

Vaccinia-related kinase 1 (VRK1) is a histone kinase that phosphorylates histone H3 at Thr-3 and Ser-10. This study shows that mitogen-activated protein kinase phosphatase 2 regulates this phosphorylation negatively via interaction with VRK1, regardless of VRK1’s phosphatase activity.
Abstract
Mitogen-activated protein kinase phosphatase 2 (MKP2) is a member of the dual-specificity MKPs that regulate MAP kinase signaling. However, MKP2 functions are still largely unknown. In this study, we showed that MKP2 could regulate histone H3 phosphorylation under oxidative stress conditions. We found that MKP2 inhibited histone H3 phosphorylation by suppressing vaccinia-related kinase 1 (VRK1) activity. Moreover, this regulation was dependent on the selective interaction with VRK1, regardless of its phosphatase activity. The interaction between MKP2 and VRK1 mainly occurred in the chromatin, where histones are abundant. We also observed that the protein level of MKP2 and its interaction with histone H3 increased from G1 to M phase during the cell cycle, which is similar to the VRK1 profile. Furthermore, MKP2 specifically regulated the VRK1-mediated histone H3 phosphorylation at M phase. Taken together, these data suggest a novel function of MKP2 as a negative regulator of VRK1-mediated histone H3 phosphorylation.
INTRODUCTION
Mitogen-activated protein (MAP) kinase phosphatases (MKPs) are dual-specificity phosphatases known to inactivate MAP kinases (MAPKs), such as ERK, p38, and JNK, involved in proliferation, apoptosis, and differentiation in response to growth factors, hormones, stresses, and infections (Guan and Butch, 1995; Johnson and Lapadat, 2002; Theodosiou and Ashworth, 2002). The inactivation of MAPKs by MKPs is elicited via dephosphorylation of the threonine and tyrosine residues of a conserved signature T-X-Y motif within the activation loop of MAPKs (Boutros et al., 2008). MKPs share a common C-terminal catalytic domain representing phosphatase activity and an N-terminal noncatalytic domain containing a MAPK interaction motif (Farooq and Zhou, 2004). Each isoform has unique and shared features, including subcellular localization and substrate specificity (Dickinson and Keyse, 2006; Jeffrey et al., 2007). Because MKPs are related to MAPK signaling, uncontrolled MKPs have been implicated mainly in cancer (Keyse, 2008).
MKP2 (also known as DUSP4 for its gene’s name) was discovered as a human VH1 (MKP1) homologous phosphatase. Despite the important role of MKP2 in regulating MAPKs and its potential proliferation-suppressing activity, its roles remain poorly understood. MKP2 is induced by MAPK signaling for negative-feedback regulation (Brondello et al., 1997). Moreover, MKP2 was found to be increased under oxidative stress–induced apoptotic conditions to enhance the apoptotic response through p53 and E2F-1 binding to the MKP2 promoter region (Shen et al., 2006; Wang et al., 2007). Additionally, MKP2-mediated inactivation of ERK2 was found to be a key event in the establishment of replicative senescence in human fibroblasts (Torres et al., 2003; Tresini et al., 2007). Furthermore, deregulation of MKP2 inducing abnormal MAPK signaling causes the development and progression of human cancers (Keyse, 2008; Britson et al., 2009). Recently it was reported that MKP2 has a nonredundant role related to proliferation and cell survival affecting G2/M-phase transition in mouse embryonic fibroblasts (MEFs) derived from DUSP4-deficient mice (Lawan et al., 2011). However, there is still a lack of information about the roles of MKP2.
During mitosis, histone H3 is phosphorylated at Ser-10 in most eukaryotes (Hendzel et al., 1997; Wei et al., 1999). Histone H3 phosphorylation at Ser-10 during interphase has been shown to correlate with chromatin condensation prior to mitosis, whereas chromosome decondensation is observed when dephosphorylation is prematurely induced in mitosis (Ajiro et al., 1996a, b). In the late G2 phase, histone H3 phosphorylation at Ser-10 occurs only on pericentromeric chromatin. As mitosis proceeds, this phosphorylation spreads along the chromosomes and is complete at prophase; by the end of mitosis, it has disappeared (Van Hooser et al., 1998; Sauve et al., 1999). Moreover, histone H3 could also be dephosphorylated as a part of the stress response under such conditions as DNA damage and oxidative stress (Ozawa, 2008; Banerjee and Chakravarti, 2011).
Three histone H3 kinases in mitosis are well known in eukaryotes: haspin for phosphorylation at Thr-3, Aurora B for phosphorylation at Ser-10 and Ser-28, and vaccinia-related kinase 1 (VRK1) for phosphorylation at Thr-3 and Ser-10 (Crosio et al., 2002; Prigent and Dimitrov, 2003; Dai and Higgins, 2005; Baek, 2011). Because sequential histone H3 phosphorylation and dephosphorylation are regulated in a sophisticated manner, identifying the regulation mechanisms of the mitotic histone kinases could be important. Inhibition of PP1 and PP2A is already known to enhance Aurora B–mediated phosphorylation of histone H3 (Sugiyama et al., 2002). Recently it has also been reported that haspin accumulation at centromeres and histone H3 phosphorylation at Thr-3 in mitosis is regulated by Aurora B with a positive-feedback loop (Wang et al., 2011).
The VRK family is composed of three members: VRK1, VRK2, and VRK3 (Nichols and Traktman, 2004). Among the three isoforms, VRK1 has been studied more than the others, but the functions of all VRKs are still largely unknown. VRK1 can phosphorylate several transcription factors, such as p53, ATF2, and c-Jun (Sevilla et al., 2004a, b; Vega et al., 2004) and also can phosphorylate Caenorhabditis elegans barrier-to-autointegration factor-1 (BAF-1), which is involved in nuclear envelope formation (Gorjanacz et al., 2007). Moreover, recent studies have revealed that VRK1 has a function as a histone kinase that phosphorylates histone H3 (Kang et al., 2007; Baek, 2011) and it also regulates induction of cyclin D1 expression by phosphorylating CREB (Kang et al., 2008). Furthermore, studies with VRK1-deficient mice demonstrated that VRK1 is necessary for gametogenesis, spermatogonia cell maintenance, and oogenesis (Choi et al., 2010; Wiebe et al., 2010; Schober et al., 2011). Therefore the enzymatic activity of VRK1 regarding histone H3 phosphorylation must be regulated very delicately. Small GTPase Ran-GDP was known for inhibiting VRK1 activity on autophosphorylation and on histone H3 phosphorylation (Sanz-Garcia et al., 2008). Recently it was also reported that macrohistone H2A1.2 (MacroH2A1.2), a type of core histone variant containing a histone domain and a large nonhistone domain called the macro domain, regulates VRK1-mediated histone H3 phosphorylation during interphase (Kim et al., 2012). However, the mechanism for regulation of VRK1 in histone H3 phosphorylation is still largely unknown. Therefore studies regarding the interaction between MKP2 and VRK1 in histone H3 phosphorylation are worthy of elucidating a novel mechanism in regulation of VRK1.
In this paper, we provide molecular evidence that MKP2 is increased in oxidative stress conditions and is related to histone H3 dephosphorylation. Moreover, MKP2 negatively regulates histone H3 phosphorylation by VRK1, and this regulation is not dependent on the phosphatase activity of MKP2. MKP2 interacts with VRK1, and their interaction actually occurs in the chromatin region. Moreover, the level of MKP2 oscillates during the cell cycle, similar to the fluctuating level of VRK1, which is reported to be high in the M phase (Kang et al., 2007). Furthermore, MKP2 down-regulation causes an increase in histone H3 phosphorylation. We demonstrate here that MKP2 regulates VRK1-mediated histone H3 phosphorylation in a phosphatase activity–independent manner.
RESULTS
MKP2 regulates histone H3 phosphorylation in response to oxidative stress
There is a report that MKP1 is related to histone H3 dephosphorylation when thrombin and vascular endothelial growth factor (VEGF) stimulate human endothelial cells (ECs; Kinney et al., 2009). Therefore we wondered whether MKP2 could also affect histone H3 phosphorylation. Because MKP2 is an inducible protein that has a low basal protein expression level, we tried to find conditions that could induce MKP2 expression. Some reports have shown that oxidative stress can induce expression of MKP2 to elicit the apoptotic process in MEF cells (Shen et al., 2006; Wang et al., 2007). We therefore tested induction of MKP2 by oxidative stress using hydrogen peroxide (H2O2) in SH-SY5Y human neuroblastoma cells. As shown in Figure 1A, MKP2 was induced by oxidative stress, and histone H3 phosphorylation gradually decreased, along with the incubation time of H2O2. For the next step, we determined whether knockdown of MKP2 with small interfering RNA (siRNA) against MKP2 could influence the phosphorylation pattern. Surprisingly, knockdown of MKP2 led to sustained histone H3 phosphorylation under oxidative stress conditions (Figure 1B). Furthermore, there is a report that MKP2 induction by H2O2 is dependent on p53 (Shen et al., 2006). Therefore we examined whether knockdown of p53 with siRNA could affect MKP2 induction and histone H3 phosphorylation. The knockdown of p53 resulted in marked reduction and delay of MKP2 induction and sustained elevation of histone H3 phosphorylation (Supplemental Figure S1). This means that sustained histone H3 phosphorylation related with MKP2 induction by H2O2 is dependent on p53. To check the relevance of cell cycle progression and histone H3 phosphorylation, we also checked MPM-2 level as a mitotic marker (Russell et al., 2012). MPM-2 level was not significantly changed by p53 down-regulation, implying that sustained histone H3 phosphorylation is not related to cell cycle progression. Taken together, these data led us to believe MKP2 could regulate histone H3 phosphorylation under oxidative stress conditions.
FIGURE 1:
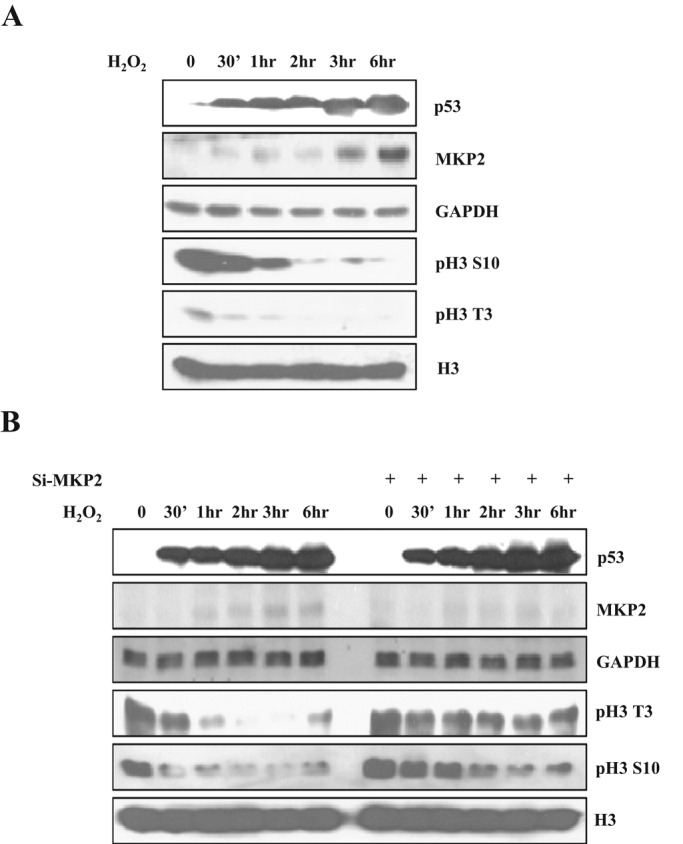
MKP2 is involved in histone H3 dephosphorylation in oxidative stress conditions. (A) SH-SY5Y cells were treated with 200 μM H2O2 for indicated times and harvested. The cell lysates were subjected to immunoblotting with the indicated antibodies. p53 was used as an indicator of oxidative stress. (B) SH-SY5Y cells were transfected with siRNA targeting MKP2 or scrambled control siRNA and cultured for 48 h. After treatment with 200 μM H2O2 for the indicated times, cells were harvested and lysed. The cell lysates were then subjected to immunoblotting with the indicated antibodies.
MKP2 regulates histone H3 phosphorylation mediated by VRK1
As previously mentioned, there are three representative mitotic histone H3 kinases: haspin for phosphorylation at Thr-3, Aurora B for phosphorylation at Ser-10 and Ser-28, and VRK1 for phosphorylation at Thr-3 and Ser-10 (Baek, 2011). Therefore we tried to examine whether MKP2 could affect histone H3 phosphorylation induced by VRK1, haspin, and Aurora B. First, we checked whether these kinases are involved in histone H3 phosphorylation by down-regulation of the kinases with si-RNAs (Figure S2A). As expected, knockdown of VRK1 decreased histone H3 phosphorylation at Thr-3 and Ser-10. Knockdown of haspin also decreased histone H3 phosphorylation at Thr-3. However, knockdown of Aurora B decreased histone H3 phosphorylation at Ser-10 as well as at Thr-3. This would be due to the function of Aurora B, which phosphorylates haspin to promote generation of inner centromeric histone H3 phosphorylation at Thr-3 (Wang et al., 2011). Second, we tested purified recombinant wild-type MKP2 (WT-MKP2 or MKP2) and catalytically inactive MKP2 (CI-MKP2) proteins carrying a mutation from cysteine to serine at position 280 in the dual-specificity phosphatase domain (Figure S2B) for their phosphatase activity toward phospho-ERK2 (Robinson et al., 2001; Cadalbert et al., 2005). As expected, WT-MKP2 could dephosphorylate phospho-ERK2, but CI-MKP2 had no phosphatase activity (Figure S2C). These proteins were coincubated in the kinase assay using purified kinases and core histone complex as the kinase and substrate, respectively (Figure 2, A–C). Addition of WT-MKP2 decreased VRK1-mediated histone H3 phosphorylation at Thr-3 and Ser-10 in a concentration-dependent manner. Similar to WT-MKP2, CI-MKP2 also inhibited VRK1-mediated histone H3 phosphorylation. In contrast with VRK1, the activities of haspin and Aurora B on histone H3 phosphorylation were not significantly changed by WT-MKP2 and CI-MKP2. Additionally, results from kinase assays also showed that both autophosphorylation of VRK1 and phosphorylation of core histone were decreased when incubated with WT-MKP2 and CI-MKP2 proteins (Figure 2D). Because BAF, mammalian homologue of BAF-1, is also known to be a substrate of VRK1, we tested whether MKP2 proteins could inhibit phosphorylation of BAF by VRK1. Similar to histone H3 phosphorylation, BAF phosphorylation was also inhibited by addition of WT-MKP2 or CI-MKP2 (Figure S2D). These results indicate that VRK1 activity is suppressed by MKP2 regardless of its phosphatase activity. Moreover, these data suggest that MKP2 specifically regulates VRK1 among mitotic histone H3 kinases.
FIGURE 2:

MKP2 inhibits VRK1-mediated histone H3 phosphorylation. (A) GST-MKP2 or GST-CI-MKP2 inhibits phosphorylation of histone H3 in a VRK1 kinase reaction in a concentration-dependent manner. GST-VRK1, purified core histone, and MKP2 or CI-MKP2 proteins were coincubated in a kinase reaction for 30 min as indicated. Purified MKP2 or CI-MKP2 proteins were used, with the following concentrations: 250 ng for lanes 3 and 8; 500 ng for lanes 4 and 9. Immunoblotting was performed using the indicated antibodies to analyze the phosphorylation of histone H3. (B and C) MKP2 does not regulate histone H3 phosphorylation by other mitotic histone kinases, such as haspin (B) or Aurora B (C). Histone kinase, core histone, and MKP2 or CI-MKP2 proteins were coincubated in a kinase reaction for 30 min as indicated. Purified MKP2 or CI-MKP2 proteins were used, with the following concentrations: 250 ng for lanes 4 and 9; 500 ng for lanes 4 and 9. Immunoblotting was performed using indicated antibodies to analyze phosphorylation of histone H3. (D) Purified GST, GST-VRK1, GST-MKP2, or GST-CI-MKP2 proteins were coincubated with histone H3 as a substrate for VRK1 in a kinase reaction containing [γ-32P] ATP for 30 min as indicated. Purified MKP2 or CI-MKP2 proteins were used, with the following concentrations: 100 ng for lanes 2 and 5; 250 ng for lanes 3 and 6; 500 ng for lanes 4 and 7. Phosphorylations were determined by autoradiography. The protein quantities were determined by SYPRO-Ruby staining.
In addition to the in vitro assays, we also tested whether MKP2 action on histone H3 phosphorylation exists in mammalian cells. WT-MKP2 overexpression in A549 human lung carcinoma cells, which are known to significantly express VRK1, caused a decrease in histone H3 phosphorylation at Thr-3 and Ser-10 (Figure 3A). Overexpression of CI-MKP2 and WT-MKP2 induced a decrease in histone H3 phosphorylation, which was similar to the in vitro results. To find the relationship between MKP2 and VRK1, we checked histone H3 phosphorylation after overexpression of MKP2 or VRK1 (Figure 3B). VRK1-mediated phosphorylation of histone H3 at Thr-3 and Ser-10 were decreased to basal level by overexpression of WT-MKP2 or CI-MKP2. Moreover, we found that the MKP2-mediated regulation of histone H3 phosphorylation did not result from any change of cell cycle progression; this was done by checking that mitotic marker MPM-2 levels were not significantly changed, as shown in Figure 3, A and B. Furthermore, we compared the degree of histone H3 phosphorylations when the VRK1 level was suppressed using si-VRK1 or when MKP2 was overexpressed. Histone H3 phosphorylation was decreased when VRK1 was down-regulated (Figure 3C). MKP2 overexpression also induced a decrease in histone H3 phosphorylation to a level similar to that seen with VRK1 knockdown. Moreover, The MKP2 overexpression did not further decrease histone H3 phosphorylation when VRK1 was down-regulated. Taken together, these results imply that MKP2 regulates the histone H3 phosphorylation through inhibition of VRK1.
FIGURE 3:

MKP2 regulates VRK1-mediated histone H3 phosphorylation regardless of its phosphatase activity. (A) A549 cells were transfected with EGFP or EGFP-MKP2 or EGFP-CI-MKP2 for 24 h. Immunoblotting was performed with indicated antibodies. (B) A549 cells were transfected with HA or HA-VRK1 and EGFP-MKP2 or EGFP-CI-MKP2 for 24 h. Immunoblotting was performed with indicated antibodies. (C) A549 cells were transfected with siRNA targeting VRK1 or scrambled control siRNA and EGFP or EGFP-MKP2, and then were harvested 48 h after transfection. Cell lysates were immunoblotted with indicated antibodies.
Furthermore, to clarify the effect of MKP2 on histone H3 phosphorylation, we examined phosphorylation patterns of histone H3 using nocodazole, which is known to cause M-phase arrest by stress-inducing depolymerization of microtubules. As shown in Figure S3, A and B, histone H3 phosphorylation gradually diminished after nocodazole release. Interestingly, this declining pattern of histone H3 phosphorylation was accelerated, and the basal level of phosphorylation was decreased, when MKP2 was overexpressed. These results indicated that MKP2 induction conditions could cause rapid histone H3 dephosphorylation.
MKP2 does not dephosphorylate histone H3 directly
Although the above data show that MKP2 regulates histone H3 phosphorylation through interaction with VRK1, there is also the possibility that MKP2 directly dephosphorylates histone H3. Therefore we examined whether MKP2 could act on histone H3 directly. First, when we added MKP2 in the middle of the kinase reaction with core histone and VRK1, both MKP2 and CI-MKP2 protein attenuated VRK1 autophosphorylation and core histone phosphorylation without decreasing these phosphorylations (Figure 4A). We further examined direct dephosphorylation of histone H3 in the MKP2 phosphatase reaction. Phosphorylated histone H3 at Thr-3 and Ser-10 induced by VRK1 showed no decrease in the phosphorylation pattern compared with the control by the addition of WT-MKP2 or CI-MKP2 (Figure 4B). Similarly, neither MKP2 nor CI-MKP2 could decrease phosphorylated histone H3 produced by haspin or Aurora B (Figure 4, C and D). Therefore we suggest that the inhibition of VRK1-mediated histone H3 phosphorylation by MKP2 is not due to its direct phosphatase action on histone H3.
FIGURE 4:

MKP2 cannot directly dephosphorylate histone H3. (A) GST-VRK1 and core histone were coincubated in a kinase reaction for 30 min. After 30 min, MKP2 or CI-MKP2 was added to the reaction, and the kinase reaction was continuously performed for 30 min, as indicated. The amounts of phosphorylation were determined by autoradiography. The protein quantities were determined by SYPRO-Ruby staining. (B–D) Phosphorylated histone H3 was obtained by coincubating histone H3 with the indicated histone kinases (B, VRK1; C, haspin; D, Aurora B) in the kinase reaction, and histone H3 was pulled down by immunoprecipitation using a ChIP-formulated histone H3 antibody. The phosphorylated histone H3 was coincubated with MKP2 or CI-MKP2 in the phosphatase reaction. Immunoblotting was performed using indicated antibodies to analyze the dephosphorylation of phosphorylated histone H3. Unphosphorylated histone H3 was used as a control for each kinase reaction.
MKP2 interacts with VRK1 regardless of its phosphatase activity
Because MKP2 regulates histone H3 phosphorylation by VRK1 (Figures 2 and 3), we also checked that MKP2 could interact with VRK1. MKP2 is an inducible phosphatase; we therefore examined the interaction between MKP2 and VRK1 by using immunoprecipitation and glutathione S-transferase (GST) pull-down assays after overexpression of phosphatases in HeLa cells. MKP2 protein interacted with VRK1, with its bands being detected by specific antibodies against Flag or enhanced green fluorescent protein (EGFP; Figures 5A and S4A). To address whether the interaction was due to the phosphatase activity of MKP2, we examined similar experiments that used CI-MKP2 rather than WT-MKP2. Interestingly, CI-MKP2 also showed similar results to WT-MKP2 regarding its interaction with VRK1 (Figures 5B and S4B). Furthermore, we examined localization of MKP2 and VRK1 in HeLa cells through overexpression of VRK1, WT-MKP2, and CI-MKP2. As shown in Figure 5C, they colocalized partially but almost in the nucleus. The colocalization of endogenous MKP2 and VRK1 was also checked. They also showed strong colocalization in the nucleus. Together these results suggest that MKP2 interacts with VRK1 in the nucleus, regardless of its phosphatase activity, similar to VRK1 regulation by MKP2.
FIGURE 5:

MKP2 interacts with VRK1 in the nucleus. (A and B) HeLa cells were transfected with Flag-VRK1 and EGFP-MKP2 or Flag-VRK1 and EGFP-CI-MKP2. After 24 h of expression, HeLa cells were harvested, and the cell lysates were subjected to immunoprecipitation with control IgG or FLAG antibody and sequential immunoblotting with the indicated antibodies. (C) EGFP-MKP2 or EGFP-CI-MKP2 was transfected into HeLa cells with pDsRed-Monomer-VRK1 (i). After 24 h of expression, immunocytochemistry was performed with nuclear staining using Hoechst (blue) and analyzed by fluorescence microscopy. Endogenous VRK1 and MKP2 were applied to immunocytochemistry by using antibodies against VRK1 and MKP2 (ii).
MKP2 and VRK1 interact in the chromatin fraction
Because MKP2 and VRK1 showed nuclear colocalization in the above data, we examined the subnuclear localization of MKP2 to further clarify its localization. VRK1 is known to localize in the nucleoplasm, euchromatin, and heterochromatin regions of the nucleus (Kang et al., 2007). In addition to VRK1’s subnuclear localization, MKP2 was also localized in the nuclear matrix and heterochromatin regions (Figure 6A). Furthermore, immunocytochemical analysis revealed localization of MKP2 in heterochromatin by showing the colocalization of MKP2 with heterochromatin protein 1γ (HP1γ) foci, a heterochromatin marker (Figure 6B). To clarify the interaction between MKP2 and VRK1, we performed GST pull-down assays, using subnuclear fractions from HeLa cells. MKP2 was precipitated with GST-VRK1 in the crude chromatin fraction but was not precipitated in the nucleoplasm fraction, which did not contain chromatin (Figure 6C). Moreover, we also performed GST pull-down assays using purified GST, GST-VRK1, core histone complex, and WT-MKP2 or CI-MKP2 (Figure S4, C and D). The binding between VRK1 and WT-MKP2 or CI-MKP2 was not apparent, but they could interact with one another when core histone was present. Thus these results suggest that MKP2 mainly interacts with VRK1 in the heterochromatin fraction, where histones are abundant and MKP2 and VRK1 exist together.
FIGURE 6:

Interaction between MKP2 and VRK1 occurs in the chromatin region. (A) EGFP-MKP2 was transfected into HeLa cells. After 24 h, subcellular and subnuclear fractionations were performed, and each fraction was subjected to immunoblotting with indicated antibodies. Antibodies against GAPDH, lamin B, or histone H3 were used as markers for the various fractions. (B) EGFP-HP1γ and pDsRed-Monomer-MKP2 were transfected into HeLa cells for 24 h. Immunocytochemistry was performed and analyzed by fluorescence microscopy. (C) Subcellular fractionation was performed on harvested HeLa cells, and a crude chromatin fraction was obtained from the pellet after nucleoplasm extraction by incubation with micrococcal nuclease. Nucleoplasm not containing chromatin fractions and crude chromatin fractions was pulled down with GST or GST-VRK1. The interaction was analyzed by immunoblotting with indicated antibodies. C.P., cytoplasm; N.P., nucleoplasm; E.C., euchromatin; N.M., nuclear matrix; H.C., heterochromatin; C.C., crude chromatin.
MKP2 regulates VRK1-mediated histone H3 phosphorylation at M phase
As previously mentioned, the VRK1 protein level is gradually increased to induce chromatin condensation through cell cycle progression, particularly from the S to M phases. Considering the interaction between VRK1 and MKP2, we wondered whether VRK1-mediated histone H3 phosphorylation could be affected by MKP2 following cell cycle progression. First, we compared the MKP2 protein level with the VRK1 level throughout G1 to M phases. For the analyses, A549 cells were arrested in the G1, S, and M phases by treatment with mimosine, thymidine, and nocodazole, respectively (Figure 7A). Indeed, MKP2 protein gradually increased in the S and M phases, similar to the profile of VRK1 (Figure 7B). We then checked the interaction pattern of MKP2 and VRK1 with histone H3 in chromatin throughout the G1 to M phases. Interestingly, MKP2 showed increased interaction with histone H3 at the M phase (Figure 7C). Similar to MKP2, VRK1 interaction with histone H3 also peaked at the M phase. Furthermore, we checked whether MKP2 down-regulation in M phase–arrested cells could affect histone H3 phosphorylation (Figure 7D). MKP2 knockdown in M phase–arrested control cells caused increase of histone H3 phosphorylation at Thr-3 and Ser-10. This increase was not detected by VRK1 down-regulation. Although the phosphorylations at Thr-3 in haspin or Aurora B down-regulation were decreased to an undetectable level, we did find that phosphorylations at Ser-10 in haspin or Aurora B down-regulation were still increased by MKP2 knockdown. This result suggests that MKP2 regulates only histone H3 phosphorylation mediated by VRK1, not by haspin or Aurora B. Together these results indicate that induced MKP2 is recruited to block extreme VRK1 action on histone H3 phosphorylation, which peaks at the M phase.
FIGURE 7:

Differential interaction between MKP2 and VRK1 during the cell cycle. (A) A549 cells were arrested in G1, S, and M phases with 50 μM mimosine, 100 μM thymidine, and 50 ng/μl nocodazole, respectively. After 16 h, cells were harvested and subjected to flow cytometry. Propidium iodide–stained DNA content was analyzed for verification of cell cycle synchronization by flow cytometry. (B) Immunoblotting was performed using indicated antibodies to analyze the profile of MKP2 and VRK1 following cell cycle progression. (C) Synchronized cells were fractionated to crude chromatin fractions. Those chromatin fractions were subjected to immunoprecipitation with the ChIP-formulated histone H3 antibody and sequential immunoblotting with the indicated antibodies. (D) Indicated siRNAs were transfected into A549 cells. After 36 h, cells were treated with 50 ng/μl nocodazole for 12 h. A549 cells were harvested, and the cell lysates were subjected to immunoblotting with indicated antibodies.
DISCUSSION
In this work, we suggest that MKP2, a member of the MKP family of dual-specificity phosphatases, interacts with VRK1 in the nucleus, mainly in the heterochromatin fraction (Figure 5C). Similar to the specific interaction between MKP2 and VRK1 shown in our study, the interactions of other mitotic histone kinases with protein phosphatase have also been reported. Protein phosphatase 1 (PP1) has a counteracting role against Aurora B kinase in mitotic phosphorylation of histone H3 by association with Aurora B (Hsu et al., 2000; Murnion et al., 2001; Emanuele et al., 2008). Aurora B activity is also known to be negatively regulated by associated PP1. Because VRK1 was only recently reported to be a histone H3 kinase, studies regarding VRK1 regulation related to histone H3 phosphorylation are fewer in number than those relating to Aurora B regulation. Therefore the regulation of VRK1-mediated histone H3 phosphorylation by MKP2 is a significantly novel observation. Interestingly, we also showed that MKP2 binding to histone H3 is increased at M phase (Figure 7C) but MKP2 does not regulate histone H3 phosphorylation mediated by haspin or Aurora B (Figure 7D). These results imply that MKP2 regulation of histone H3 phosphorylation is not due to masking histone H3 from kinases; it is due only to specific inhibition of VRK1 activity.
MKP2 is a member of the dual-specificity MKPs. Even though the major targets of MKPs are MAPKs, the interaction between MKP2 and VRK1 gives more insight into the function of MKP2. MKP1, which is one of the MKPs, has been demonstrated to be related to histone H3 dephosphorylation when thrombin and VEGF stimulate human ECs (Kinney et al., 2009). Moreover, oxidative stress–induced DNA damage caused reductions of histone H3 phosphorylations at M phase as a part of the DNA damage response, allowing cells time for DNA repair (Ozawa, 2008). In addition, it was recently reported that MKP2 knockdown caused retention at M phase and growth slowdown of fibroblasts in proliferative conditions (Lawan et al., 2011). In this study, we showed that MKP2 can be induced under oxidative stress, and the MKP2 induction is related to histone H3 dephosphorylation at Thr-3 and Ser-10. Interestingly, we also showed here that endogenous MKP2 protein level and its interaction with histone H3 were increased in M phase. Furthermore, MKP2 knockdown in M phase increased VRK1-mediated histone H3 phosphorylation, suggesting that MKP2 is responsible for regulation of VRK1-mediated histone H3 phosphorylation in M phase. Taken together, these results suggested that MKP2 might be responsible for the reduction of histone H3 phosphorylations in DNA damage responses under oxidative stress. Moreover, those results also imply that MKP2 might be related to histone H3 dephosphorylation in the transition period of M phase.
Previously we reported that VRK1 is a histone H3 kinase that phosphorylates histone H3 at Thr-3 and Ser-10 (Kang et al., 2007). In this study, we showed that MKP2 negatively regulates VRK1 activity, regardless of its phosphatase activity, by confirming inhibition of VRK1 with inactive MKP2 constructs verified in previous studies (Robinson et al., 2001; Cadalbert et al., 2005). Although the WT-MKP2 construct, but not the CI-MKP2 construct, resulted in a decrease in ERK phosphorylation, both constructs showed an obvious decrease in phosphorylation of histone H3 via interaction with VRK1 (Figures 2 and S2C). Moreover, WT-MKP2 protein could not directly dephosphorylate phospho–histone H3 (Figure 4). These results suggested that the MKP2-mediated dephosphorylation of histone H3 is not due to direct action of MKP2 but to regulation of VRK1 activity through protein–protein interaction, regardless of phosphatase activity. Furthermore, we also recognized that the in vitro interaction between VRK1 and MKP2 was not apparent but the interaction became stronger when core histone was added. Taken together, these data suggest that MKP2 recognizes allosteric structural changes of VRK1 induced by interaction with its substrates and binds to VRK1 by forming a ternary complex. Therefore, if VRK1 interacts with histone H3 or BAF, substrate-bound VRK1 could be recognized and regulated by MKP2. Even though the gross structures of histone H3 and BAF differ, we speculate that the phosphorylated motifs of two substrates should fit into the active site of VRK1 and trigger allosteric conformation change. However, it will be necessary to verify the possibility that BAF-bound VRK1 induces MKP2 interaction. In a manner similar to allosteric regulation of VRK1 by protein–protein interaction with MKP2, Ran-GDP and MacroH2A1.2 were also revealed to interact with VRK1 and regulated VRK1 activity via protein–protein interaction (Sanz-Garcia et al., 2008; Kim et al., 2012). On the basis of these data, we suggest that nuclear VRK1 kinase activity can be regulated by protein–protein interactions depending on VRK1’s subcellular localization, resulting in an intranuclear asymmetric distribution of the kinase activity.
In this paper, we showed that MKP2 negatively regulates phosphorylation of histone H3 by VRK1. The histone H3 phosphorylation gradually increases from the S to M phase and disappears at the late M phase, implying the existence of some regulatory mechanisms. As previously mentioned, we reported that MacroH2A1.2 suppresses VRK1 activity during interphase (Kim et al., 2012). In addition to the MacroH2A1.2-mediated inhibition of VRK1, we here suggest that MKP2 has a regulatory role in dephosphorylating histone H3 under stress condition or that it regulates excessive phosphorylation of histone H3 at the M phase. Because MKP2 cannot directly dephosphorylate histone H3, it is possible that other factors may elicit down-regulation of histone H3 phosphorylation via direct dephosphorylation or other histone modifications.
In this study, we showed that MKP2 was responsible for histone H3 dephosphorylation under oxidative stress conditions. Moreover, we also found that MKP2 regulated histone H3 phosphorylation mediated by VRK1, a member of the mitotic histone kinases. Additionally, MKP2 regulated VRK1 in a phosphatase activity–independent manner and interacted with VRK1 in the chromatin region, where histones are abundant. We also demonstrated that the MKP2 level was changed during the cell cycle, similar to the VRK1 profile. Furthermore, MKP2 affected the histone H3 phosphorylation induced by VRK1 during M phase. These results suggest that MKP2 induction might regulate VRK1-mediated histone H3 phosphorylation. Although other possible mechanisms may exist to regulate VRK1 action, such as suppression by macro H2A1.2, we suggest that MKP2 is a negative regulator of VRK1-mediated histone H3 phosphorylation. Unraveling the novel function of MKP2 regulation of VRK1-mediated histone H3 phosphorylation through protein–protein interaction would help our understanding of the physiological roles of MKP2.
MATERIALS AND METHODS
Materials
DMEM, minimum essential medium (MEM), and fetal bovine serum (FBS) were purchased from Hyclone Laboratories (Logan, UT). Penicillin/streptomycin was obtained from Life Technologies (Carlsbad, CA). Mimosine, thymidine, and nocodazole were purchased from Sigma-Aldrich (St. Louis, MO). DNase I (EN0521) and micrococcal nuclease (EN0181) were purchased from Fermentas (Burlington, Ontario, Canada). VRK1 antiserum was generated in rabbit by using full-length recombinant VRK1 protein as described previously (Kang and Kim, 2006). Polyclonal FLAG (F7425) antibody was purchased from Sigma-Aldrich. Polyclonal 6xHis (A190-114A) antibody was obtained from Bethyl Laboratories (Montgomery, TX). Monoclonal GFP (B-2), GAPDH (FL-335), p53 (DO-I), and lamin B (C-12) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies specific for histone H3 (9715), phospho–Thr-3 of histone H3 (9714), phospho–Ser-10 of histone H3 (9701), phospho–Ser-28 of histone H3 (971), ERK (4696), phospho-ERK (4377), phospho-cdc2 (9111), and FoxM1 (5436) were purchased from Cell Signaling (Beverly, MA). Chromatin-immunoprecipitation (ChIP)-formulated polyclonal histone H3 (2650) antibody was also obtained from Cell Signaling. Monoclonal MKP2 (610850) and Aim-1 (Aurora B kinase; 611082) antibodies were purchased from BD Biosciences (Franklin Lakes, NJ). Polyclonal MKP2 (ab72593) antibody and antibody for phospho–Ser-10 of histone H3 (ab14955) were obtained from Abcam (Cambridge, UK). Alexa Fluor 546–conjugated antibody against rabbit immunoglobulin G (IgG; A11010) and Alexa Fluor 488–conjugated antibody against mouse IgG (A11001) were purchased from Invitrogen (Carlsbad, CA). MPM-2 antibody (05-368) was purchased from Upstate (Billerica, MA). Phospho-ERK protein was kindly provided by H. S. Yoon (Nanyang Technological University, Singapore). Purified free-core histone proteins were provided by J. B. Kwon (Ewha Womans University, Republic of Korea). Histone H3 protein was purchased from Roche (Penzberg, Germany). Western blot detection reagent (SUPEX) was obtained from Neuronex (Pohang, Korea). [γ-32P]dATP was purchased from NEN (Boston, MA).
Plasmids and siRNAs
pFLAG-CMV2-VRK1, pGEX4T-1-VRK1, pEGFP-N1-VRK1, and pEGFP-N1-HP1γ were generated as described previously (Kang et al., 2007). VRK1 cDNA was subcloned to pDsRed-Monomer-N1 (BD Biosciences) and pHA (a gift from S. K. Jang, POSTECH, Pohang, Korea). GST-tagged expression constructs for MKP2 and CI-MKP2 (Robinson et al., 2001) and Flag-tagged constructs for MKP2 and CI-MKP2 (Cadalbert et al., 2005) were kindly provided by R. Plevin (Strathclyde University, Scotland). For purification of full-length size without tag of MKP2 and CI-MKP2, MKP2 and CI-MKP2 cDNAs were subcloned to PTYB2 vector, which is an IMPACT-CN system from New England Biolabs (Ipswich, MA). MKP2 and CI-MKP2 were also subcloned to pEGFP-N1 and pDsRed-Monomer-N1. Expression constructs for 6xHis-tagged kinase domain of haspin was a kind gift from J. M. Higgins (Harvard Medical School, Boston, MA). Aurora kinase B cDNA was amplified from a human cDNA library and subcloned to pGEX4T-3. For generation of His-tagged recombinant BAF, full-length BAF was amplified from HeLa cells by using reverse transcriptase PCR (RT-PCR). BAF was cloned into pPROEX-HTa (Invitrogen). siRNAs against VRK1 and MKP2, ON-TARGETplus SMARTpool containing four pooled SMARTselected siRNA duplexes were purchased from Thermo Scientific–Dharmacon (Lafayette, CO). siRNAs against Aurora B, haspin, and p53 were synthesized from Bioneer (Daejeon, Korea) as described previously (Kim et al., 2012).
Cell cultures and transfection of plasmids and siRNAs
Human cervical carcinoma HeLa cells and human lung carcinoma A549 cells were maintained in DMEM containing heat-inactivated 10% FBS and 1% penicillin/streptomycin in a humidified 5% CO2 incubator at 37°C. Human neuroblastoma SH-SY5Y cells were grown in MEM containing heat-inactivated 10% FBS and 1% penicillin/streptomycin in a humidified 5% CO2 incubator at 37°C. Transient transfections for HeLa cells were performed by using METAFECTENE from Biontex (Munich, Germany) according to the manufacturer’s instructions. Transient transfections for A549 and SH-SY5Y cells were performed using the Neon transfection system (Invitrogen) according to the manufacturer’s instructions. Cells were collected at 24 h after transfection with plasmids or 48 h after transfection with siRNAs for immunoblotting and immunoprecipitation or were treated with indicated drugs for further experiments.
Western blotting, GST pull down, and immunoprecipitation
The Western blotting procedure was performed as described previously (Lee et al., 2008). For Western blot analysis, bands were visualized using the Western blot detection kit (Pohang, Korea). For GST pull down, GST or GST-VRK1 proteins coupled to glutathione-Sepharose beads were incubated with solubilized lysates. Similarly, indicated antibodies coupled to protein G–agarose beads were incubated with solubilized lysates for immunoprecipitation. The beads were washed four times with TNE lysis buffer, and the remaining proteins bound to beads were resolved by SDS–PAGE and analyzed by Western blotting.
Immunocytochemistry
Immunocytochemistry was performed as described previously (Lee et al., 2008). Slides were visualized by confocal laser-scanning microscopy (Fluoview FV1000; Olympus, Tokyo, Japan)
Subcellular and subnuclear fractionation
Subcellular and subnuclear fractions were prepared as described previously (Kang et al., 2007). The pellet obtained following the extraction of the nucleoplasm, which contained the subnuclear organelles and chromatins, was treated with micrococcal nuclease for 1 h at room temperature in order to extract the crude chromatin fraction, which was isolated as the soluble fraction.
Purification of recombinant proteins
The purifications of GST fusion proteins were carried out as described previously (Kang and Kim, 2006). Monomeric BAF protein was prepared from the insoluble fraction by tobacco etch virus protease treatment to cleave the hexameric His tag as described previously (Nichols et al., 2006). PTYB2-MKP2 and PTYB2-CI-MKP2 proteins were purified according to the manufacturer’s instructions. His-tagged kinase domain of haspin was purified as previously described (Dai et al., 2005). GST-tagged Aurora kinase B was also purified as previously described (Girdler et al., 2008).
In vitro kinase assay
The standard procedure for the in vitro kinase assay was carried out as described previously (Kang and Kim, 2006). When the [γ-32P] ATP was used as the source of phosphate in the reaction, radioactivity was measured by autoradiography. In the case in which unlabeled ATP was used, phosphorylation was measured by Western blotting by using the indicated phospho-residue–specific antibodies. The quantities of proteins used in kinase assays were measured by using Coomassie brilliant blue G-250 (Bio-Rad, Hercules, CA) or SYPRO Ruby (Invitrogen) according to manufacturers’ instructions.
In vitro phosphatase assay
The standard procedure was carried out as described previously (Sloss et al., 2005), except that ATP was used in place of [γ-32P]ATP. The phosphatase activity was measured by Western blotting using phospho-ERK–specific antibody. For determination of the MKP2 phosphatase activity on phospho–histone H3, H3 proteins were phosphorylated by the indicated mitotic histone H3 kinases and then immunoprecipitated by using ChIP-formulated histone H3 antibody (Cell Signaling) with protein G agarose. After two washes with phosphatase reaction buffer, MKP2 and CI-MKP2 proteins were added as indicated. Phosphorylation and dephosphorylation of histone H3 were measured by Western blotting by using the indicated phospho-residue–specific antibodies.
Cell cycle synchronization and flow cytometry
Cell cycle synchronization and flow cytometry were performed as described previously (Kang et al., 2007). For analysis of mitotic-phase cells, fixed cells were incubated with antibody against phospho–Aurora A/B/C and stained with Alexa Fluor 647–conjugated antibody. Then, cellular DNA was stained with 50 μg/ml of propidium iodide (Sigma-Aldrich) and 100 μg/ml RNase A (Sigma-Aldrich) in PBS, and samples were analyzed on a FACSCalibur system (BD Biosciences).
Statistical analysis
All experiments, including the immunoblots and kinase assays, were independently repeated at least three times. All assays and blots presented are representative of more than three separate experiments. All quantitative data are presented as means ± SD. Comparisons between two groups were analyzed via Student’s t test, and values of p < 0.05 were considered to be significant.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea (grants: 20110027957, 20110031234, 20120005830, 20110031517; and WCU program R31-10105). This work was also supported by the Brain Korea 21 program of the Korean Ministry of Education, Science and Technology.
Abbreviations used:
- BAF-1
barrier-to-autointegration factor-1
- ChIP
chromatin-immunoprecipitation
- CI-MKP2
catalytically inactive MKP2
- EC
endothelial cell
- EGFP
enhanced green fluorescent protein
- FBS
fetal bovine serum
- GST
glutathione S-transferase
- IgG
immunoglobulin G
- MacroH2A1.2
macrohistone H2A1.2
- MAP
mitogen-activated protein
- MAPK
MAP kinase
- MEF
mouse embryonic fibroblast
- MEM
minimum essential medium
- MKP
MAP kinase phosphatase
- PP1
protein phosphatase 1
- siRNA
small interfering RNA
- VEGF
vascular endothelial growth factor
- VRK1
vaccinia-related kinase 1
- WT-MKP2
wild-type MKP2
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0456) on December 5, 2012.
REFERENCES
- Ajiro K, Yasuda H, Tsuji H. Vanadate triggers the transition from chromosome condensation to decondensation in a mitotic mutant (tsTM13) inactivation of p34cdc2/H1 kinase and dephosphorylation of mitosis-specific histone H3. Eur J Biochem. 1996a;241:923–930. doi: 10.1111/j.1432-1033.1996.00923.x. [DOI] [PubMed] [Google Scholar]
- Ajiro K, Yoda K, Utsumi K, Nishikawa Y. Alteration of cell cycle-dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J Biol Chem. 1996b;271:13197–13201. doi: 10.1074/jbc.271.22.13197. [DOI] [PubMed] [Google Scholar]
- Baek SH. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell. 2011;42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- Banerjee T, Chakravarti D. A peek into the complex realm of histone phosphorylation. Mol Cell Biol. 2011;31:4858–4873. doi: 10.1128/MCB.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- Britson JS, Barton F, Balko JM, Black EP. Deregulation of DUSP activity in EGFR-mutant lung cancer cell lines contributes to sustained ERK1/2 signaling. Biochem Biophys Res Commun. 2009;390:849–854. doi: 10.1016/j.bbrc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- Brondello JM, Brunet A, Pouyssegur J, McKenzie FR. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J Biol Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- Cadalbert L, Sloss CM, Cameron P, Plevin R. Conditional expression of MAP kinase phosphatase-2 protects against genotoxic stress-induced apoptosis by binding and selective dephosphorylation of nuclear activated c-jun N-terminal kinase. Cell Signal. 2005;17:1254–1264. doi: 10.1016/j.cellsig.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Choi YH, Park CH, Kim W, Ling H, Kang A, Chang MW, Im SK, Jeong HW, Kong YY, Kim KT. Vaccinia-related kinase 1 is required for the maintenance of undifferentiated spermatogonia in mouse male germ cells. PLoS One. 2010;5:e15254. doi: 10.1371/journal.pone.0015254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Higgins JM. Haspin: a mitotic histone kinase required for metaphase chromosome alignment. Cell Cycle. 2005;4:665–668. doi: 10.4161/cc.4.5.1683. [DOI] [PubMed] [Google Scholar]
- Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19:472–488. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CS, Stukenberg PT. Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol. 2008;181:241–254. doi: 10.1083/jcb.200710019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16:769–779. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S. Molecular basis of drug resistance in Aurora kinases. Chem Biol. 2008;15:552–562. doi: 10.1016/j.chembiol.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Gorjanacz M, Klerkx EP, Galy V, Santarella R, Lopez-Iglesias C, Askjaer P, Mattaj IW. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. EMBO J. 2007;26:132–143. doi: 10.1038/sj.emboj.7601470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Butch E. Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J Biol Chem. 1995;270:7197–7203. doi: 10.1074/jbc.270.13.7197. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Hsu JY, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kang TH, Kim KT. Negative regulation of ERK activity by VRK3-mediated activation of VHR phosphatase. Nat Cell Biol. 2006;8:863–869. doi: 10.1038/ncb1447. [DOI] [PubMed] [Google Scholar]
- Kang TH, Park DY, Choi YH, Kim KJ, Yoon HS, Kim KT. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol Cell Biol. 2007;27:8533–8546. doi: 10.1128/MCB.00018-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Park DY, Kim W, Kim KT. VRK1 phosphorylates CREB and mediates CCND1 expression. J Cell Sci. 2008;121:3035–3041. doi: 10.1242/jcs.026757. [DOI] [PubMed] [Google Scholar]
- Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- Kim W, Chakraborty G, Kim S, Shin J, Park CH, Jeong MW, Bharatham N, Yoon HS, Kim KT. Macro histone H2A1.2 (MacroH2A1) protein suppresses mitotic kinase VRK1 during interphase. J Biol Chem. 2012;287:5278–5289. doi: 10.1074/jbc.M111.281709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney CM, Chandrasekharan UM, Yang L, Shen J, Kinter M, McDermott MS, DiCorleto PE. Histone H3 as a novel substrate for MAP kinase phosphatase-1. Am J Physiol Cell Physiol. 2009;296:C242–C249. doi: 10.1152/ajpcell.00492.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawan A, Al-Harthi S, Cadalbert L, McCluskey AG, Shweash M, Grassia G, Grant A, Boyd M, Currie S, Plevin R. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J Biol Chem. 2011;286:12933–12943. doi: 10.1074/jbc.M110.181370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Jeong MW, Kim W, Choi YH, Kim KT. Cooperative roles of c-Abl and Cdk5 in regulation of p53 in response to oxidative stress. J Biol Chem. 2008;283:19826–19835. doi: 10.1074/jbc.M706201200. [DOI] [PubMed] [Google Scholar]
- Murnion ME, Adams RR, Callister DM, Allis CD, Earnshaw WC, Swedlow JR. Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J Biol Chem. 2001;276:26656–26665. doi: 10.1074/jbc.M102288200. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Traktman P. Characterization of three paralogous members of the mammalian vaccinia related kinase family. J Biol Chem. 2004;279:7934–7946. doi: 10.1074/jbc.M310813200. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Wiebe MS, Traktman P. The vaccinia-related kinases phosphorylate the N′ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol Biol Cell. 2006;17:2451–2464. doi: 10.1091/mbc.E05-12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K. Reduction of phosphorylated histone H3 serine 10 and serine 28 cell cycle marker intensities after DNA damage. Cytometry A. 2008;73:517–527. doi: 10.1002/cyto.a.20559. [DOI] [PubMed] [Google Scholar]
- Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for. J Cell Sci. 2003;116:3677–3685. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Sloss CM, Plevin R. Inactivation of JNK activity by mitogen-activated protein kinase phosphatase-2 in EAhy926 endothelial cells is dependent upon agonist-specific JNK translocation to the nucleus. Cell Signal. 2001;13:29–41. doi: 10.1016/s0898-6568(00)00121-2. [DOI] [PubMed] [Google Scholar]
- Russell P, Hennessy BT, Li J, Carey MS, Bast RC, Freeman T, Venkitaraman AR. Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest. Oncogene. 2012;31:2450–2460. doi: 10.1038/onc.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Garcia M, Lopez-Sanchez I, Lazo PA. Proteomics identification of nuclear Ran GTPase as an inhibitor of human VRK1 and VRK2 (vaccinia-related kinase) activities. Mol Cell Proteomics. 2008;7:2199–2214. doi: 10.1074/mcp.M700586-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve DM, Anderson HJ, Ray JM, James WM, Roberge M. Phosphorylation-induced rearrangement of the histone H3 NH2-terminal domain during mitotic chromosome condensation. J Cell Biol. 1999;145:225–235. doi: 10.1083/jcb.145.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober CS, Aydiner F, Booth CJ, Seli E, Reinke V. The kinase VRK1 is required for normal meiotic progression in mammalian oogenesis. Mech Dev. 2011;128:178–190. doi: 10.1016/j.mod.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Sevilla A, Santos CR, Barcia R, Vega FM, Lazo PA. c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK) Oncogene. 2004a;23:8950–8958. doi: 10.1038/sj.onc.1208015. [DOI] [PubMed] [Google Scholar]
- Sevilla A, Santos CR, Vega FM, Lazo PA. Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J Biol Chem. 2004b;279:27458–27465. doi: 10.1074/jbc.M401009200. [DOI] [PubMed] [Google Scholar]
- Shen WH, Wang J, Wu J, Zhurkin VB, Yin Y. Mitogen-activated protein kinase phosphatase 2: a novel transcription target of p53 in apoptosis. Cancer Res. 2006;66:6033–6039. doi: 10.1158/0008-5472.CAN-05-3878. [DOI] [PubMed] [Google Scholar]
- Sloss CM, Cadalbert L, Finn SG, Fuller SJ, Plevin R. Disruption of two putative nuclear localization sequences is required for cytosolic localization of mitogen-activated protein kinase phosphatase-2. Cell Signal. 2005;17:709–716. doi: 10.1016/j.cellsig.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Sugiyama K, Sugiura K, Hara T, Sugimoto K, Shima H, Honda K, Furukawa K, Yamashita S, Urano T. Aurora-B associated protein phosphatases as negative regulators of kinase activation. Oncogene. 2002;21:3103–3111. doi: 10.1038/sj.onc.1205432. [DOI] [PubMed] [Google Scholar]
- Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-reviews3009. reviews3009-1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres C, Francis MK, Lorenzini A, Tresini M, Cristofalo VJ. Metabolic stabilization of MAP kinase phosphatase-2 in senescence of human fibroblasts. Exp Cell Res. 2003;290:195–206. doi: 10.1016/s0014-4827(03)00309-4. [DOI] [PubMed] [Google Scholar]
- Tresini M, Lorenzini A, Torres C, Cristofalo VJ. Modulation of replicative senescence of diploid human cells by nuclear ERK signaling. J Biol Chem. 2007;282:4136–4151. doi: 10.1074/jbc.M604955200. [DOI] [PubMed] [Google Scholar]
- Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111:3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Vega FM, Sevilla A, Lazo PA. p53 Stabilization and accumulation induced by human vaccinia-related kinase 1. Mol Cell Biol. 2004;24:10366–10380. doi: 10.1128/MCB.24.23.10366-10380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Ulyanova NP, van der Waal MS, Patnaik D, Lens SM, Higgins JM. A positive feedback loop involving Haspin and Aurora B promotes CPC accumulation at centromeres in mitosis. Curr Biol. 2011;21:1061–1069. doi: 10.1016/j.cub.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shen WH, Jin YJ, Brandt-Rauf PW, Yin Y. A molecular link between E2F-1 and the MAPK cascade. J Biol Chem. 2007;282:18521–18531. doi: 10.1074/jbc.M610538200. [DOI] [PubMed] [Google Scholar]
- Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 1999;97:99–109. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- Wiebe MS, Nichols RJ, Molitor TP, Lindgren JK, Traktman P. Mice deficient in the serine/threonine protein kinase VRK1 are infertile due to a progressive loss of spermatogonia. Biol Reprod. 2010;82:182–193. doi: 10.1095/biolreprod.109.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.