenchymal neoplasms. Mod Pathol. 2009;22:914–921. doi: 10.1038/modpathol.2009.48. [DOI] [PubMed] [Google Scholar]
- 11.Janeway KA, Zhu MJ, Barretina J, Perez-Atayde A, Demetri GD, Fletcher JA. Strong expression of IGF1R in pediatric gastrointestinal stromal tumors without IGF1R genomic amplification. Int J Cancer. 2010;127:2718–2722. doi: 10.1002/ijc.25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 13.Pink D, Schoeler D, Lindner T, et al. Severe hypoglycemia caused by paraneoplastic production of IGF-II in patients with advanced gastrointestinal stromal tumors: a report of two cases. J Clin Oncol. 2005;23:6809–6811. doi: 10.1200/JCO.2005.02.4828. [DOI] [PubMed] [Google Scholar]
- 14.Rikhof B, Van Den Berg G, Van Der Graaf WT. Non-islet cell tumour hypoglycaemia in a patient with a gastrointestinal stromal tumour. Acta Oncol. 2005;44:764–766. doi: 10.1080/02841860500267816. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda I, Hizuka N, Ishikawa Y, et al. Clinical features of insulin-like growth factor-II producing non-islet-cell tumor hypoglycemia. Growth Horm IGF Res. 2006;16:211–216. doi: 10.1016/j.ghir.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Guiteau J, Fanucchi M, Folpe A, et al. Hypoglycemia in the setting of advanced gastrointestinal stromal tumor. Am Surg. 2006;72:1225–1230. doi: 10.1177/000313480607201216. [DOI] [PubMed] [Google Scholar]
- 17.Hamberg P, de Jong FA, Boonstra JG, van Doorn J, Verweij J, Sleijfer S. Non-islet-cell tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor possibly worsened by imatinib. J Clin Oncol. 2006;24:e30–31. doi: 10.1200/JCO.2006.06.5318. [DOI] [PubMed] [Google Scholar]
- 18.Escobar GA, Robinson WA, Nydam TL, et al. Severe paraneoplastic hypoglycemia in a patient with a gastrointestinal stromal tumor with an exon 9 mutation: a case report. BMC Cancer. 2007;7:13. doi: 10.1186/1471-2407-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zapf J. Insulin-like growth factor binding proteins and tumor hypoglycemia. Trends Endocrinol Metab. 1995;6:37–42. doi: 10.1016/1043-2760(94)00144-s. [DOI] [PubMed] [Google Scholar]
- 20.Zafar H, Takimoto CH, Weiss G. Doege-Potter syndrome: hypoglycemia associated with malignant solitary fibrous tumor. Med Oncol. 2003;20:403–408. doi: 10.1385/MO:20:4:403. [DOI] [PubMed] [Google Scholar]
- 21.Chao W, D’Amore PA. IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 2008;19:111–120. doi: 10.1016/j.cytogfr.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajdu M, Singer S, Maki RG, Schwartz GK, Keohan ML, Antonescu CR. IGF2 over-expression in solitary fibrous tumours is independent of anatomical location and is related to loss of imprinting. J Pathol. 2010;221:300–307. doi: 10.1002/path.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leick MB, Shoff CM, Wang EC, Congress JL, Gallicano GI. Loss of imprinting of IGF2 and the epigenetic progenitor model of cancer. Am J Stem Cell. 2012;1:59–74. [PMC free article] [PubMed] [Google Scholar]
- 24.Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–3337. [PubMed] [Google Scholar]
- 25.Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–1759. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 26.Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10:3282–3290. doi: 10.1158/1078-0432.CCR-03-0715. [DOI] [PubMed] [Google Scholar]
- 27.Lawson EA, Zhang X, Crocker JT, Wang WL, Klibanski A. Hypoglycemia from IGF2 overexpression associated with activation of fetal promoters and loss of imprinting in a metastatic hemangiopericytoma. J Clin Endocrinol Metab. 2009;94:2226–2231. doi: 10.1210/jc.2009-0153. [DOI] [PubMed] [Google Scholar]
- 28.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. doi: 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 30.Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen LL, Zhu J, Prieto VG, Velasco MA, et al. Gastrointestinal Cancers Symposium. KIT signal induces membrane-bound endothelin 3 suggestive of a paracrine regulatory role of KIT and endothelin 3 in coordination of ICC and myenteric plexus ganglions in GI peristalsis. Gastrointestinal Cancers Symposium. 2007 abstract 413. [Google Scholar]
- 32.Martin JL, Willetts KE, Baxter RC. Purification and properties of a novel insulin-like growth factor-II binding protein from transformed human fibroblasts. J Biol Chem. 1990;265:4124–4130. [PubMed] [Google Scholar]
- 33.Bach LA. Insulin-like growth factor binding protein-6: the “forgotten” binding protein? Horm Metab Res. 1999;31:226–234. doi: 10.1055/s-2007-978723. [DOI] [PubMed] [Google Scholar]
- 34.Sueoka N, Lee HY, Wiehle S, et al. Insulin-like growth factor binding protein-6 activates programmed cell death in non-small cell lung cancer cells. Oncogene. 2000;19:4432–4436. doi: 10.1038/sj.onc.1203813. [DOI] [PubMed] [Google Scholar]
- 35.Leng SL, Leeding KS, Whitehead RH, Bach LA. Insulin-like growth factor (IGF)-binding protein-6 inhibits IGF-II-induced but not basal proliferation and adhesion of LIM 1215 colon cancer cells. Mol Cell Endocrinol. 2001;174:121–127. doi: 10.1016/s0303-7207(00)00444-5. [DOI] [PubMed] [Google Scholar]
- 36.Koike H, Ito K, Takezawa Y, Oyama T, Yamanaka H, Suzuki K. Insulin-like growth factor binding protein-6 inhibits prostate cancer cell proliferation: implication for anticancer effect of diethylstilbestrol in hormone refractory prostate cancer. Br J Cancer. 2005;92:1538–1544. doi: 10.1038/sj.bjc.6602520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grellier P, De Galle B, Babajko S. Expression of insulin-like growth factor-binding protein 6 complementary DNA alters neuroblastoma cell growth. Cancer Res. 1998;58:1670–1676. [PubMed] [Google Scholar]
- 38.Gallicchio MA, Kneen M, Hall C, Scott AM, Bach LA. Overexpression of insulin-like growth factor binding protein-6 inhibits rhabdomyosarcoma growth in vivo. Int J Cancer. 2001;94:645–651. doi: 10.1002/ijc.1519. [DOI] [PubMed] [Google Scholar]
- 39.Yao R, Wang Y, Lubet RA, You M. Differentially expressed genes associated with mouse lung tumor progression. Oncogene. 2002;21:5814–5821. doi: 10.1038/sj.onc.1205422. [DOI] [PubMed] [Google Scholar]
- 40.Reik W, Constancia M, Dean W, et al. Ig/2 imprinting in development and disease. Chromosomes today. 2000;13:93. [Google Scholar]
- 41.Sasaki HK, Ishihara K, Kato R. Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. Journal of Biochemistry. 2000;127:711. doi: 10.1093/oxfordjournals.jbchem.a022661. [DOI] [PubMed] [Google Scholar]
- 42.Arney KL. H19 and Igf2-enhancing the confusion? Trends in Genetics. 2003;19:17–23. doi: 10.1016/s0168-9525(02)00004-5. [DOI] [PubMed] [Google Scholar]
- 43.Engel N, Bartolomei MS. Mechanisms of insulator function in gene regulation and genomic imprinting. International review of cytology. 2003;232:89–127. doi: 10.1016/s0074-7696(03)32003-0. [DOI] [PubMed] [Google Scholar]
- 44.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nature genetics. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 45.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 46.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 47.Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, Ohlsson R, Lobanenkov VV. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Current Biology. 2000;10:853–856. doi: 10.1016/s0960-9822(00)00597-2. [DOI] [PubMed] [Google Scholar]
- 48.Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS. Examination of IGF2 and H19 loss of imprinting in bladder cancer. Cancer Res. 2007;67:10753–8. doi: 10.1158/0008-5472.CAN-07-0329. [DOI] [PubMed] [Google Scholar]
- 49.Dupart JJ, Trent JC, Lee HY, Hess KR, Godwin AK, Taguchi T, Zhang W. Insulin-like growth factor binding protein-3 has dual effects on gastrointestinal stromal tumor cell viability and sensitivity to the anti-tumor effects of imatinib mesylate in vitro. Mol Cancer. 2009;8:99. doi: 10.1186/1476-4598-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]