

Human C7orf30, an uncharacterized protein of the DUF143/iojap family, interacts with and is necessary for the incorporation of L14 into the large mitochondrial ribosomal subunit. Loss of either C7orf30 or MRPL14 impairs ribosome biogenesis and monosome formation, resulting in a specific mitochondrial translation defect.
Abstract
Mammalian mitochondria harbor a dedicated translation apparatus that is required for the synthesis of 13 mitochondrial DNA (mtDNA)-encoded polypeptides, all of which are essential components of the oxidative phosphorylation (OXPHOS) complexes. Little is known about the mechanism of assembly of the mitoribosomes that catalyze this process. Here we show that C7orf30, a member of the large family of DUF143 proteins, associates with the mitochondrial large ribosomal subunit (mt-LSU). Knockdown of C7orf30 by short hairpin RNA (shRNA) does not alter the sedimentation profile of the mt-LSU, but results in the depletion of several mt-LSU proteins and decreased monosome formation. This leads to a mitochondrial translation defect, involving the majority of mitochondrial polypeptides, and a severe OXPHOS assembly defect. Immunoprecipitation and mass spectrometry analyses identified mitochondrial ribosomal protein (MRP)L14 as the specific interacting protein partner of C7orf30 in the mt-LSU. Reciprocal experiments in which MRPL14 was depleted by small interfering RNA (siRNA) phenocopied the C7orf30 knockdown. Members of the DUF143 family have been suggested to be universally conserved ribosomal silencing factors, acting by sterically inhibiting the association of the small and large ribosomal subunits. Our results demonstrate that, although the interaction between C7orf30 and MRPL14 has been evolutionarily conserved, human C7orf30 is, on the contrary, essential for mitochondrial ribosome biogenesis and mitochondrial translation.
INTRODUCTION
Eukaryotic cells maintain both cytoplasmic and organellar (mitochondrial, chloroplast) translation machineries. Although cytosolic translation is responsible for the majority of cellular protein synthesis, the organellar systems are required for the translation of the proteins encoded in the mitochondrial and chloroplast genomes, which comprise a relatively small number of proteins involved in energy-transducing systems. In mammals, mitochondrial DNA (mtDNA) codes for 13 polypeptides, all essential subunits of the oxidative phosphorylation (OXPHOS) complexes. The basic elements of the mitochondrial translational apparatus resemble those in prokaryotes, reflecting their evolutionary origin; however, many proteins are specific to the mitochondrial ribosome, having no obvious bacterial orthologues (Sharma et al., 2003).
The investigation of the structure and assembly of organellar ribosomes has lagged far behind that of bacterial and cytosolic eukaryotic ribosomes. Studies almost a century ago (Jenkins, 1924) reported a mutant in Zea mays called iojap (after the maize variety Iodent japonica), characterized by an albino striped leaf phenotype. The biochemical abnormality in the iojap mutant was identified as a defect in plastid ribosomal biogenesis (Walbot and Coe, 1979), due to a mutation in a nuclear gene that is a member of a widely expressed family of proteins containing a DUF143 domain (Han et al., 1992). Phylogenetic analysis of the DUF143 family shows that plants have two genes, encoding both chloroplast and mitochondrial DUF143 proteins, whereas bacteria and other eukaryotes have a single DUF143 family gene (Wanschers et al., 2012). DUF143 proteins are among the most conserved uncharacterized proteins in bacteria (Galperin and Koonin, 2004). Investigation of YbeB, an Escherichia coli DUF143 protein, showed that it associated with the bacterial large ribosomal subunit (LSU), suggesting a role in ribosome biogenesis and protein synthesis (Jiang et al., 2007). Deletion strains did not, however, exhibit an obvious growth phenotype. We investigated an uncharacterized mouse gene in the iojap family as a candidate gene for a quantitative trait locus associated with tissue-specific differential segregation of mtDNA sequence variants (Battersby et al., 2003), but ruled it out, and proceeded to investigate the role of the human orthologue, C7orf30, in ribosome biogenesis and mitochondrial translation.
Two recent studies of C7orf30 demonstrated that, like the bacterial orthologue, it associates with the mitochondrial large ribosomal subunit (mt-LSU) and appears to be necessary for mitochondrial translation, but the molecular basis for the translation defect was not resolved (Rorbach et al., 2012; Wanschers et al., 2012). In striking contrast, another investigation of C7orf30 and YbeB concluded that members of the DUF143 protein family are universally conserved ribosomal silencing factors, a completely opposite function (Hauser et al., 2012).
Here we show that human C7orf30 interacts with mitochondrial ribosomal protein (MRP)L14 in the mt-LSU and that this interaction is required for normal ribosomal biogenesis and mitochondrial translation.
RESULTS
Human C7orf30 is a soluble mitochondrial protein required for mitochondrial translation
To determine the cellular localization of C7orf30, we stably expressed FLAG-tagged C7orf30 in immortalized human fibroblasts, then performed immunofluorescence studies. The FLAG-tagged protein colocalized with cytochrome c in mitochondria, confirming previous reports (Rorbach et al., 2012; Wanschers et al., 2012). After alkaline carbonate extraction of isolated mitochondria, C7orf30 was detected in the soluble supernatant, and not in the insoluble pellet fraction, where membrane proteins such as COXI are located, indicating that C7orf30 is a soluble mitochondrial protein (unpublished data).
We used lentiviral short hairpin RNA (shRNA) constructs to stably deplete expression of C7orf30 in two different immortalized human fibroblast lines and investigated the effect on the mitochondrial OXPHOS complexes. The effectiveness of the knockdown was verified at the transcript level by real-time quantitative RT-PCR (qRT-PCR; Supplemental Table 1) and by immunoblot analysis (Figure 1A). One of the shRNA constructs was not effective in depleting C7orf30 (shRNA-A1), so this was used as a control in subsequent experiments. The other shRNA construct (shRNA-A2) knocked down expression to undetectable levels in both fibroblast lines, as determined by immunoblot analysis (Figure 1A and unpublished data). We established two stable clonal lines from a bulk culture of shRNA-A2; shRNA-A2-C1 (clone 1) and shRNA-A2-C2 (clone 2). There was a marked decrease in the steady-state levels of mitochondrially encoded subunits of OXPHOS enzymes (ND1 of complex I, and COXI and COXII of complex IV) in both clonal knockdown lines. Nuclear-encoded proteins such as SDHA of complex II and UQCRC1 of complex III were unaffected in shRNA-A2-C2 cells compared with the loading control VDAC1 (Figure 1A). The steady-state level of nuclear-encoded COX4 was reduced, particularly in clone 2, which is typical of a complex IV assembly defect (Weraarpachai et al., 2009, 2012). Because of the stronger OXPHOS phenotype of clone 2, we continued our investigations into C7orf30 function with this line.
FIGURE 1:

Stable knockdown of C7orf30 in human fibroblasts results in OXPHOS assembly defects and mitochondrial fragmentation. (A) SDS–PAGE immunoblot analysis of OXPHOS proteins in fibroblasts transduced with lentiviral shRNA constructs targeting C7orf30. The shRNA-A1 construct, which was not effective in knocking down C7orf30, was used as a control; shRNA-A2-C1 and -C2 are two clonal lines established from the shRNA-A2 construct. ND1, subunit of complex I; COX, subunits of complex IV; UQCRC1, subunit of complex III; SDHA, subunit of complex II; VDAC1, loading control. (B) BN–PAGE analysis of control or C7orf30-shRNA lines alone or expressing C7orf30-FLAG. Antibodies against individual subunits of OXPHOS complexes I–V were used for immunoblotting. (C) Northern blot analysis of RNA from control and C7orf30-shRNA fibroblasts. Hybridization was performed with probes specific for mitochondrial 12S and 16S rRNA (mt-rRNA), and the mitochondrial mRNAs encoding ATP6 and ATP8 of complex V, COX1 of complex IV, and ND5 of complex I. (D) Control and C7orf30-shRNA fibroblast lines stained with MitoTracker Green. Knockdown of C7orf30 (shRNA-A2) disrupts the mitochondrial network.
As anticipated, there was also a decrease in fully assembled OXPHOS complexes in C7orf30-shRNA-A2-C2 fibroblasts, as determined by Blue Native-PAGE. Complexes I, III, IV, and V were all lower in the knockdown line compared with parental or shRNA-A1 controls, whereas complex II was unaffected (Figure 1B). Complex IV activity was similarly reduced in the knockdown line to 50% of controls (unpublished data). Northern blot analysis of mitochondrial transcripts revealed no obvious change in mRNA levels compared with controls (Figure 1C). Analysis of all 13 mRNAs and the two rRNAs by qRT-PCR confirmed that there were no major changes in steady-state levels in C7orf30 knockdown cells (Supplemental Table 1). Mitochondrial morphology was also altered in the knockdown line (Figure 1D), in that the mitochondrial network appeared much more fragmented. Together these data suggested a role for C7orf30 in mitochondrial protein translation or in OXPHOS stability/assembly.
To investigate whether C7orf30 was required for mitochondrial translation, we pulse-labeled cells with [35S]methionine/cysteine in the presence of emetine, a cytosolic protein synthesis inhibitor, as previously described (Sasarman and Shoubridge, 2012). There was a clear synthesis defect for many mitochondrial polypeptides in the shRNA-A2-C2 line, the most severely affected being ND6 and ND4 (Figure 2; ratio over control is 0.2 for ND6 and 0.3 for ND4). Interestingly, other polypeptides were translated normally in the shRNA line (ND1, ND4L, ATP6, and ATP8), suggesting that C7orf30 is not essential for translation of all mitochondrially encoded polypeptides and that only a subset depends on C7orf30 for optimal translation.
FIGURE 2:
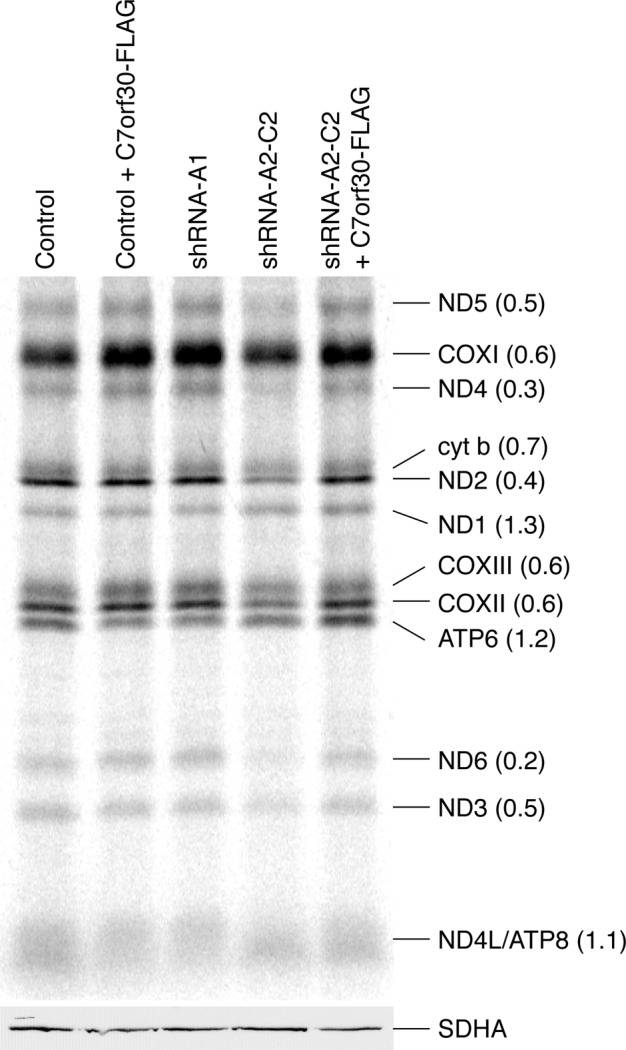
Stable knockdown of C7orf30 results in a mitochondrial translation defect. Pulse-chase labeling of newly synthesized mitochondrial polypeptides (at right; ND, subunits of complex I; CO, subunits of complex IV; cyt b, subunit of complex III; ATP, subunits of complex V) in control and C7orf30-shRNA-A2-C2 fibroblasts alone or expressing C7orf30-FLAG. The number in parentheses is the ratio of individual band intensities of the shRNA-A2-C2 lane over the shRNA-A1 (control) lane. SDHA is used as a loading control on an SDS–PAGE immunoblot of the same samples.
C7orf30 associates with the 39S mitoribosome large subunit and the 55S monosome
Because of the requirement of C7orf30 for efficient translation, we next investigated whether C7orf30 might be associated with the mitoribosome. To this end, we separated mitochondrial lysates on 10–30% discontinuous sucrose density gradients and collected 14 equal fractions from the top. The majority of C7orf30 sedimented at the same density as the 39S mt-LSU proteins MRPL11 (Figure 3, A and B), MRPL14, and MRPL44 (Figure 3B), although some remained in less dense fractions on top of the gradient. We also detected C7orf30 in the 55S monosome fractions 10 and 11 (Figure 3B), in which we found immunoreactive signals for all MRPs we have examined so far, in both the LSU (MRPL11, MRPL14, MRPL44, and unpublished data for MRPL32) and the SSU (MRPS18B, MRPS27, MRPS22 [see Figure 5B later in the paper] and unpublished data for MRPS35). We verified that the presence of C7orf30 in the same fractions as MRPL11 was due to mitoribosome association by treating mitochondrial lysates with ribonuclease A (RNaseA) before sedimentation on sucrose gradients. RNaseA treatment digests mt-rRNA and thus compromises mitoribosomal integrity. RNaseA treatment shifted the mt-LSU protein MRPL11 to less dense fractions near the top of the density gradient, and C7orf30 similarly shifted to less dense fractions following this treatment (Figure 3A). Other mt-LSU proteins, such as MRPL44 and MRPL14, were also shifted to the top fractions when pretreated with RNaseA (unpublished data).
FIGURE 3:

C7orf30 is associated with the 39S large subunit (LSU) of the mitochondrial ribosome, and stable knockdown of C7orf30 reduces the amount of the LSU, the 28S small subunit (SSU), and the 55S monosome. SDS–PAGE immunoblot analysis was performed on fractions sampled from isokinetic sucrose gradients loaded with mitochondrial lysates from the indicated cell lines. (A) Control fibroblasts alone or pretreated with RNaseA. (B) Control, C7orf30-shRNA-A2-C2, or C7orf30-shRNA-A2-C2 expressing C7orf30-FLAG (shRNA-A2-C2 +) fibroblast lines probed with antibodies against LSU and SSU proteins as indicated.
FIGURE 5:

Transient siRNA-mediated knockdown of MRPL14 in control fibroblasts phenocopies the C7orf30 knockdown. MRPL14 was knocked down using a Stealth RNAi construct, and a BLOCK-iT-Alexa Fluor oligonucleotide was used as a control. (A) SDS–PAGE immunoblot analysis of mitochondrial lysates using antibodies against MRPL14, C7orf30, OXPHOS proteins, and MRPs as indicated. (B) SDS–PAGE immunoblot analysis of isokinetic sucrose gradients loaded with mitochondrial lysates from Alexa control or siRNA-MRPL14 (si-L14) probed with antibodies against MRPS as indicated. (C) Pulse-chase labeling of newly synthesized mitochondrial polypeptides (at right; ND, subunits of complex I; COX, subunits of complex IV; cyt b, subunit of complex III; ATP, subunits of complex V) in control and siRNA-MRPL14 cells. (D) Quantification of the amount of labeled newly synthesized mitochondrial polypeptides in shRNA-C7orf30-A2-C2 and siRNA-MRPL14 fibroblast lines expressed as a percentage of their respective controls.
We next investigated whether there was any difference in the sedimentation of the mt-LSU, mitochondrial small ribosomal subunit (mt-SSU), or 55S monosome on sucrose gradients in cells lacking C7orf30. The sedimentation profiles of the mt-LSU and mt-SSU were unchanged in the shRNA-A2-C2 knockdown line; mt-LSU proteins (MRPL11, MRPL14, and MRPL44) and mt-SSU proteins (MRPS18B and MRPS27) were detected in the same fractions as in control gradients (Figure 3B), but the overall amount of some of these proteins was reduced. Furthermore, formation of the 55S monosome (fractions 10 and 11) was reduced in the knockdown line; MRPs no longer sedimented in the monosome fractions, with the exception of a small amount of MRPL44 (Figure 3B). To compare the steady-state levels of mitoribosome proteins, we performed immunoblot analysis for 11 MRPs in mitochondrial lysates from control or shRNA-A2-C2 cell lines. Many of the mitoribosome proteins investigated were reduced in the C7orf30 knockdown line compared with control (Figure 4A), particularly MRPL14, MRPL28, MRPL32, ICT1, and MRPS35, whereas other proteins, such as MRPL44, MRPS18B, and MRPS27, were less affected.
FIGURE 4:

Stable knockdown of C7orf30 decreases the steady-state levels of several MRPs. (A) SDS–PAGE immunoblot analysis of MRPs in control and C7orf30-shRNA-A2-C2 cells alone or expressing C7orf30-FLAG. VDAC1 and SDHA are loading controls. (B) Immunoprecipitation of C7orf30-FLAG and MRPL14 using anti-FLAG or anti-MRPL14–coated Protein A agarose beads, respectively. Cells were treated with the reversible cross-linker DSP before lysis and extraction. Input (IN) and eluate (E) fractions were analyzed by SDS–PAGE followed by immunoblotting with antibodies against C7orf30 and MRPL14.
To verify that these changes in mitoribosome protein expression were a specific effect of the knockdown of C7orf30 and not an off-target effect of the shRNA construct, we sought to rescue the knockdown phenotype by generating a FLAG-tagged version of C7orf30 with synonymous mutations in the shRNA target sequence, enabling the mRNA to escape small interfering RNA (siRNA)-mediated degradation. Expression of C7orf30-FLAG in the shRNA-A2-C2 background rescued all of the mitoribosome protein defects of the knockdown line (Figure 4A, shRNA-A2-C2+C7orf30-FLAG). The data in Figure 4A also demonstrate the tight regulation of C7orf30 protein levels in fibroblasts. When C7orf30-FLAG was expressed from a mammalian retroviral vector (pBABE), endogenous C7orf30 expression was markedly decreased; however, the total amount of C7orf30 (endogenous and epitope-tagged) was similar to that of the endogenous amount alone in control cells, despite the fact that steady-state levels of C7orf30 mRNA, determined by qRT-PCR, were more than 35 times control levels (Supplemental Table 1). In line with this observation, retroviral expression of the FLAG-tagged version in the shRNA-A2-C2 line, in which endogenous C7orf30 was undetectable, was comparable to that seen for the endogenous protein in control cells. This observation contrasts with the relatively high levels of expression of other translation factors that we usually observe when expressed from retroviral vectors (Smeitink et al., 2006; Weraarpachai et al., 2009, 2012). This contrast may reflect the fact that the stability of C7orf30 depends on interacting protein partners, consistent with the observation that C7orf30 is not immunodetectable in rho0 cells (Wanschers et al., 2012; unpublished data).
The mitochondrial translation defects in shRNA-A2-C2 cells were concordantly rescued upon expression of C7orf30-FLAG (Figure 2), as were the defects in OXPHOS enzyme assembly (Figure 1B). Sucrose density sedimentation analysis demonstrated an association of C7orf30-FLAG with the mt-LSU, but, surprisingly, the majority of the protein was in the less dense fractions at the top of the gradient (Figure 3B). This finding is in contrast to that of endogenous C7orf30, which cosediments predominantly with the mt-LSU (Figure 3, A and B), with only a small amount in the lighter fractions. Although these data suggest that the FLAG tag might interfere somewhat with mitoribosome integration of C7orf30, mitoribosome function, assessed by mitochondrial translation assay, was normal in the rescued line, even with this relatively diminished association of C7orf30 with the mt-LSU. The defect in monosome formation in the knockdown line was also partially rescued by C7orf30-FLAG expression, as seen by the reappearance of MRPL14, MRPL11, and MRPS18B in fractions 10 and 11 of the sucrose gradient in Figure 3B.
C7orf30 interacts with MRPL14
To confirm the interaction of C7orf30 with the mt-LSU and to uncover interacting proteins, we immunoprecipitated FLAG-tagged C7orf30 from mitochondrial lysates of HEK293 cells expressing C7orf30-FLAG and identified coimmunoprecipitating proteins by liquid chromatography–tandem mass spectrometry (LC/MS/MS) and immunoblot analyses. Mass spectrometry identified the mt-LSU protein MRPL14 in the eluates of three independent immunoprecipitation experiments (Supplemental Table 2). We also cross-linked mitochondria with either disuccinimidyl glutarate (DSG) or dithiobis(succinimidyl propionate) (DSP) and were able to consistently identify MRPL14 in the eluates (three independent experiments). Supporting our conclusions, a direct interaction of C7orf30 with MRPL14 in human cells was also demonstrated using a split Venus fluorescence complementation assay (Hauser et al., 2012). The reciprocal immunoprecipitation using an MRPL14 antibody successfully pulled down endogenous C7orf30 in HEK293 cells (Figure 4B). Immunoprecipitation of cross-linked mitochondria followed by glycine elution from the FLAG beads (instead of elution with the FLAG peptide) resulted in the detection of MRPL14 and many other mitoribosomal proteins (Supplementary Table 2), both LSU and SSU components, corroborating the association of C7orf30 with the mitochondrial monosome through its interaction with MPRL14.
Further support for an interaction between C7orf30 and MRPL14 came from our sucrose density sedimentation analysis of the shRNA-A2-C2 knockdown line rescued with C7orf30-FLAG. As mentioned previously, the FLAG-tagged protein behaved differently than native C7orf30 in that a large proportion of the protein remained in the less dense (top three) fractions of the sucrose gradient rather than being associated with the mt-LSU in heavier fractions (Figure 3B). Immunoblot detection of MRPL14 in the same experiment clearly detected an expanded pool of MRPL14 in the less dense top three fractions compared with the sedimentation pattern of MRPL14 in control cells (Figure 3B). MRPL44 also displayed this change in sedimentation pattern in the rescued knockdown line, whereas MRPL11 did not, suggesting that C7orf30 may form a pre-LSU mitoribosomal complex with MRPL14, MRPL44, and perhaps other MRPs, although this awaits further investigation. MRPL14 was also the most down-regulated mitoribosome protein of the 11 we analyzed in shRNA-A2-C2 mitochondria (Figure 4A).
Depletion of MRPL14 phenocopies the C7orf30-shRNA mitoribosome and translation defects
Because MRPL14 expression and its assembly into the mt-LSU were dependent, in part, on the presence of C7orf30, we next evaluated whether the converse was also true–that is, whether expression of C7orf30 was dependent on the presence of MRPL14. To this end, we depleted MRPL14 in human fibroblasts by siRNA and investigated the resultant phenotype. Treatment of immortalized human fibroblasts with a Stealth siRNA oligomer targeted against MRPL14 mRNA effectively decreased protein levels of MRPL14 by greater than 90% (Figure 5A). Knockdown of MRPL14 also led to a marked decrease in C7orf30 protein levels, along with a decrease in the steady-state levels of all mitochondrially encoded proteins analyzed (ND1, COXI, and COXII). Nuclear-encoded COX4 was also substantially decreased, whereas UQCRC1 and SDHA were only slightly reduced compared with the loading control VDAC1 (Figure 5A). This expression profile of OXPHOS proteins in siRNA-MRPL14 cells was nearly identical to that of C7orf30-shRNA-A2-C2 cells. In line with this observation, the decreases in mitoribosomal proteins seen in C7orf30 knockdown cells were recapitulated in MRPL14-depleted cells; the mt-LSU proteins MRPL11, L12, L32, and ICT1 were dramatically decreased, whereas MRPL44 was decreased to a lesser extent (Figure 5A). The mt-SSU proteins MRPS18B and MRPS27 were unaffected compared with the loading control SLIRP, whereas MRPS22 was reduced (Figure 5A).
We next performed sucrose density gradient sedimentation analysis on siRNA-MRPL14 mitochondrial lysates to investigate mitoribosome assembly. Despite the depletion of MRPL14 and other mt-LSU proteins in the knockdown cells, MRPL44 was still detectable in the 39S LSU fractions 8 and 9 in the knockdown cells, but its immunoreactivity was markedly reduced in the 55S monosome fractions 10 and 11 (Figure 5B). The amount of “free” MRPL44 (fractions 1–4) was similar in the control and knockdown lines, despite a decrease of MRPL44 in the LSU and a near complete lack of monosome in the knockdown line, suggesting impaired LSU assembly or stability in the knockdown line and subsequent reduction in monosome formation. The sedimentation pattern of the SSU proteins MRPS18B, MRPS22, and MRPS27 confirms the lack of monosome formation in siRNA-MRPL14 cells; there is a striking absence of immunoreactive protein in monosome fractions 10 and 11, whereas the pattern and amount of these proteins in SSU fractions 6–8 were relatively unchanged.
To complete our phenotypic characterization of siRNA-MRPL14 cells, we performed mitochondrial translation assays and compared the results to our previous data for C7orf30-shRNA-A2-C2. Mitochondrial translation was clearly compromised in siRNA-MPRL14 cells (Figure 5C), and the pattern of the translation defect phenocopied that observed in C7orf30-shRNA cells (Figure 5D). Together these data strongly suggest an interaction between MRPL14 and C7orf30. The expression of one depends on the presence of the other, and depletion of either causes nearly identical defects in mitoribosome assembly/function, and hence OXPHOS protein expression.
DISCUSSION
In this study we demonstrate that C7orf30, a member of the widely expressed DUF143 family, is a soluble mitochondrial protein that specifically associates with MRPL14 in the mt-LSU. C7orf30 promotes incorporation of MRPL14 into the mt-LSU, and the loss of C7orf30 results in impaired ribosomal biogenesis, a translation defect, and an ensuing inability to assemble normal amounts of the OXPHOS complexes that contain mtDNA-encoded subunits. We further show that the stability and association of C7orf30 with the mt-LSU is completely dependent on the presence of MRPL14. C7orf30 thus appears to be an essential ribosome biogenesis factor in mammals.
A recent study of YbeB, the E. coli orthologue of C7orf30, showed that it also interacts with bacterial L14 (Hauser et al., 2012); however, binding of YbeB to L14 appears to sterically hinder the association of the large and small subunit, resulting in decreased monosome formation and a consequent decrease in translation. YbeB was thus suggested to function as a translational repressor in E. coli. Competition experiments showed that a YbeB deletion strain was at a disadvantage during stationary growth phase and in a switch from rich to poor medium conditions, due to an inability to repress translation under conditions of nutrient deprivation. In vitro experiments with bovine mitoribosomes, in which addition of purified C7orf30 led to a reduction in the translation of oligo(phenylalanine) from a synthetic polyU transcript, led to the conclusion that proteins in the DUF143 family are universally conserved translational silencers (Hauser et al., 2012). However, no one has yet succeeded in reconstituting a complete mammalian mitochondrial translation system in vitro, so the extent to which assays with mitochondrial ribosomes and synthetic mRNAs reflect authentic rates of mitochondrial translation remains unknown.
The data we present here, using validated assays of in vivo mitochondrial translation, show that the biological function of DUF143 family members is not evolutionarily conserved. Knockdown of C7orf30 produces a clear loss-of-function phenotype in which the synthesis of the majority of mtDNA-encoded polypeptides is markedly reduced and the assembly of the OXPHOS complexes containing mtDNA-encoded polypeptides is severely impaired as a consequence. This conclusion is in line with the two previous reports on the human protein (Rorbach et al., 2012; Wanschers et al., 2012) and unequivocally demonstrates that although the interaction with a large ribosomal subunit protein L14 is evolutionarily conserved, DUF143 proteins have evolved new functions in mitochondrial protein translation. The absence of chloroplast ribosomal biogenesis in Zea mays iojap mutants also suggests that the DUF143 proteins targeted to plastids are not translation silencers, but rather are required for protein synthesis (Walbot and Coe, 1979).
L14 is a remarkably well-conserved protein during evolution; MRPL14 is 47% similar and 28% identical to the E. coli protein. The structure of L14 was first solved by x-ray crystallography at 1.5 Å resolution in Bacillus stearothermophilus (Davies et al., 1996). The L14 structure comprises a five-stranded beta barrel and two small alpha helices in the C-terminal region with one protein-binding site and two RNA-binding sites. It is situated in a central location of the bacterial 50S ribosome and forms a dynamic, bridge structure (B8) at the interface of the large and small subunit that is conserved in bacteria (Yusupov et al., 2001; Gao et al., 2003). The assembly maps of the bacterial LSU, depicting the thermodynamic protein-binding dependencies, categorize L14 as a tertiary binding protein (Shajani et al., 2011). The predicted structure of human MRPL14 is similar to that of the bacterial protein, and the amino acid residues in L14 that reportedly make contact with YbeB (Hauser et al., 2012) are also completely conserved in human MRPL14. It is difficult to predict exactly how C7orf30 docks onto MRPL14 in the mt-LSU, because only a low (13.5 Å)-resolution cryo-electron microscopy (cryo-EM) structure exists for the mammalian mitochondrial ribosome (Sharma et al., 2003); however, it appears that this interaction does not hinder monosome formation, as we find C7orf30 associated with monosomes on sucrose gradients and by mass spectrometry analysis of cross-linked C7orf30 immunoprecipitates. Although we cannot conclude from these observations that C7orf30 is necessary for monosome formation, it is required for efficient insertion of MRPL14 into the mt-LSU; failing this, monosome formation is markedly curtailed, likely because of the reduction in the number of fully assembled mt-LSUs.
How might we explain the difference between the results in the human and bacterial systems? Despite their common evolutionary origins and drug sensitivities, mitochondrial and bacterial ribosomes are in fact different structures. Most strikingly, the RNA-to-protein ratio, which is ∼2:1 in bacteria, is nearly completely reversed (1:2) in mammalian mitochondria (Sharma et al., 2003). In addition, 44% of the 79 identified MRPs of the mammalian mitoribosome have no bacterial orthologues. Fewer than half of these specific mammalian MRPs have orthologues in yeast, indicating a rapid evolution of the mitochondrial ribosomal components in the eukaryotes (Christian and Spremulli, 2011). Although the mitoribosome has a greater mass than does its bacterial counterpart, it sediments at a lower density because it is more porous than the bacterial ribosome (Sharma et al., 2003), as only ∼28% of the missing rRNA elements in mitochondrial ribosomes have been compensated by proteins. In addition, of the 15 intersubunit bridges identified in the cryo-EM structure of the bovine mitochondrial ribosome, only six (not including B8), are conserved with their counterparts in bacterial ribosomes (Sharma et al., 2003). Thus it seems possible that the reduction in packing density of the mitochondrial ribosome, and the different structure of the subunit bridges, has alleviated any steric hindrance problems associated with the docking of C7orf30 on MRPL14, permitting it to acquire novel functions in mammalian ribosome biogenesis.
How is it that the synthesis of some mitochondrial polypeptides is not affected by the loss of C7orf30? Although there is a reduction in the amount of several MRPLs in the mt-LSU on sucrose gradients, and marked reductions in their levels by immunoblot analyses, the mt-LSU in C7orf30 knockdown cells nevertheless sediments in a manner indistinguishable from controls. This finding would suggest a reduced population of fully assembled mt-LSUs and subsequently a reduced population of monosomes containing the full complement of MRPs. However, our data also show that MRPL44, the expression level of which is less affected in both the C7orf30 and MRPL14 knockdown lines, is clearly assembled into a relatively abundant, normally sedimenting mt-LSU, and is also still present in monosome fractions. Thus it is possible that there exist separate populations of mitoribosomes composed of more limited MRP complements which are able to form monosomes for the dedicated synthesis of specific polypeptides, a process that has been demonstrated in yeast, albeit on ribosomes with a full complement of MRPs (Gruschke et al., 2011). Another possibility is that the efficiency of translation is polypeptide specific. We have previously reported that, although mutations in mitochondrial translation factors, such as the elongation factor EFG1 (Antonicka et al., 2006), result in reduced synthesis of many mitochondrial polypeptides, some are actually synthesized at levels higher than control. This finding suggests that there is competition among mRNAs for available mitoribosomes and that when overall translation is impaired by an imbalance in the components of the translation apparatus some mRNAs are preferentially translated. Nothing is known, however, about the factors that might be involved in the regulation of such a process.
Ribosomal biogenesis is a complex multistep pathway in which rRNA plays a crucial role. The process is thought to involve modification and progressive stabilization of rRNA structures based on the successive binding of the structural protein subunits (Shajani et al., 2011). The assembly of the bacterial ribosome has been studied for decades, as has that of the cytosolic ribosome, largely in Saccharomyces cerevisiae, but little is known about the factors or pathways that are required for mitochondrial ribosome assembly. The number of protein factors necessary for bacterial ribosome assembly is about two orders of magnitude lower than that for the assembly of yeast cytosolic ribosome (Hage and Tollervey, 2004), and, on the basis of their evolutionary relationship with eubacterial ribosomes, one might expect similar simplicity, but not necessarily conservation, in the mitochondrial system.
Only four other factors have so far been implicated in the assembly of the mt-LSU viz.: Pet56 (now known as Mrm1) (Sirum-Connolly and Mason, 1993), MTERF4 (Camara et al., 2011), MTG1 (Barrientos et al., 2003), and the m-AAA protease of the inner mitochondrial membrane (Nolden et al., 2005). Mrm1 is an RNA methyl transferase that catalyzes the formation of a 2′-O-methylguanosine at an evolutionarily conserved site in the peptidyl transferase center of the yeast 21S rRNA (Sirum-Connolly and Mason, 1993). Deletion of Mrm1 leads to defective assembly of the mt-LSU, suggesting that this modification produces a crucial assembly intermediate. The function of the human homologue, MRM1, has apparently not been investigated, but it may be responsible for methylating the equivalent site in the 16s rRNA, a site which is known to be methylated in hamster LSU rRNA (Baer and Dubin, 1981). MTERF4 complexes with the RNA dimethyl transferase, NSUN4, which is also thought to methylate a residue (not yet identified) in the 16S rRNA. A tissue-specific knockdown of Mterf4 results in a marked increase in the steady-state levels of mitochondrial mRNAs due to a translation defect that results from the inability to form monosomes (Camara et al., 2011). The GTPase MTG1 is thought to play a role in mt-LSU assembly, based on its ability to partially rescue a deletion of the yeast orthologue, but its precise role has not been defined (Barrientos et al., 2003). Proteolytic processing of MRPL32 by the m-AAA protease is necessary for mitoribosomal function in yeast and mammals (Nolden et al., 2005).
To our knowledge, C7orf30 is the only identified protein that is responsible for chaperoning or stabilizing the insertion of a protein subunit into the mammalian mitochondrial ribosome, but the precise molecular mechanism will require additional investigation. In summary, our results show that DUF143 family members do not behave like translational silencers in human cells as has been recently proposed (Hauser et al., 2012), but are in fact essential for mitoribosome biogenesis and translation.
MATERIALS AND METHODS
Cloning of C7orf30-FLAG expression plasmid
Human C7orf30-FLAG cDNA was generated by RT-PCR (One-Step RT-PCR kit; Qiagen, Chatsworth, CA) using total RNA from control human fibroblasts as the template. AttB recombination site sequences were added to the forward and reverse primers (bold) for Gateway Cloning into pDONR201 (Invitrogen, Carlsbad, CA), and the FLAG sequence was added to the reverse primer for C-terminal tagging (italics): C7orf30 forward: (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGGGGCCGGGCGGCCGTGTGGCGCGG-3′), C7orf30 reverse (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTTACTTGTCATCGTCATCCTTGTAATCTTCACATTTTAACTCCACTGGAGTC-3′). The PCR product was recombined into pDONR201 using BP Clonase II and then transferred into pBabe mammalian expression vector by recombination using LR Clonase II (Invitrogen). All plasmids were verified by DNA sequencing.
Transduction of C7orf30-FLAG into human cell lines
Transduction of C7orf30-FLAG-pBabe into HEK293 and human fibroblast cell lines was performed as described previously (Weraarpachai et al., 2009) using the Phoenix packaging cell line. Transduced lines were maintained in DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS) and selected with puromycin (2.5 μg/ml).
Lentiviral shRNA and siRNA depletion of C7orf30 and MRPL14
Human fibroblasts were depleted for C7orf30 by lentiviral transduction. Lentiviral hairpin-pLKO.1 plasmids from the RNAi Consortium targeted against C7orf30 (A1: GCTTCCAGAAACCAGAGAAAT, A2: CGTGACCCTCATGTTAAGATA) were cotransfected with a packaging plasmid (pCMV-R8.74psPAX2) and an envelope plasmid (VSV-G/pMD2.G). Transduced cells were maintained in DMEM supplemented with 10% (vol/vol) FBS and puromycin selection.
Stealth siRNA was carried out as per the manufacturer’s protocol (Invitrogen) using Lipofectamine RNAiMAX and a Stealth siRNA duplex against MRPL14 (HSS127899) at 12 nM. Cells were transfected with the siRNA duplex twice (at days 1 and 3) before harvesting on day 6.
Immunofluorescence experiments
Cells were paraformaldehyde-fixed, solubilized with 0.1% Triton X-100, and immunostained using antibodies against FLAG (Sigma, St. Louis, MO) and cytochrome c (BD PharMingen, San Jose, CA). The secondary antibodies, anti-mouse-ALEXA 488 and anti-rabbit-ALEXA 594 (Molecular Probes, Eugene, OR), were used for immunofluorescence detection. MitoTracker Green (Invitrogen) was used to visualize the mitochondrial network. A working solution (1 mM) was prepared in dimethyl sulfoxide. MitoTracker was added to cells growing in DMEM containing 10% FBS at a final concentration of 0.1 μM, and the cells were incubated at 37°C for 10–15 min. After removing the MitoTracker, the cells were washed with regular medium twice. The cells were incubated in regular medium for another 15 min at 37°C and washed once with phosphate-buffered saline (PBS) before visualization on an inverted fluorescence microscope.
C7orf30 antibody production
A polyclonal antibody was raised against the peptide VGAAFCRACQTPNFVRGLHSEPGLEERA-EG by 21st Century Biochemicals (Marlboro, MA) and affinity purified.
Blue Native and SDS–PAGE
Blue Native-PAGE was used to separate digitonized (digitonin/protein ratio 0.8 using whole cells) mitochondrial samples on 6–15% polyacrylamide gradient gels as described (Klement et al., 1995). For SDS–PAGE, 12% polyacrylamide gels were used to separate whole-cell extracts prepared with 1.5% n-dodecyl β-d-maltoside (DDM). Proteins were transferred to nitrocellulose, blocked with 5% milk, incubated with indicated primary antibodies, and detected by enhanced chemiluminescence using LumiGLO reagent (Cell Signaling Technology, Danvers, MA).
Pulse labeling of mitochondrial translation products for translation assay
Cells at 80–90% confluence were pulse labeled for 60 min at 37°C in methionine/cysteine-free DMEM supplemented with a [35S]methionine/cysteine mix (200 µCi/ml; Perkin Elmer, Waltham, MA) and emetine at 100 μg/ml. The cells were chased for 10 min in regular DMEM/10% FBS. Cellular protein (50 μg) was resuspended in loading buffer containing 93 mM Tris-HCl, pH 6.7, 7.5% glycerol, 1% SDS, 0.25 mg/ml bromophenol blue, and 3% β-mercaptoethanol. Cells were sonicated for 5–10 s before loading, and run on a 15–20% polyacrylamide gradient gel for 17 h. The labeled translation products were detected by direct autoradiography (Leary and Sasarman, 2009).
Northern blotting analysis
Total RNA (10 μg; RNeasy Kit, Qiagen) was separated on a denaturing 3-morpholinopropane-1-sulfonic acid/formaldehyde agarose gel and transferred to a nylon membrane. Probes specific for mitochondrial mRNAs were labeled with [32P]-labeled deoxycytidine triphosphate ([32P]-dCTP; GE Healthcare, Waukesha, WI) using the MegaPrime DNA labeling kit (GE Healthcare). Hybridization was conducted according to the manufacturer’s manual using ExpressHyb Hybridization Solution (Clontech, Palo Alto, CA), and the radioactive signal was detected using a Phosphorimager.
Cross-linking of mitochondrial proteins
Mitochondria were isolated in ice-cold 250 mM sucrose/10 mM HEPES-KOH, pH 7.4/1 mM EDTA. For DSP and DSG cross-linking, mitochondrial pellets were resuspended in 800 μl of PBS and either DSP or DSG (Thermo Fisher Scientific, Waltham, MA) were added to a final concentration of 1 mM. Tubes were kept on ice for 2 h, and the reaction was terminated by the addition of 160 μl of 0.5 M glycine for 15 min on ice. The cross-linked mitochondria were lysed with the appropriate lysis buffer for either sucrose density gradient or immunoprecipitation. To reverse DSP cross-linking, samples were boiled at 95ºC in Laemmli sample buffer containing 5% β-mercaptoethanol.
Sucrose density gradients
Mitochondria (600 μg) were lysed in lysis buffer (260 mM sucrose, 100 mM KCl, 20 mM MgCl2, 10 mM Tris-Cl, pH 7.5, 1% Triton X-100, 5 mM β-mercaptoethanol, protease inhibitor cocktail without EDTA) on ice for 20 min. For RNase A treatment, RNase A (Qiagen) was added to the mitochondrial lysate to a final concentration of 600 U/ml and the lysate was incubated at room temperature for 30 min. Lysates were cleared before loading onto the sucrose density gradient by centrifugation at 9400 × g for 45 min at 4°C. Lysates were loaded on a 1-ml, 10–30% discontinuous sucrose gradient (50 mM Tris-Cl, 100 mM KCl, 10 mM MgCl2) and centrifuged at 32,000 rpm for 130 min in a Beckman SW60-Ti rotor (Beckman Coulter, Brea, CA). After centrifugation, 14 fractions were collected from the top and analyzed by Western blot.
Immunoprecipitation of C7orf30-FLAG for LC/MS/MS
Mitochondria from cells expressing C7orf30-FLAG were lysed (at 4 mg/ml) with lysis buffer (50 mM HEPES, pH 7.6, 150 mM NaCl, 10 mM MgCl2, 1% sodium taurodeoxycholate, 1x Protease Inhibitor Cocktails without EDTA) for 45 min on ice. After lysis, the lysate was centrifuged at 20,000 × g for 30 min at 4ºC. Lysates were precleared with Protein G agarose beads for 60 min at 4°C and then incubated with anti-FLAG M2 affinity gel (Sigma) overnight at 4°C with rotation. The beads were washed six times with modified lysis buffer containing 0.08% Tween and 0.05% sodium taurodeoxycholate. Elution was performed in elution buffer (Tris-buffered saline, pH 7.0, with 3xFLAG peptide at 150 ng/μl or 0.1 M glycine with 0.05% DDM, pH 2.5) with rotation for 15 min at room temperature, repeated once. The eluates were pooled and precipitated with 15% (vol/vol) 6 M trichloroacetic acid, washed twice with 1 ml of ice-cold acetone, and dried for LC/MS/MS analysis (Orbitrap; Thermo Fisher Scientific) at the Institut de Recherches Cliniques de Montreal. For immunoprecipitation of MRPL14, Protein A Dynabeads (Invitrogen) were coated with anti-MRPL14 antibody (Sigma) and cross-linked using 20 mM dimethyl pimelimidate dihydrochloride (Thermo Fisher Scientific). Immunoprecipitation was then performed as mentioned earlier in the text.
Supplementary Material
Acknowledgments
This study was supported by an operating grant (FRN 15460) from the Canadian Institutes of Health Research to E.A.S., who is an International Scholar of the Howard Hughes Medical Institute.
Glossary
Abbreviations used:
- cryo-EM
cryo-electron microscopy
- DDM
n-dodecyl β-d-maltoside
- DSG
disuccinimidyl glutarate
- DSP
dithiobis(succinimidyl propionate)
- LC/MS/MS
liquid chromatography–tandem mass spectrometry
- LSU
large bacterial ribosomal subunit
- MRP
mitochondrial ribosomal protein
- mt-SSU
mitochondrial small ribosomal subunit
- mt-LSU
mitochondrial large ribosomal subunit
- OXPHOS
oxidative phosphorylation
- qRT-PCR
quantitative RT-PCR
- RNaseA
ribonuclease A
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0651) on November 21, 2012.
*Co–first authors.
REFERENCES
- Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- Baer RJ, Dubin DT. Methylated regions of hamster mitochondrial ribosomal RNA: structural and functional correlates. Nucleic Acids Res. 1981;9:323–337. doi: 10.1093/nar/9.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A, Korr D, Barwell KJ, Sjulsen C, Gajewski CD, Manfredi G, Ackerman S, Tzagoloff A. MTG1 codes for a conserved protein required for mitochondrial translation. Mol Biol Cell. 2003;14:2292–2302. doi: 10.1091/mbc.E02-10-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby BJ, Loredo-Osti JC, Shoubridge EA. Nuclear genetic control of mitochondrial DNA segregation. Nat Genet. 2003;33:183–186. doi: 10.1038/ng1073. [DOI] [PubMed] [Google Scholar]
- Camara Y, et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–539. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta. 2011;1819:1035–1054. doi: 10.1016/j.bbagrm.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C, White SW, Ramakrishnan V. The crystal structure of ribosomal protein L14 reveals an important organizational component of the translational apparatus. Structure. 1996;4:55–66. doi: 10.1016/s0969-2126(96)00009-3. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. “Conserved hypothetical” proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, et al. Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell. 2003;113:789–801. doi: 10.1016/s0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Gruschke S, Kehrein K, Rompler K, Grone K, Israel L, Imhof A, Herrmann JM, Ott M. Cbp3-Cbp6 interacts with the yeast mitochondrial ribosomal tunnel exit and promotes cytochrome b synthesis and assembly. J Cell Biol. 2011;193:1101–1114. doi: 10.1083/jcb.201103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage AE, Tollervey D. A surfeit of factors: why is ribosome assembly so much more complicated in eukaryotes than bacteria. RNA Biol. 2004;1:10–15. [PubMed] [Google Scholar]
- Han CD, Coe EH, Jr., Martienssen RA. Molecular cloning and characterization of iojap (ij), a pattern striping gene of maize. EMBO J. 1992;11:4037–4046. doi: 10.1002/j.1460-2075.1992.tb05497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser R, et al. RsfA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet. 2012;8:e1002815. doi: 10.1371/journal.pgen.1002815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MT. Heritable characters of maize. XX iojap-striping, a chlorophyl defect. J. Heredity. 1924;15:467–472. [Google Scholar]
- Jiang M, Sullivan SM, Walker AK, Strahler JR, Andrews PC, Maddock JR. Identification of novel Escherichia coli ribosome-associated proteins using isobaric tags and multidimensional protein identification techniques. J Bacteriol. 2007;189:3434–3444. doi: 10.1128/JB.00090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement P, Nijtmans LG, Van den Bogert C, Houstek J. Analysis of oxidative phosphorylation complexes in cultured human fibroblasts and amniocytes by blue-native-electrophoresis using mitoplasts isolated with the help of digitonin. Anal Biochem. 1995;231:218–224. doi: 10.1006/abio.1995.1523. [DOI] [PubMed] [Google Scholar]
- Leary SC, Sasarman F. Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol Biol. 2009;554:143–162. doi: 10.1007/978-1-59745-521-3_10. [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Rorbach J, Gammage PA, Minczuk M. C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res. 2012;40:4097–4109. doi: 10.1093/nar/gkr1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, Shoubridge EA. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol Biol. 2012;837:207–217. doi: 10.1007/978-1-61779-504-6_14. [DOI] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem. 2011;80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- Sirum-Connolly K, Mason TL. Functional requirement of a site-specific ribose methylation in ribosomal RNA. Science. 1993;262:1886–1889. doi: 10.1126/science.8266080. [DOI] [PubMed] [Google Scholar]
- Smeitink JA, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walbot V, Coe EH. Nuclear gene iojap conditions a programmed change to ribosome-less plastids in Zea mays. Proc Natl Acad Sci USA. 1979;76:2760–2764. doi: 10.1073/pnas.76.6.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanschers BF, Szklarczyk R, Pajak A, van den Brand MA, Gloerich J, Rodenburg RJ, Lightowlers RN, Nijtmans LG, Huynen MA. C7orf30 specifically associates with the large subunit of the mitochondrial ribosome and is involved in translation. Nucleic Acids Res. 2012;40:4040–4051. doi: 10.1093/nar/gkr1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weraarpachai W, et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- Weraarpachai W, Sasarman F, Nishimura T, Antonicka H, Aure K, Rotig A, Lombes A, Shoubridge EA. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.