

A genome-wide RNA interference screen using Caenorhabditis elegans LRP-1/megalin as a model for LDLR transport was employed to identify factors critical to LDLR uptake. We provide evidence that epsin1 promotes LDLR internalization via a FxNPxY-independent pathway. We complement C. elegans in vivo approaches with loss-of-function and biochemical analyses, using mammalian cell culture systems to evaluate epsin1’s mode of action in LDLR endocytosis.
Abstract
Low-density lipoprotein receptor (LDLR) internalization clears cholesterol-laden LDL particles from circulation in humans. Defects in clathrin-dependent LDLR endocytosis promote elevated serum cholesterol levels and can lead to atherosclerosis. However, our understanding of the mechanisms that control LDLR uptake remains incomplete. To identify factors critical to LDLR uptake, we pursued a genome-wide RNA interference screen using Caenorhabditis elegans LRP-1/megalin as a model for LDLR transport. In doing so, we discovered an unanticipated requirement for the clathrin-binding endocytic adaptor epsin1 in LDLR endocytosis. Epsin1 depletion reduced LDLR internalization rates in mammalian cells, similar to the reduction observed following clathrin depletion. Genetic and biochemical analyses of epsin in C. elegans and mammalian cells uncovered a requirement for the ubiquitin-interaction motif (UIM) as critical for receptor transport. As the epsin UIM promotes the internalization of some ubiquitinated receptors, we predicted LDLR ubiquitination as necessary for endocytosis. However, engineered ubiquitination-impaired LDLR mutants showed modest internalization defects that were further enhanced with epsin1 depletion, demonstrating epsin1-mediated LDLR endocytosis is independent of receptor ubiquitination. Finally, we provide evidence that epsin1-mediated LDLR uptake occurs independently of either of the two documented internalization motifs (FxNPxY or HIC) encoded within the LDLR cytoplasmic tail, indicating an additional internalization mechanism for LDLR.
INTRODUCTION
Clearance of cholesterol-laden low-density lipoprotein (LDL) from circulation occurs via endocytosis of the LDL receptor (LDLR; Anderson et al., 1977). Following receptor-ligand internalization, the complex is targeted to the endosomal pathway, the acidity of which dissociates LDL from the receptor. LDL is then targeted to the degradative pathway, while the receptor is recycled back to the plasma membrane to promote additional rounds of LDL endocytosis. Maintaining robust LDLR internalization rates is critical because failure to do so can result in hypercholesterolemia and lead to atherosclerosis (Soutar and Naoumova, 2007).
LDLR uptake is a highly coordinated process that is dependent on a clathrin-mediated internalization mechanism. Clathrin coat assembly drives plasma membrane invagination and formation of cargo-containing endocytic vesicles, but it is not sufficient for LDLR uptake (Maldonado-Baez and Wendland, 2006). Also required are endocytic adaptor proteins that act as intermediates, linking clathrin to membranes and cargo destined for internalization. In particular, LDLR endocytosis relies on AP-2 and autosomal recessive hypercholesterolemia (ARH) or disabled homologue 2 (Dab2). AP-2 is a heterotetrameric adaptor protein complex consisting of two large subunits (α, β2) that mediate interaction with clathrin, phospholipids, and a host of accessory factors; a medium subunit (μ2) that directly engages endocytic cargo; and a small subunit (σ2) that appears to stabilize the complex (Robinson, 2004; Edeling et al., 2006). AP-2 serves a general requirement in the formation of endocytic clathrin coats and is essential to internalization of myriad receptors, including LDLR, epidermal growth factor receptor (EGFR), and the transferrin receptor (Motley et al., 2003; Boucrot et al., 2010). By contrast, ARH and Dab2 are related monomeric adaptor proteins that perform more selective, tissue-specific roles in receptor endocytosis (Morris and Cooper, 2001; Eden et al., 2002; Mishra et al., 2002; Sirinian et al., 2005; Mettlen et al., 2010).
ARH and Dab2 are modular scaffolding proteins containing an amino-terminal phosphotyrosine-binding (PTB) domain that engages both phospholipids and the FxNPxY internalization motif present within the cytoplasmic tail of target receptors such as LDLR (Yun et al., 2003). The carboxy-terminal region encompasses several small protein interaction modules that facilitate recruitment of core endocytic machinery, such as AP2 and clathrin, as well as additional accessory factors. For example, Dab2 interacts with Eps15 homology (EH) domain proteins, such as Eps15 and intersectin, to coordinate β1-integrin endocytosis (Teckchandani et al., 2012), and FCH only domain-2 (Henne et al., 2010), to promote AP-2–independent LDLR uptake (Mulkearns and Cooper, 2012). These extended interactions, beyond the core endocytic machinery, implicate a dynamic protein-interaction network, the activity of which is likely a key regulatory step in governing receptor internalization efficiency.
To gain additional insight into the regulatory steps that control LDLR endocytosis and identify additional factors critical for receptor internalization, we used an unbiased genome-wide RNA interference (RNAi) screen, using a Caenorhabditis elegans LDLR superfamily member and megalin homologue, LRP-1, as a model for LDLR transport. In doing so, we discovered a requirement for epsin1 in LDLR internalization.
Epsin family members are clathrin-associated proteins conserved from yeast to mammals (Legendre-Guillemin et al., 2004), where they coordinate internalization of signaling receptors and their ligands (Wang and Struhl, 2004; Dores et al., 2009; Kazazic et al., 2009; Sorensen and Conner, 2010; Xie et al., 2012), control synaptic vesicle recycling (Jakobsson et al., 2008), and perform a key role in influenza virus uptake (Chen and Zhuang, 2008). In this paper, we provide evidence that epsin1 promotes LDLR internalization via a FxNPxY-independent pathway. We complement C. elegans in vivo approaches with loss-of-function and biochemical analyses, using mammalian cell culture systems to evaluate epsin1’s mode of action in LDLR endocytosis.
RESULTS
Genome-wide screen identifies dab-1 and epn-1 in LRP-1 transport
To identify factors that promote LDLR internalization, we pursued a genome-wide RNAi feeding strategy in C. elegans to screen for genes involved in endocytosis of LRP-1, an LDLR superfamily member that possesses two conserved [F/V]xNPxY internalization motifs (FTNPVY4658, VDNPLY4744; Yochem and Greenwald, 1993). Given that animals lacking LRP-1 arrest during larval development and have trouble molting (Figure 1; Yochem et al., 1999), we reasoned that defects in LRP-1 trafficking should mimic lrp-1 loss-of-function (lf) phenotypes. Indeed, RNAi-induced clathrin-heavy chain (CHC-1) depletion leads to molting defects and larval arrest (Figure 1). This indicated that lrp-1(lf) phenotypes could serve as an initial visual screen to identify LRP-1 transport genes. As with animal viability, molting is orchestrated by myriad processes, many of which are unrelated to LRP-1 transport defects (Frand et al., 2005). Therefore, to focus on genes involved in LRP-1 trafficking, we examined RNAi-fed animals that phenocopy lrp-1(lf) for alterations in LRP-1 localization. To facilitate the assessment of LRP-1 localization, we used LH191, an lrp-1(ku156); rrf-3(pk1426) strain with a genome-integrated lrp-1::gfp transgenic array (eqIs1) that fully rescues the lrp-1(lf) phenotypes (Figure 1). LRP-1::GFP is enriched in hyp7, a large hypodermal syncytial cell that encompasses the animal body and spans the anterior/posterior axis, and localizes to the apical region of the cell (Figure 1), similar to endogenous LRP-1 (Yochem et al., 1999).
FIGURE 1:
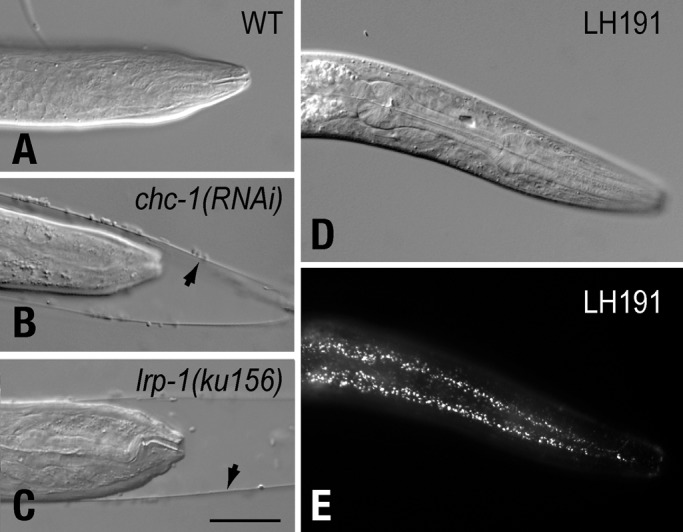
Depleting endocytic machinery phenocopies lrp-1(lf) molting defects. Wild-type C. elegans animals (A) fail to molt when fed chc-1 RNAi (B), similar to lrp-1(ku156) animals (C). lrp-1(ku156) molting defects and animal viability are rescued with an lrp-1::gfp transgene (eqIs1, D). LRP-1::GFP can be detected along the anterior-posterior axis within hyp7 (E). Arrowheads indicate unshed cuticle of animals exhibiting molting defects. Scale bar: 20 μm.
Using the described two-step strategy, we screened a bacterial RNAi library that encompasses more than 80% of the 19,000 C. elegans open reading frames (Fraser et al., 2000; Kamath and Ahringer, 2003). We identified 235 genes that alter LPR-1::GFP distribution within hyp7 when expression of these genes is reduced by RNAi feeding (Supplemental Table 1). Consistent with published studies, 31% of these identified genes are critical for molting (Frand et al., 2005), while 26% are involved in oocyte yolk uptake, a process dependent on receptor-mediated endocytosis (Figure 2A; Balklava et al., 2007). Given the nature of our screening strategy, the largest group of genes identified (∼22%) was those implicated in intracellular transport (Figure 2B and Table S2).
FIGURE 2:
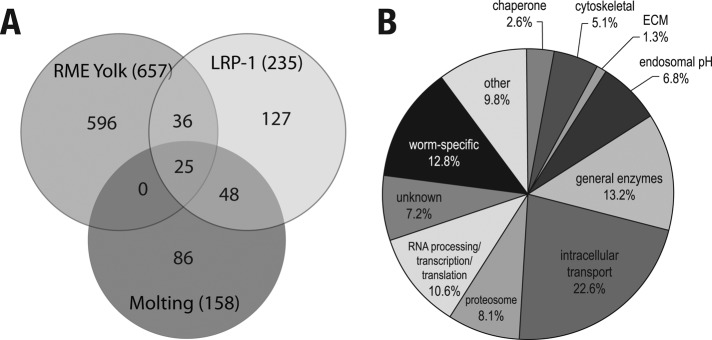
Distribution of genes identified using the two-step LRP-1::GFP transport screen. (A) Venn diagram indicating the number of unique and overlapping genes identified from C. elegans screens for genes involved in molting (Frand et al., 2005) and yolk uptake in oocytes (Balklava et al., 2007). Numbers in parentheses indicate the total number of genes identified. (B) Functional grouping of genes identified in the screen (see Table S2).
As LDLR internalization occurs via clathrin-mediated endocytosis, it is not surprising that the screen identified genes encoding DYN-1/dynamin 1 and the major coat constituents of the clathrin-mediated endocytic pathway, including CHC-1/clathrin heavy chain and subunits of the adaptor protein complex AP-2. As seen with CHC-1 depletion, LRP-1::GFP redistributed to the plasma membrane following RNAi-induced depletion of DYN-1/dynamin, DPY-23/μ2 adaptin, or APS-2/σ2 adaptin (Figure 3), although the extent of localization defect was somewhat variable between animals, likely reflecting knockdown efficiency. Surprisingly, RNAi-induced depletion of the single β-adaptin subunit, APB-1, resulted in a distinct, albeit abnormal, LRP-1::GFP distribution reminiscent of endosomal accumulation. This putative endosomal distribution of LRP-1::GFP is consistent with AP1 functioning in receptor sorting decisions within the endosome (Reusch et al., 2002; Gravotta et al., 2012; Shafaq-Zadah et al., 2012; Xu et al., 2012; Zhang et al., 2012). Given that C. elegans encodes a single β-adaptin subunit, it is possible that the spatial and functional distinctions observed for mammalian AP-1 and AP-2 (Robinson, 2004) are not as well resolved in nematodes. Alternatively, this distinct LRP-1::GFP distribution may reflect cross-reactivity of the RNAi clone, resulting in the knockdown of other gene(s).
FIGURE 3:
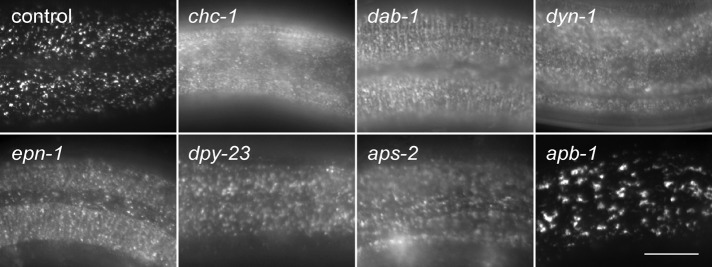
LRP-1::GFP accumulates on the apical surface of hyp7 in epn-1 RNAi-fed animals. LRP-1::GFP was analyzed by epifluorescence in LH191 (control) animals fed with RNAi directed against each indicated gene. Scale bar: 10 μm.
As anticipated, we also identified dab-1, the single C. elegans homologue of ARH and Dab2. Both ARH and Dab2 are essential for efficient LDLR endocytosis in mammals and promote LDLR uptake by coupling the receptor to other components of the endocytic machinery (Garcia et al., 2001). dab-1 RNAi frequently resulted in molting defects (unpublished data; Kamikura and Cooper, 2006), as well as significant plasma membrane accumulation of LRP-1::GFP (Figure 3), consistent with impaired LRP-1 endocytosis. These observations were also confirmed in dab-1(gk291)–null animals (Supplemental Figure S1). Given the critical role of mammalian ARH and Dab2 in LDLR endocytosis (Garcia et al., 2001; Maurer and Cooper, 2006), the identification of dab-1 validates our strategy of using C. elegans LRP-1 trafficking defects to elucidate genes involved in LDLR transport. In addition to the aforementioned genes, the screen also uncovered epn-1 as a candidate gene required for LRP-1 endocytosis. Indeed, RNAi-depletion of EPN-1 resulted in a redistribution of LRP-1 to the plasma surface, similar to the distribution seen following DAB-1 depletion (Figure 3).
Epsin1 promotes LDLR endocytosis
epn-1 is the sole gene in C. elegans to encode epsin (Chen et al., 1998), a clathrin-binding protein thought to serve as an endocytic adaptor to coordinate internalization of some receptors (Maldonado-Baez and Wendland, 2006). To determine whether the role for epsin in LRP-1 internalization is also conserved with mammalian LDLR, we first examined uptake of DiI-labeled LDL (DiI-LDL) in HeLa cells following epsin1 small interfering RNA (siRNA)-mediated depletion. Relative to control cells, a marked reduction in internalized DiI-LDL was observed following a reduction in epsin1 expression, similar to that observed following clathrin heavy chain depletion (Figure 4A). By comparison, epsin1 knockdown did not alter fluorescein isothiocyanate (FITC)-labeled transferrin internalization in the same cells, consistent with published reports that epsin1 is not necessary for transferrin uptake (Huang et al., 2004). We interpret these observations to indicate that epsin1 is essential for robust internalization of endogenous LDLR.
FIGURE 4:

LDLR endocytosis requires epsin1. tTA HeLa cells were transfected with control siRNA or siRNAs targeting the indicated gene. LDL uptake was then qualitatively evaluated by epifluorescence (A) or measured by tracking internalization of a CD8-LDLR chimera (B and C). (A) Control cells or those depleted of CHC or epsin1 were incubated with FITC-labeled transferrin (5 μg/ml) and Dil-labeled LDL (10 μg/ml) for 20 min at 37°C to analyze ligand uptake. Panels include cells (arrowheads) we conclude were not depleted of either CHC or epsin1, thus serving as internal controls for ligand uptake. LDLR chimera uptake was qualitatively measured by 51.1 mAb uptake using epifluorescence in tTA HeLa cells treated with control siRNA (B) or siRNA targeting epsin1 (C). (D and E) tTA HeLa cells were treated with siRNA targeting the indicated factor and infected with CD8-LDLR adenovirus to quantitatively measure internalization kinetics. Control and epsin1 data were split into two graphs to facilitate visualization. Uptake experiments for each factor were performed in tandem. Scale bar: 20 μm; error bars indicate ±SD of at least three independent experiments.
To measure changes in LDLR internalization following epsin1 knockdown, we used a CD8-LDLR chimera, which has been successfully used to quantitate LDLR internalization kinetics (Motley et al., 2003) by tracking uptake of an antibody directed at the extracellular CD8 epitope (51.1; Martin et al., 1984). Consistent with the DiI-LDL uptake data, epsin1 depletion impaired CD8-LDLR uptake relative to controls when evaluated qualitatively by immunofluorescence after CD8-LDLR–expressing cells were incubated for 5 min with 51.1 antibody (Figure 4, B and C). Likewise, quantitative analysis revealed a significant reduction in CD8-LDLR internalization rate following epsin1 depletion, similar to that observed following depletion of clathrin heavy chain (Figure 4D) or Dab2 (Figure 4E). By comparison, ARH depletion showed an intermediate internalization defect in HeLa cells, in line with published reports (Maurer and Cooper, 2006). Collectively, these observations strongly support the conclusion that epsin1 plays a role in promoting LDLR endocytosis.
Epsin1 mediates LDLR endocytosis via an FxNPxY-independent mechanism
To resolve the mechanism by which epsin1 mediates LDLR endocytosis, we evaluated the possibility that it serves in concert with ARH and Dab-2 via the FxNPxY mechanism. To test this, we generated a CD8-LDLR mutant in which the NPxY region of the FxNPxY internalization motif was deleted (CD8-LDLRΔNPxY) to eliminate the contribution of Dab2 and ARH in CD8-LDLR uptake. As expected, the CD8-LDLRΔNPxY internalization rate was markedly reduced relative to that of the CD8-LDLR control (Figure 5). Moreover, the CD8-LDLRΔNPxY internalization rate was not significantly altered following Dab2 depletion, consistent with the critical role of Dab2 binding to the FxNPxY motif in LDLR endocytosis.
FIGURE 5:
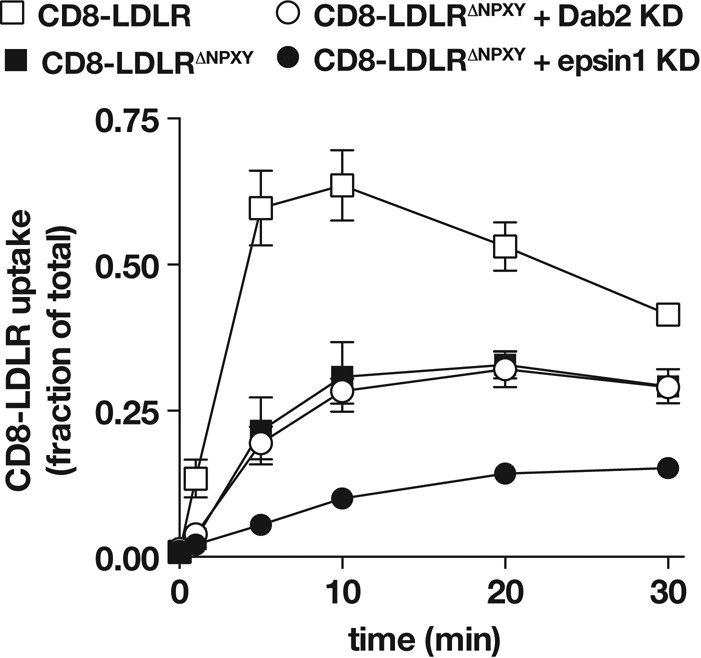
Epsin1 promotes FxNPxY-independent LDLR internalization. tTA HeLa cells were infected with adenovirus encoding the indicated CD8-LDLR chimera, and internalization was quantitatively measured and compared with Dab2- or epsin1-depleted cells. Error bars indicate ±SD of at least three independent experiments.
Interestingly, an appreciable degree of CD8-LDLRΔNPxY uptake was observed (Figure 5), raising the possibility that FxNPxY-independent LDLR internalization mechanisms exist, as previously reported (Michaely et al., 2007). Alternatively, LDLR could dimerize (van Driel et al., 1987). Thus the observed CD8-LDLRΔNPxY uptake might result from dimerization and cointernalization with endogenous LDLR, similar to published reports (Yoshida et al., 1999; Zou and Ting, 2011). We ruled out the latter possibility by testing CD8-LDLRΔNPxY internalization kinetics in CHO ldlA cells that lack functional LDLR (Kingsley and Krieger, 1984). As in HeLa cells, a similar degree of CD8-LDLRΔNPxY uptake was also observed in CHO ldlA cells, as well as in the parental cell line (Figure S2). This indicated that the observed uptake for CD8-LDLRΔNPxY did not arise from dimerization with endogenous receptor, the expression of which is significantly less than that of recombinant CD8-LDLR. We thus conclude that internalization observed for CD8-LDLRΔNPxY occurred independent of the mechanism requiring the FxNPxY motif.
We next evaluated how epsin mediates LDLR internalization. If, like Dab2, epsin1 also relies on the FxNPxY motif, epsin1 depletion should not further impair CD8-LDLRΔNPxY uptake. Surprisingly and in contrast to Dab2 depletion, epsin1 siRNA decreased CD8-LDLRΔNPxY internalization even further, thus revealing epsin1 promotes LDLR endocytosis via an FxNPxY-independent mechanism.
The ubiquitin-interacting motif is critical for receptor internalization
Epsin is a modular protein (Figures 6A and 7A) consisting of an epsin N-terminal homology (ENTH) domain that directly binds phosphatidylinositol-4,5-bisphosphate (Itoh et al., 2001) and Cdc42 GAPs (Aguilar et al., 2006); a middle region encoding multiple ubiquitin-interacting motifs (UIMs) that engage ubiquitinated endocytic cargo (Hawryluk et al., 2006; Kazazic et al., 2009); and a series of small motifs that support interaction with clathrin, AP-2, and EH domain–containing proteins, such as Eps15 (Chen et al., 1998; Drake et al., 2000). To further resolve how epsin1 controls LDLR transport, we pursued a functional dissection of EPN-1 in C. elegans, given that epsin overexpression in mammalian cells is dominant negative for endocytosis of multiple receptors (Sugiyama et al., 2005; Kazazic et al., 2009; Sorensen and Conner, 2010). Animals homozygous for a strong loss-of-function epn-1 allele tm3357 (Figure S3) arrest as larvae at a stage before molting defects are apparent but can be fully rescued by an epn-1::mChr extrachromosomal array (Figure 6A).
FIGURE 6:

C. elegans EPN-1 UIM is essential for receptor internalization. (A) Diagram indicating the domain structure of EPN-1 and the constructs used for the functional dissection in eqIs1; epn-1(tm3357) animals. The associated table indicates the capacity of each EPN-1 mutant form to rescue epn-1(tm3357) lethality. The number of independent animal lines and LRP-1::GFP localization data for each line is also indicated. (B–G) LRP-1::GFP localization analysis in hyp7 using TIRF microscopy for each indicated line or RNAi-fed LH191 animal. Animals expressing EPN-1ΔUIM rescue viability defects, but LRP-1::GFP accumulates on the surface of hyp7, like that for dab-1(gk291) animals (F) and epn-1 RNAi-fed LH191 (G). Scale bar: 20 μm.
FIGURE 7:
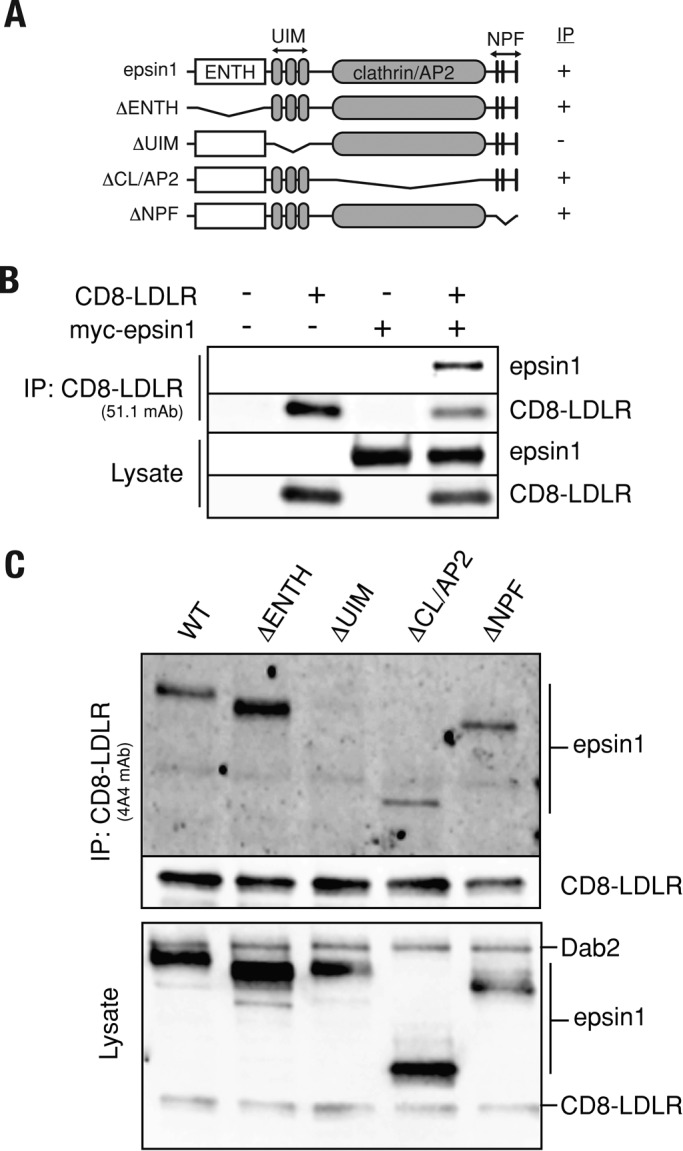
The UIM stabilizes protein complexes containing epsin1 and LDLR. (A) Diagram illustrating the mammalian epsin1 domain structure and constructs used for immunoprecipitation analysis. (B) tTA HeLa cells expressing the indicated recombinant protein were lysed and subjected to coimmunoprecipitation with the 51.1 mAb, which recognizes the extracellular CD8 epitope of CD8-LDLR. Bottom two panels indicate CD8-LDLR and epsin1 expression levels. (C) CD8-LDLR–expressing tTA HeLa cells were transfected with plasmid encoding each epsin1 construct shown in (A). CD8-LDLR was then immunoprecipitated with the mAb 4A4 antibody that recognizes the LDLR cytoplasmic tail. Protein samples were then evaluated for epsin1 binding by immunoblot analysis.
To establish EPN-1 regions essential for LRP-1 internalization, we generated a series of deletion mutants and tested their ability to rescue larval lethality. In combination, we also imaged LRP-1::GFP distribution in hyp7 in epn-1(tm3357) animals with an lrp-1(ku156) eqIs1(lrp-1::gfp) background by total internal reflective fluorescence (TIRF) microscopy to selectively visualize LRP-1::GFP localization differences at the plasma membrane. Full-length epn-1::mChr fully rescues the epn-1 larval lethality, resulting in healthy adult animals that exhibit LRP-1::GFP distribution in hyp7 indistinguishable from that of control LH191 lrp-1(ku156) eqIs1(lrp-1::gfp) animals (Figure 6). On the other hand, an epn-1 construct lacking the ENTH (EPN-1ΔENTH) fails to rescue epn-1(tm3357) larval lethality (Figure 6A). By contrast, engineered versions of EPN-1 lacking specific motifs that support interaction with AP-2 or eps15 fully rescue epn-1 larval lethality. In each case, LRP-1::GFP localization is indistinguishable from that of control animals (Figure 6D; unpublished data), indicating the AP-2– and EH-binding motifs in EPN-1 are not required for viability or LRP-1 trafficking.
Remarkably, an EPN-1 mutant lacking the UIM (EPN-1ΔUIM) fully rescues larval lethality, yet LRP-1::GFP localization is diffuse at the apical cell surface of hyp7 (Figure 6E). In addition to displaying diffuse LRP-1::GFP localization, epn-1ΔUIM animals also resemble dab-1 mutant animals in that they are viable, fertile, and Dpy (short and fat), and exhibit partial molting defects (Kamikura and Cooper, 2003). Interestingly, rescue of epn-1 lethality by EPN-1ΔUIM is lost in the dab-1–null background. This synthetic lethality of dab-1;epn-1ΔUIM animals indicates epn-1 and dab-1 function in parallel pathways and is reminiscent of our mammalian analysis showing epsin1 and Dab2 act by distinct mechanisms to internalize LDLR. To determine which of the two EPN-1 UIMs is required for LRP-1 internalization, we tested the ability of engineered EPN-1 forms lacking only one UIM to rescue LRP-1 distribution in epn-1 mutant animals. As expected, both constructs rescue epn-1 larval lethality. Animals expressing EPN-1ΔUIM2 (lacking the second UIM) displayed apparently wild-type LRP-1::GFP distribution. However, animals expressing EPN-1ΔUIM1 exhibited diffuse cell surface accumulation of LRP-1::GFP (unpublished data) that was similarly observed in LH191 animals following epn-1 RNAi feeding or in dab-1(gk291) animals in a lrp-1(ku156) eqIs1(lrp-1::gfp) background (Figure 6, F and G). Taken together, these observations reveal a striking requirement for the first EPN-1 UIM in promoting internalization of LDLR superfamily members, similar to that recently reported for Notch transport in Drosophila (Xie et al., 2012). Remarkably, this requirement is not coupled to the as-yet-uncharacterized role of epsin that is essential for animal viability.
The UIM stabilizes epsin1 complex formation with LDLR
Robust receptor uptake necessitates tight spatiotemporal assembly of protein complexes that couple cargo to the endocytic machinery. Like epsin1, LDLR colocalizes with clathrin in coated pits at the cell surface (Anderson et al., 1977; Chen et al., 1998). In support of epsin1 mediating LDLR endocytosis, TIRF imaging revealed that epsin1 and LDLR colocalize at the cell surface in mammalian cells (Figure S4). Given the critical role of the UIM in receptor internalization, we postulated that the UIM might influence epsin1 targeting to clathrin-coated pits. However, no significant alteration in epsin1 recruitment to clathrin-coated structures was observed when comparing epsin1-mChr and epsin1ΔUIM-mChr using live-cell imaging (Figure S4), a result that is in agreement with published reports (Chen and Zhuang, 2008).
Given that epsin1 recruitment to clathrin-coated pits occurs independently of the UIM domain, we postulated that the UIM domain might function as a protein–protein interaction platform to promote assembly of an endocytic network that drives receptor internalization, similar to one recently suggested for yeast epsins (Dores et al., 2009). To test this idea, we evaluated complex formation via coimmunoprecipitation assays in cells that coexpressed CD8-LDLR together with either wild-type or epsin1 deletion mutants (Figure 7A). Wild-type epsin1 readily coimmunoprecipitated with CD8-LDLR (Figure 7B), indicating epsin1 and CD8-LDLR are present in a complex. Similarly, engineered versions of epsin1 lacking the ENTH domain, the region spanning the clathrin- and AP-2–binding region, or the single amino acid code (NPF) motifs also coimmunoprecipitated with CD8-LDLR. By contrast, epsin1ΔUIM did not coimmunoprecipitate with CD8-LDLR (Figure 7C), indicating that the UIM is critical for epsin1 incorporation into CD8-LDLR complexes.
Epsin-mediated LDLR endocytosis is independent of ubiquitin modification
Drosophila and mammalian epsins can bind and promote internalization of ubiquitinated cargo (Chen et al., 2002; Sigismund et al., 2005). Given the important role of the epsin1 UIM in receptor uptake, we postulated that LDLR ubiquitination might be essential for epsin1-mediated LDLR endocytosis. To test this notion, we first evaluated the internalization kinetics of a ubiquitination-impaired form of CD8-LDLR (CD8-LDLRUb-mut; Figure 8A), using established mutations that block LDLR ubiquitination (K790R,K795R,K809R and C818A; Zelcer et al., 2009). We reasoned that, if ubiquitination was critical for epsin1 activity, internalization of the CD8-LDLRUb-mut should be impaired. Moreover, CD8-LDLRUb-mut internalization defects should not be further impacted following epsin1 depletion. Relative to the CD8-LDLR control, the internalization rate of CD8-LDLRUb-mut was appreciably reduced, although this defect was not as severe as that observed for CD8-LDLR following epsin1 depletion (Figure 8B). Moreover, contrary to our prediction, CD8-LDLRUb-mut uptake rate was further decreased following epsin1 knockdown. Taken together, these results indicate epsin1-mediated LDLR uptake can occur independent of LDLR ubiquitination status.
FIGURE 8:

Epsin1 promotes HIC-independent LDLR uptake. (A) Schematic amino acid sequence of the human cytoplasmic domain (Lys-790 to Ala-839) for the CD8-LDLR chimera (wild-type) and the various mutant forms tested. Numbers indicate amino acid positions relative to full-length human LDLR. (B–E) tTA HeLa cells were infected with adenovirus encoding the indicated CD8-LDLR chimera and internalization was quantitatively measured and compared with cells treated with control siRNA or siRNA targeting epsin1. Error bars indicate ±SD of three or more independent experiments.
Epsin1 and FxNPxY-independent LDLR internalization mechanisms
Our observations clearly indicate that epsin1 can promote LDLR uptake via a mechanism independent of the FxNPxY motif (Figure 5). Consistently, published observations reveal that LDLR internalization can occur via the single amino acid code (HIC) motif (Michaely et al., 2007), positioned C-terminal of the FxNPxY motif within the LDLR cytoplasmic tail (Figure 8A). Indeed, mutating the HIC motif (CD8-LDLRHIC-mut, HIC mutated to AAA) reduced receptor internalization rates relative to control CD8-LDLR (Figure 8C). Interestingly, the cysteine residue within the HIC motif (Cys-818; Figure 8A) is also deemed essential for the ubiquitin modification of LDLR (Zelcer et al., 2009). Hence, it was mutated in CD8-LDLRUb-mut. This raised the possibility that the internalization defects observed for CD8-LDLRUb-mut might not result from changes in LDLR ubiquitination status, but might instead result from a defective HIC motif. To test this, we compared the internalization kinetics of CD8-LDLRUb-mut with those of CD8-LDLRHIC-mut and a CD8-LDLR form in which only the lysines were mutated (CD8-LDLR3K-mut, K790R,K795R,K809R). CD8-LDLR3K-mut internalization was similar to that of control CD8-LDLR (Figure 8C). By comparison, indistinguishable defects in receptor uptake rate were observed for CD8-LDLRHIC-mut and CD8-LDLRUb-mut (Figure 8C).
The striking similarity in the internalization kinetics for both CD8-LDLRHIC-mut and CD8-LDLRUb-mut raised the possibility that the mechanisms driving their endocytosis might be linked. To address this possibility, we tested the uptake of a CD8-LDLR form that was mutant for HIC and ubiquitination (CD8-LDLRUb-HIC-mut). We reasoned that if both mechanisms were linked, the CD8-LDLRUb-HIC-mut internalization rate should be similar to the rates of CD8-LDLR forms that cannot be ubiquitinated or those lacking the HIC motif. Indeed, CD8-LDLRUb-HIC-mut uptake was indistinguishable from either CD8-LDLRUb-mut or CD8-LDLRHIC-mut (Figure 8C). Combined with the observation that CD8-LDLR3K-mut endocytosis is normal, the latter findings indicate ubiquitination is not critical for LDLR uptake. Instead, the impaired internalization observed for CD8-LDLRUb-mut likely arises from mutating the cysteine residue, which is critical for HIC-mediated LDLR uptake.
Consistent with published observations, our findings reinforce a model in which at least two independent mechanisms drive LDLR endocytosis—one reliant on the FxNPxY motif and another on the HIC motif. However, our findings also suggest that epsin1-mediated LDLR uptake can occur independent of either internalization motif. To better test the latter notion, we measured the internalization kinetics of a ubiquitination-defective form of CD8-LDLR that is incapable of FxNPxY- or HIC-mediated uptake (CD8-LDLRΔNPXY;HIC-mut) following epsin1 depletion. Consistent with two distinct internalization mechanisms, CD8-LDLRΔNPXY;HIC-mut internalization rate was reduced relative to CD8-LDLRΔNPXY. Moreover, further reductions in CD8-LDLRΔNPXY;HIC-mut internalization rate were observed following epsin1 depletion (Figure 8D), similar to reductions observed for a CD8-LDLR form lacking a functional FxNPxY that cannot be ubiquitinated (CD8-LDLRΔNPXY;Ub-mut; Figure 8E) and in agreement with an additional epsin-dependent LDLR uptake mechanism. To determine whether this epsin-mediated internalization is reliant on clathrin, we next measured CD8-LDLRΔNPXY;HIC-mut uptake following clathrin depletion. Following clathrin heavy chain knockdown, CD8-LDLRΔNPXY;HIC-mut was not impaired, as was also observed for CD8-LDLR uptake (Figure 8). Instead, CD8-LDLRΔNPXY;HIC-mut internalization was markedly increased (Figure S5), indicating that when clathrin-mediated uptake is impaired, alternative internalization pathways can be stimulated, similar to those previously reported (Damke et al., 1995). Taken together, these findings reveal a distinct epsin1-mediated pathway that promotes LDLR uptake via a clathrin-independent pathway.
DISCUSSION
Epsin1 requirement in LDLR endocytosis
In this study, we describe a genome-wide RNAi screen identifying EPN-1/epsin as an essential factor in promoting internalization of the C. elegans LDLR superfamily member LRP-1. We provide genetic, cell-based, and biochemical evidence that demonstrate a conserved activity for mammalian epsin1 in LDLR endocytosis. The requirement for epsin family members in promoting internalization of the LDLR family members is further underscored by the identification of EPN-1/epsin in a previous C. elegans screen for genes involved in yolk uptake into oocytes, a process also mediated by an LDLR superfamily member, RME-2 (Balklava et al., 2007).
Interestingly, these findings contrast two studies in which siRNA-mediated epsin1 depletion in human HeLa SS6 cells or monkey BSC-1 cells was not found to disrupt LDLR internalization (Keyel et al., 2006; Chen and Zhuang, 2008). This apparent discrepancy may reflect differences in siRNA-induced epsin1 depletion or in the experimental conditions and approach used to measure LDLR internalization, similar to the experimental conditions and approach used recently for AP-2 (Boucrot et al., 2010).
FxNPxY- and HIC-independent LDLR uptake
The cytoplasmic LDLR tail encodes multiple internalization signals critical for maintaining robust receptor internalization. The canonical FxNPxY motif (Chen et al., 1990) is recognized by the PTB domain–containing endocytic adaptor proteins ARH and Dab-2 (Garcia et al., 2001; Morris and Cooper, 2001; Mishra et al., 2002) that couple the receptor to core endocytic machinery to drive LDLR endocytosis. In the absence of a functional FxNPxY motif, LDLR internalization can occur via an HIC-dependent mechanism (Michaely et al., 2007), although the identity of endocytic adaptors essential for HIC-mediated endocytosis are currently unknown. By comparison, our loss-of-function analyses indicate that epsin1 can promote LDLR internalization via a mechanism independent of clathrin or either internalization motif. An FxNPxY-independent mechanism for epsin is also supported by our observations in C. elegans. Despite LRP-1 internalization defects, dab-1 animals, as well as epn-1ΔUIM–expressing epn-1 mutants, are viable and fertile. However, EPN-1ΔUIM–expressing dab-1;epn-1 double mutant animals exhibit synthetic larval lethality, consistent with both genes acting in parallel pathways. While our data demonstrate that epsin1 promotes LDLR endocytosis independent of the FxNPxY and HIC mechanisms, our observations do not enable us to resolve the potential role for epsin1 in also facilitating LDLR uptake via an FxNPxY- or HIC-dependent route.
Our in vivo dissection of EPN-1 to rescue epn-1(tm3357) lethality provided us the ability to distinguish protein motifs that are critical for EPN-1 function from protein domains that have regulatory roles. The inability of EPN-1ΔENTH to rescue epn-1(tm3357) lethality highlights the necessity of the ENTH domain to epsin function, a result that was also uncovered by functional assays in other systems (Wendland et al., 1999; Overstreet et al., 2003; Brady et al., 2008; Xie et al., 2012). In contrast with the ENTH domain, EPN-1 transgenes that lack the UIM or protein modules important for interaction with AP-2 or EH domain-containing factors rescue epn-1(tm3357) lethality, revealing these motifs as having regulatory or redundant roles. However, only EPN-1 forms lacking the UIM show abnormal LRP-1::GFP accumulation at the plasma membrane, pinpointing the UIM as essential for LRP-1 internalization. Reinforcing the importance of the UIM, our coimmunoprecipitation analyses in mammalian cells also defined the UIM as necessary for epsin1 to stabilize complex formation with the LDL receptor.
The requirement for the UIM in epsin1-mediated receptor transport is echoed in a recent in vivo dissection of Drosophila epsin that also revealed the importance of the UIM in Notch signaling (Xie et al., 2012), as well as in previous studies that implicate a critical role for the UIM in receptor transport (Shih et al., 2002; Overstreet et al., 2003; Sugiyama et al., 2005; Dores et al., 2009; Kazazic et al., 2009). However, the mechanistic role for the UIM remains unclear. Our observations, combined with those of others (Chen and Zhuang, 2008), indicate that epsin1 recruitment to clathrin-coated pits is UIM independent. By contrast, epsin1 interaction with ubiquitinated EGF receptor relies on the UIM; UIM-mediated binding to ubiquitinated EGFR is thought to promote receptor recognition and packaging for endocytosis (Sigismund et al., 2005; Hawryluk et al., 2006; Kazazic et al., 2009).
By comparison, ubiquitination is known to direct LDLR for degradation within the lysosome (Zelcer et al., 2009). However, our observations indicate that LDLR ubiquitination is not essential for epsin-mediated receptor endocytosis; epsin1 depletion further reduced the modestly impaired CD8-LDLRUb-mut internalization rate. These findings, which are consistent with those in yeast, in which ubiquitinated cargo can be endocytosed in the absence of epsin binding to ubiquitin (Dores et al., 2009), raise the possibility that the epsin1 UIM serves as a protein–protein interaction platform, as previously suggested (Dores et al., 2009). We do not favor a direct interaction between epsin1 and LDLR, given that in vitro binding and yeast two-hybrid assays failed to detect a robust interaction (unpublished data). Instead, it is possible that epsin1 interacts with other factors that may be ubiquitin-modified to coordinate LDLR uptake. Alternatively, the UIM might be important for regulating epsin1 endocytic activity. For example, the UIM is essential for epsin1 ubiquitination within the ENTH domain (Oldham et al., 2002). Additional analysis is required to resolve these possibilities.
Role of epsins in animal viability
Our studies reveal that epsin function is required for viability in C. elegans, similar to Drosophila and mice. Indeed, epsin loss in Drosophila and mice results in embryonic lethality, although the precise defect that leads to lethality remains to be determined (Tian et al., 2004; Chen et al., 2009; Xie et al., 2012). Likewise, the cause for epn-1 larval lethality in C. elegans is not known, although one possibility may be LRP-1 trafficking defects. Indeed, lrp-1–null animals exhibit early larval lethality (Yochem et al., 1999). epn-1 larval lethality is rescued by EPN-1ΔUIM as a result of a partial suppression of epn-1 LRP-1 trafficking defects. The synthetic lethality of dab-1;epn-1ΔUIM animals is suggestive of enhanced LRP-1 trafficking defects, which is reminiscent of our data indicating that epsin1 promotes LDLR internalization in parallel with the FxNPxY mechanism that relies on Dab2. Alternatively, epn-1 lethality may be LRP-1 independent and a result of more pleiotropic defects in epn-1–mediated transport of unidentified factors or the disruption of critical signaling pathways that rely on receptor transport. While the cause for epn-1 lethality is unknown, it is clear from the dab-1;epn-1ΔUIM synthetic lethality that epn-1 acts in parallel with dab-1 in C. elegans, as do epsin1 and Dab2 in mammalian LDLR internalization.
MATERIALS AND METHODS
Animal strains
C. elegans strains were grown on nematode growth medium plates at 20°C as described by Brenner (1974). The following alleles were used: lrp-1(ku156), dab-1(gk291) and epn-1(tm3357). Bristol N2 served as the wild-type C. elegans strain. The following alleles were used:
LGI: lrp-1(ku156)
LGII: dab-1(gk291), rrf-3(pk1426)
LGX: epn-1(tm3357), unc-7(35)
The epn-1(tm3357) strain was kindly provided by Shohei Mitani (National Bioresource Project, Tokyo Women’s Medical University School of Medicine). We obtained a twice-outcrossed epn-1(tm3357) strain from Erik Jorgensen (University of Utah) that we outcrossed seven additional times.
C. elegans expression vectors.
epn-1 genomic DNA was kindly provided by Erik Jorgensen (University of Utah). All epn-1 expression constructs were cloned in the pGEM-3Zf(+)vector. epn-1 wild-type and deletion DNA was cloned using engineered BglII and NotI restriction sites. mCherry was inserted in frame at the 3′ end of epn-1 just prior to the stop codon using engineered NotI and BamHI restriction sites.
Pepn-1::epn-1::mCherry (epn-1 genomic DNA, including 1551 base pairs of sequences upstream of the start codon and 1659 base pairs of sequences downstream of the epn-1 coding region)
Pepn-1::epn-1∆ENTH::mCherry (epn-1 genomic DNA, deletion of aa 19–140)
Pepn-1::epn-1∆UIM::mCherry (epn-1 genomic DNA, deletion of aa 193–232)
Pepn-1::epn-1∆UIM1::mCherry (epn-1 genomic DNA, deletion of aa 193–207)
Pepn-1::epn-1∆UIM2::mCherry (epn-1 genomic DNA, deletion of aa 218–232)
Pepn-1::epn-1∆AP2::mCherry (epn-1 genomic DNA, deletion of aa 285–332)
Pepn-1::epn-1∆NPF::mCherry (epn-1 genomic DNA, deletion of aa 425–469)
Generated C. elegans transgenic strains
LH191: lrp-1(ku156) eqIs1; rrf-3(pk1426)
LH801: lrp-1(ku156) eqIs1; dab-1(gk291)
LH757: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1::mCherry
LH764: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1∆UIM::mCherry
LH819: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1∆UIM1::mCherry
LH824: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1∆UIM2::mCherry
LH770: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1∆AP2::mCherry
LH761: lrp-1(ku156) eqIs1; epn-1(tm3357); eqEx Pepn-1::epn-1∆NPF::mCherry
eqIs1 is a spontaneously integrated transgene containing lrp-1::gfp that rescues lrp-1 mutations. It was generated in the following manner: Two plasmids, MH#jy209b and MH#jy207b, contain overlapping DNA from the lrp-1 locus that can result in an intact gene to rescue lrp-1 mutant animals (Yochem et al., 1999). GFP was inserted in frame with the cytoplasmic tail of LRP-1 in MH#jy207b at the PacI/SalI restriction sites. The resulting LRP-1::GFP-pBluescript(SK+) plasmid was injected into unc-13 lrp-1(ku156); nDp4/+ animals, together with MH#jy209b and a coinjection marker, pRF4, which confers dominant rolling. Rescue of ku156 is indicated by the production of healthy, viable unc-13 progeny, which were picked for further propagation. One such animal and its progeny appeared to have a spontaneous insertion at or near the lrp-1(ku156) locus and showed GFP expression in hyp7 with a pattern identical to that observed in wild-type animals stained with anti–LRP-1 antibodies (Yochem et al., 1999). The proximity to the lrp-1 locus and an absence of rolling are consistent with gene conversion, but this has not been examined at the DNA level. eqIs1 was then separated from the unc-13 marker by standard genetic recombination, and offspring homozygous for the eqIs1 were then isolated. eqIs1 was crossed into rrf-3(pk1426) and dab-1(gk291) animals to generate the LH191 and LH801 strains, respectively.
All epn-1 expression constructs were injected at 1 ng/μl with 70 ng/μl str-1::gfp as the coinjection marker into lrp-1(ku156) eqIs1; epn-1(tm3357)/unc-7(e5) animals.
C. elegans RNAi screen.
We used an established bacterial RNAi library that covers an estimated 86% of the C. elegans genome and adapted an established RNAi by feeding protocol (Kamath and Ahringer, 2003). Briefly, we placed three gravid LH191 animals on each plate seeded with a single bacterial RNAi clone. Gravid animals were used in the screen to bypass any embryonic requirement of the gene targeted by RNAi. After 3 d, the progeny of the RNAi-treated adults were examined for lrp-1–null phenotypes, which include larval arrest and/or defective molting. These animals were then further examined under light microscopy for alterations in LRP-1::GFP distribution, as compared with control LH191 animals.
Reagents
The monoclonal antibodies TD.1, E7, and 4A4 were used to recognize clathrin heavy chain, β-tubulin, and LDLR. The mouse hybridomas 4A4 (CRL-1898), 51.1 (HB-230), and TD.1 (CRL-2232) were obtained from the American Type Culture Collection (Manassas, VA). Rabbit antibody against CD8-α (H-160) and mouse antibody against Dab2 (610464) were from Santa Cruz Biotechnology (Santa Cruz, CA) and BD Biosciences (Franklin Lakes, NJ), respectively. Rabbit polyclonal antiserum against Epsin1 was a generous gift from Sandra Schmid (University of Texas, Southwestern Medical Center). BSC-1 cells stably expressing CLC-GFP were a generous gift from Tom Kirchhausen (Harvard University). Lipofectamine LTX and Plus Reagent and Lipofectamine RNAiMAX were used for DNA and siRNA transfection following the manufacturer’s instructions (Invitrogen, Carlsbad, CA). CHO ldlA cells lacking functional LDLR and the parental CHO cell line were a gift from Monty Krieger (Massachusetts Institute of Technology).
Plasmid used to generate adenovirus coding for mouse CD8-LDLR chimera was a gift from Margaret Robinson (Cambridge University) and were used in experiments for Figures 4 and 7. Human cDNA for LDLR was a gift from Peter Tontonoz (University of California, Los Angeles) and was used to generate adenovirus encoding the extracellular domain of human CD8 fused to LDLR, starting with the LDLR transmembrane domain (WT, ∆NPxY, Ub-mut, HIC-mut, 3K-mut, Ub-HIC-mut, ∆NPxY-HIC-mut, ∆NPxY-Ub-mut) and was used in experiments for Figures 5, 8, and S2. Rat epsin1 sequence was based on NM_057136. myc-Tagged Rat epsin1 cDNA was kindly provided by Pietro De Camilli (Yale University). Epsin1 truncations were engineered using PCR-based mutagenesis and subcloned to the EcoRI/XhoI site of pcDNA3-myc plasmid. Epsin1 domain deletions spanned the following amino acids: ENTH (19–132), UIM (184–248), CL/AP-2 (257–484), and NPF (501–575). For carboxy-terminal fusions, mCherry was subcloned into the XhoI/XbaI site of pcDNA containing myc-epsin1 or myc-epsin1∆UIM.
Coimmunoprecipitation
tTA HeLa cells were infected with CD8-LDLR adenovirus and transfected with myc-tagged epsin1 wild-type or deletion constructs 4–6 h after seeding in 60-mm dishes. Cells were harvested 18 h after transfection and lysed in IP buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 2 mM EDTA, and 1% NP-40) with protease inhibitor complex (P8340; Sigma-Aldrich, St. Louis, MO). CD8-LDLR containing protein complexes were immunoprecipitated using either 51.1 or 4A4 antibody; this was followed by incubation with protein G agarose beads (EMD Bio, San Diego, CA). The resulting IP products were subjected to immunoblot analysis.
Immunolocalization
tTA HeLa cells grown on coverslips were fixed with ice-cold acetone for 10 min, which was followed by ice-cold methanol for 2 min. Cells were then washed with 1X PBS and incubated with primary antibody at room temperature for 1 h. After three washes with 1X PBST (PBS containing 0.1% Tween-20), cells were incubated with secondary antibodies conjugated with either Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen) at room temperature for 1 h. Samples were imaged with a Zeiss TIRF microscope (Jena, Germany). For live-cell imaging, 8 × 104 BSC-1 cells stably expressing clathrin-light-chain-eGFP were seeded in 35-mm dishes and transfected with either myc-epsin1-mCherry or myc-epsin1 ∆UIM-mCherry. Samples were visualized with the Zeiss TIRF microscope.
siRNA-mediated knockdown
tTA HeLa cells (3.1 × 105) were plated in six-well plates and transfected with siRNA on days 1 and 2. For the internalization assay, cells were then infected with CD8-LDLR expressing adenovirus on day 3 for an additional 18 h. Knockdown siRNAs for each target included: ARH, AACAGCATGATTCTGACAGGGTTGG; CHC, TAATCCAATTCGAAGACCAAT; epsin1, GGAAGACGCCGGAGTCATT; and Dab2, AGGTTGGAACCAGCCTTCACCCTTT. Each siRNA was obtained from Invitrogen. Silencer negative control siRNA (#1) was from Ambion (Austin, TX). The extent of expression knockdown was evaluated by immunoblot (Figure S6).
Internalization assay
For quantitative uptake assays, tTA HeLa cells were resuspended in DMEM/10% fetal bovine serum with CD8 mAb (51.1); this was followed by incubation of [125I]protein A (Perkin Elmer-Cetus, Waltham, MA) in DMEM/0.5% bovine serum albumin at 4°C. Cells were transferred to a 37°C water bath to allow for internalization for each indicated time point. Internalization was stopped by moving cells to 4°C. Surface-bound ligands were stripped with acid wash (0.2 M acetic acid, 0.5 M NaCl). The amount of internalized CD8-LDLR was determined by γ counting and expressed as a percentage of total surface-bound γ count.
For epifluorescence analysis, tTA HeLa cells grown on coverslips were transfected with either control or the indicated siRNA. After 70 h of knockdown, cells were incubated with 10 μg/ml DiI-LDLR (Biomedical Technologies, Stoughton, MA) and 5 μg/ml Alexa Fluor 488–labeled transferrin (Invitrogen) at 37°C for 20 min. Coverslips were then transferred to ice, washed once with 1X PBS, and fixed with ice-cold 3.7% formaldehyde. Cells were mounted and imaged by epifluorescence using a Zeiss Axio Imager M1 and captured with a monochrome Jenoptik (Jena, Germany) CCD camera. Images were imported, cropped and illustrated using the Adobe Creative Suite CS3 (San Jose, CA).
Supplementary Material
Acknowledgments
We thank Caleb Schimdt and Karla Opperman for technical support in performing the genome-wide screen and in generation of transgenic animals. We also thank the C. elegans Genetic Center for providing strains. This work was supported in part by National Institutes of Health grants to S.D.C. (GM085029) and L.C. (NS045873), a predoctoral fellowship from the American Heart Association to Y.-L.K. (12PRE8480006), and a Developmental Biology Training grant to E.B.S. (2T32-HD007480-11A1).
Abbreviations used:
- ARH
autosomal recessive hypercholesterolemia
- Dab2
disabled homologue 2
- EGFR
epidermal growth factor receptor
- EH
Eps15 homology
- ENTH
epsin N-terminal homology
- FxNPxY
single amino acid code, where x can be any amino acid
- HIC
single amino acid code
- FITC
fluorescein isothiocyanate
- LDL
low-density lipoprotein
- LDLR
LDL receptor
- lf phenotype
loss-of-function phenotype
- NPF
single amino acid code
- PTB
phosphotyrosine-binding
- RNAi
RNA interference
- siRNA
small interfering RNA
- TIRF
total internal reflective fluorescence
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0163) on December 14, 2012.
REFERENCES
- Aguilar RC, et al. Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc Natl Acad Sci USA. 2006;103:4116–4121. doi: 10.1073/pnas.0510513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG, Goldstein JL, Brown MS. A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature. 1977;270:695–699. doi: 10.1038/270695a0. [DOI] [PubMed] [Google Scholar]
- Balklava Z, Pant S, Fares H, Grant BD. Genome-wide analysis identifies a general requirement for polarity proteins in endocytic traffic. Nat Cell Biol. 2007;9:1066–1073. doi: 10.1038/ncb1627. [DOI] [PubMed] [Google Scholar]
- Boucrot E, Saffarian S, Zhang R, Kirchhausen T. Roles of AP-2 in clathrin-mediated endocytosis. PLoS One. 2010;5:e10597. doi: 10.1371/journal.pone.0010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady RJ, Wen Y, O’Halloran TJ. The ENTH and C-terminal domains of Dictyostelium epsin cooperate to regulate the dynamic interaction with clathrin-coated pits. J Cell Sci. 2008;121:3433–3444. doi: 10.1242/jcs.032573. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhuang X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci USA. 2008;105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci USA. 2009;106:13838–13843. doi: 10.1073/pnas.0907008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, De Camilli P. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 1998;394:793–797. doi: 10.1038/29555. [DOI] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- Chen X, Zhang B, Fischer JA. A specific protein substrate for a deubiquitinating enzyme: liquid facets is the substrate of fat facets. Genes Dev. 2002;16:289–294. doi: 10.1101/gad.961502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Baba T, van der Bliek AM, Schmid SL. Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J Cell Biol. 1995;131:69–80. doi: 10.1083/jcb.131.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dores MR, Schnell JD, Maldonado-Baez L, Wendland B, Hicke L. The function of yeast epsin and ede1 ubiquitin-binding domains during receptor internalization. Traffic. 2009;11:151–160. doi: 10.1111/j.1600-0854.2009.01003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MT, Downs MA, Traub LM. Epsin binds to clathrin by associating directly with the clathrin-terminal domain. Evidence for cooperative binding through two discrete sites. J Biol Chem. 2000;275:6479–6489. doi: 10.1074/jbc.275.9.6479. [DOI] [PubMed] [Google Scholar]
- Edeling MA, Mishra SK, Keyel PA, Steinhauser AL, Collins BM, Roth R, Heuser JE, Owen DJ, Traub LM. Molecular switches involving the AP-2 β2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev Cell. 2006;10:329–342. doi: 10.1016/j.devcel.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Eden ER, Patel DD, Sun XM, Burden JJ, Themis M, Edwards M, Lee P, Neuwirth C, Naoumova RP, Soutar AK. Restoration of LDL receptor function in cells from patients with autosomal recessive hypercholesterolemia by retroviral expression of ARH1. J Clin Invest. 2002;110:1695–1702. doi: 10.1172/JCI16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C. elegans molting. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Garcia CK, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–1398. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- Gravotta D, Carvajal-Gonzalez JM, Mattera R, Deborde S, Banfelder JR, Bonifacino JS, Rodriguez-Boulan E. The clathrin adaptor AP-1A mediates basolateral polarity. Dev Cell. 2012;22:811–823. doi: 10.1016/j.devcel.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk MJ, Keyel PA, Mishra SK, Watkins SC, Heuser JE, Traub LM. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic. 2006;7:262–281. doi: 10.1111/j.1600-0854.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, McMahon HT. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. 2010;328:1281–1284. doi: 10.1126/science.1188462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Khvorova A, Marshall W, Sorkin A. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279:16657–16661. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- Itoh T, Koshiba S, Kigawa T, Kikuchi A, Yokoyama S, Takenawa T. Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science. 2001;291:1047–1051. doi: 10.1126/science.291.5506.1047. [DOI] [PubMed] [Google Scholar]
- Jakobsson J, Gad H, Andersson F, Low P, Shupliakov O, Brodin L. Role of epsin 1 in synaptic vesicle endocytosis. Proc Natl Acad Sci USA. 2008;105:6445–6450. doi: 10.1073/pnas.0710267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kamikura DM, Cooper JA. Lipoprotein receptors and a disabled family cytoplasmic adaptor protein regulate EGL-17/FGF export in C. elegans. Genes Dev. 2003;17:2798–2811. doi: 10.1101/gad.1136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikura DM, Cooper JA. Clathrin interaction and subcellular localization of Ce-DAB-1, an adaptor for protein secretion in Caenorhabditis elegans. Traffic. 2006;7:324–336. doi: 10.1111/j.1600-0854.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- Kazazic M, Bertelsen V, Pedersen KW, Vuong TT, Grandal MV, Rodland MS, Traub LM, Stang E, Madshus IH. Epsin 1 is involved in recruitment of ubiquitinated EGF receptors into clathrin-coated pits. Traffic. 2009;10:235–245. doi: 10.1111/j.1600-0854.2008.00858.x. [DOI] [PubMed] [Google Scholar]
- Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol Biol Cell. 2006;17:4300–4317. doi: 10.1091/mbc.E06-05-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley DM, Krieger M. Receptor-mediated endocytosis of low density lipoprotein: somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc Natl Acad Sci USA. 1984;81:5454–5458. doi: 10.1073/pnas.81.17.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Wasiak S, Hussain NK, Angers A, McPherson PS. ENTH/ANTH proteins and clathrin-mediated membrane budding. J Cell Sci. 2004;117:9–18. doi: 10.1242/jcs.00928. [DOI] [PubMed] [Google Scholar]
- Maldonado-Baez L, Wendland B. Endocytic adaptors: recruiters, coordinators and regulators. Trends Cell Biol. 2006;16:505–513. doi: 10.1016/j.tcb.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Martin PJ, Ledbetter JA, Clark EA, Beatty PG, Hansen JA. Epitope mapping of the human surface suppressor/cytotoxic T cell molecule Tp32. J Immunol. 1984;132:759–765. [PubMed] [Google Scholar]
- Maurer ME, Cooper JA. The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH. J Cell Sci. 2006;119:4235–4246. doi: 10.1242/jcs.03217. [DOI] [PubMed] [Google Scholar]
- Mettlen M, Loerke D, Yarar D, Danuser G, Schmid SL. Cargo- and adaptor-specific mechanisms regulate clathrin-mediated endocytosis. J Cell Biol. 2010;188:919–933. doi: 10.1083/jcb.200908078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaely P, Zhao Z, Li WP, Garuti R, Huang LJ, Hobbs HH, Cohen JC. Identification of a VLDL-induced, FDNPVY-independent internalization mechanism for the LDLR. EMBO J. 2007;26:3273–3282. doi: 10.1038/sj.emboj.7601769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Keyel PA, Hawryluk MJ, Agostinelli NR, Watkins SC, Traub LM. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 2002;21:4915–4926. doi: 10.1093/emboj/cdf487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM, Cooper JA. Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic. 2001;2:111–123. doi: 10.1034/j.1600-0854.2001.020206.x. [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkearns EE, Cooper JA. FCH domain only-2 organizes clathrin-coated structures and interacts with Disabled-2 for low-density lipoprotein receptor endocytosis. Mol Biol Cell. 2012;23:1330–1342. doi: 10.1091/mbc.E11-09-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham CE, Mohney RP, Miller SL, Hanes RN, O’Bryan JP. The ubiquitin-interacting motifs target the endocytic adaptor protein epsin for ubiquitination. Curr Biol. 2002;12:1112–1116. doi: 10.1016/s0960-9822(02)00900-4. [DOI] [PubMed] [Google Scholar]
- Overstreet E, Chen X, Wendland B, Fischer JA. Either part of a Drosophila epsin protein, divided after the ENTH domain, functions in endocytosis of delta in the developing eye. Curr Biol. 2003;13:854–860. doi: 10.1016/s0960-9822(03)00326-9. [DOI] [PubMed] [Google Scholar]
- Reusch U, Bernhard O, Koszinowski U, Schu P. AP-1A and AP-3A lysosomal sorting functions. Traffic. 2002;3:752–761. doi: 10.1034/j.1600-0854.2002.31007.x. [DOI] [PubMed] [Google Scholar]
- Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Shafaq-Zadah M, Brocard L, Solari F, Michaux G. AP-1 is required for the maintenance of apico-basal polarity in the C. elegans intestine. Development. 2012;139:2061–2070. doi: 10.1242/dev.076711. [DOI] [PubMed] [Google Scholar]
- Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol. 2002;4:389–393. doi: 10.1038/ncb790. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirinian MI, et al. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein (LDL) binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem. 2005;280:38416–38423. doi: 10.1074/jbc.M504343200. [DOI] [PubMed] [Google Scholar]
- Sorensen EB, Conner SD. γ secretase-dependent cleavage initiates Notch signaling from the plasma membrane. Traffic. 2010;11:1234–1245. doi: 10.1111/j.1600-0854.2010.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–225. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Kishida S, Chayama K, Koyama S, Kikuchi A. Ubiquitin-interacting motifs of Epsin are involved in the regulation of insulin-dependent endocytosis. J Biochem. 2005;137:355–364. doi: 10.1093/jb/mvi044. [DOI] [PubMed] [Google Scholar]
- Teckchandani A, Mulkearns EE, Randolph TW, Toida N, Cooper JA. The clathrin adaptor Dab2 recruits EH domain scaffold proteins to regulate integrin β1 endocytosis. Mol Biol Cell. 2012;23:2905–2916. doi: 10.1091/mbc.E11-12-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Hansen D, Schedl T, Skeath JB. Epsin potentiates Notch pathway activity in Drosophila and C. elegans. Development. 2004;131:5807–5815. doi: 10.1242/dev.01459. [DOI] [PubMed] [Google Scholar]
- van Driel IR, Davis CG, Goldstein JL, Brown MS. Self-association of the low density lipoprotein receptor mediated by the cytoplasmic domain. J Biol Chem. 1987;262:16127–16134. [PubMed] [Google Scholar]
- Wang W, Struhl G. Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development. 2004;131:5367–5380. doi: 10.1242/dev.01413. [DOI] [PubMed] [Google Scholar]
- Wendland B, Steece KE, Emr SD. Yeast epsins contain an essential N-terminal ENTH domain, bind clathrin and are required for endocytosis. EMBO J. 1999;18:4383–4393. doi: 10.1093/emboj/18.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Cho B, Fischer JA. Drosophila Epsin’s role in Notch ligand cells requires three Epsin protein functions: the lipid binding function of the ENTH domain, a single ubiquitin interaction motif, and a subset of the C-terminal protein binding modules. Dev Biol. 2012;363:399–412. doi: 10.1016/j.ydbio.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Toops KA, Diaz F, Carvajal-Gonzalez JM, Gravotta D, Mazzoni F, Schreiner R, Rodriguez-Boulan E, Lakkaraju A. Mechanism of polarized lysosome exocytosis in epithelial cells. J Cell Sci. 2012 doi: 10.1242/jcs.109421. doi:10.1242/jcs.109421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem J, Greenwald I. A gene for a low density lipoprotein receptor-related protein in the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA. 1993;90:4572–4576. doi: 10.1073/pnas.90.10.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem J, Tuck S, Greenwald I, Han M. A gp330/megalin-related protein is required in the major epidermis of Caenorhabditis elegans for completion of molting. Development. 1999;126:597–606. doi: 10.1242/dev.126.3.597. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Yokode M, Yamamoto A, Masaki R, Murayama T, Horiuchi H, Kita T. Compensated endocytosis of LDL by hamster cells co-expressing the two distinct mutant LDL receptors defective in endocytosis and ligand binding. J Lipid Res. 1999;40:814–823. [PubMed] [Google Scholar]
- Yun M, Keshvara L, Park CG, Zhang YM, Dickerson JB, Zheng J, Rock CO, Curran T, Park HW. Crystal structures of the Dab homology domains of mouse disabled 1 and 2. J Biol Chem. 2003;278:36572–36581. doi: 10.1074/jbc.M304384200. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kim A, Abraham N, Khan LA, Hall DH, Fleming JT, Gobel V. Clathrin and AP-1 regulate apical polarity and lumen formation during C. elegans tubulogenesis. Development. 2012;139:2071–2083. doi: 10.1242/dev.077347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Ting AY. Imaging LDL receptor oligomerization during endocytosis using a co-internalization assay. ACS Chem Biol. 2011;6:308–313. doi: 10.1021/cb100361k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.