

The cholesterol biosynthetic enzyme 3-hydroxy-3-methylglutaryl CoA reductase is subjected to sterol-accelerated degradation from endoplasmic reticulum membranes. This study shows that reductase degradation is mediated by the lipid droplet–associated protein ancient ubiquitous protein-1, which facilitates binding of the E2 Ubc7 to the E3 ligases, gp78 and Trc8, that initiate reductase ubiquitination.
Abstract
Sterol-induced binding to Insigs in endoplasmic reticulum (ER) membranes triggers ubiquitination of the cholesterol biosynthetic enzyme 3-hydroxy-3-methylglutaryl CoA reductase. This ubiquitination, which is mediated by Insig-associated ubiquitin ligases gp78 and Trc8, is obligatory for extraction of reductase from lipid droplet–associated ER membranes into the cytosol for proteasome-mediated, ER-associated degradation (ERAD). In this study, we identify lipid droplet–associated, ancient, ubiquitous protein-1 (Aup1) as one of several proteins that copurify with gp78. RNA interference (RNAi) studies show that Aup1 recruits the ubiquitin-conjugating enzyme Ubc7 to lipid droplets and facilitates its binding to both gp78 and Trc8. The functional significance of these interactions is revealed by the observation that RNAi-mediated knockdown of Aup1 blunts sterol-accelerated ubiquitination of reductase, which appears to occur in lipid droplet–associated membranes and subsequent ERAD of the enzyme. In addition, Aup1 knockdown inhibits ERAD of Insig-1, another substrate for gp78, as well as that of membrane-bound precursor forms of sterol-regulatory, element-binding protein-1 and -2, transcription factors that modulate expression of genes encoding enzymes required for cholesterol synthesis. Considered together, these findings not only implicate a role for Aup1 in maintenance of intracellular cholesterol homeostasis, but they also highlight the close connections among ERAD, lipid droplets, and lipid droplet–associated proteins.
INTRODUCTION
Cholesterol synthesis in mammalian cells is controlled in part through sterol-accelerated, endoplasmic reticulum (ER)-associated degradation (ERAD) of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl CoA (HMG CoA) reductase (Brown and Goldstein, 1980). This metabolically controlled reaction results from sterol-induced binding of reductase to highly related ER membrane proteins called Insig-1 and Insig-2 (Sever et al., 2003b; Song et al., 2005a; Jo and DeBose-Boyd, 2010). Insig binding is mediated entirely by the N-terminal membrane domain of reductase, which contains eight membrane-spanning segments and precedes a large C-terminal cytosolic domain that exerts enzymatic activity (Liscum et al., 1985; Roitelman et al., 1992). The Insig-associated ubiquitin ligases gp78 and Trc8 subsequently initiate reductase ubiquitination (Sever et al., 2003a; Song et al., 2005b; Jo et al., 2011a), thereby marking reductase for recognition by the VCP/p97 ATPase complex. VCP/p97-catalyzed hydrolysis of ATP is thought to drive extraction and dislocation of ubiquitinated reductase and other substrates from ER membranes into the cytosol for proteasomal degradation (Ye et al., 2001; Song et al., 2005b; Hartman et al., 2010). While membrane extraction and cytosolic dislocation are established aspects in the ERAD of soluble and some membrane-bound substrates, a detailed understanding of mechanisms underlying these reactions is lacking (Brodsky and Skach, 2011).
Lipid droplets are ER-derived organelles composed of neutral lipids, such as triglycerides and cholesterol esters, surrounded by a phospholipid monolayer (Martin and Parton, 2006; Farese and Walther, 2009). Storage of cholesterol esters and triglycerides in lipid droplets provides a protective mechanism that guards against the overaccumulation of free cholesterol and fatty acids in cells (Fujimoto and Parton, 2011). Under conditions of high demand, these lipids are mobilized from lipid droplets for utilization in metabolic processes, including β-oxidation and synthesis of membranes, lipoproteins, or steroid hormones. Multiple lines of evidence are emerging that implicate lipid droplets in cellular functions beyond lipid metabolism (Welte, 2007; Fujimoto and Parton, 2011; Walther and Farese, 2012). For example, a model has been proposed in which lipid droplets play a role in extraction of ERAD substrates from membranes (Ploegh, 2007). Experimental support for this is provided by studies of apolipoprotein B-100, the primary protein of low-density lipoproteins. Apolipoprotein B-100 is subjected to ERAD in the absence of proper cotranslational lipidation (Fisher and Ginsberg, 2002); the protein accumulates on lipid droplets when proteasomes are inhibited, indicating localization to the organelle as an intermediate step in its ERAD (Ohsaki et al., 2006; Suzuki et al., 2012). Consistent with this, our studies show that sterols trigger translocation of reductase to lipid droplet–associated ER membranes en route to its extraction and cytosolic dislocation (from membranes into the cytosol for proteasomal degradation; Ye et al., 2001; Song et al., 2005b; Hartman et al., 2010). Ploegh and coworkers recently showed that pharmacological inhibition of lipid droplet formation blunts cytosolic dislocation of model ERAD substrates (Klemm et al., 2011). Finally, multiple proteomic studies have identified several chaperones and components of the ERAD pathway, including VCP/p97 and its membrane receptors ubiquitin regulatory x (Ubx) domain-containing proteins 2 and 8 (Ubxd2 and Ubxd8, respectively), as lipid droplet–associated proteins (Brasaemle et al., 2004; Liu et al., 2004; Sato et al., 2006; Bartz et al., 2007b; Wan et al., 2007). It should be noted that in the yeast Saccharomyces cerevisiae, lipid droplets appear to be dispensable for ERAD (Olzmann and Kopito, 2011), reflecting key differences between the ERAD pathway in yeast and mammalian cells.
Proteomic studies also identified ancient ubiquitous protein-1 (Aup1) as a protein associated with lipid droplets (Brasaemle et al., 2004; Liu et al., 2004; Sato et al., 2006; Bartz et al., 2007a; Wan et al., 2007). Aup1 contains an N-terminal hydrophobic region that orients the protein in membranes such that the N- and C-termini both face the cytosol (Spandl et al., 2011). In addition, Aup1 contains an acyl-transferase domain, a Cue domain, and a UBE2G2-binding region (G2BR). The Cue domain is known to bind ubiquitin (Kang et al., 2003; Prag et al., 2003), but its precise role in ERAD is not completely understood. The G2BR plays a well-established role in ERAD through its binding to the soluble ubiquitin-conjugating enzyme Ubc7 (or Ube2g2; Chen et al., 2006). Aup1 has been identified as a component of a large multiprotein ERAD complex and appears to play a role in the cytosolic dislocation of several ERAD substrates (Mueller et al., 2008).
In this study, Aup1 was identified among several proteins that associate with gp78. Further examination revealed that Aup1 recruits Ubc7 to lipid droplets and facilitates its binding to gp78 as well as to Trc8. Reducing Aup1 levels through RNA interference (RNAi) inhibited sterol-accelerated ubiquitination of reductase, which appears to occur in lipid droplet–associated ER membranes. In turn, the inhibition of ubiquitination blunted sterol-accelerated reductase ERAD. Considered together, these findings not only further establish a role for Aup1 in ERAD, but they also provide another line of evidence for an integral role of lipid droplets and their associated proteins in the pathway.
RESULTS
Techniques of tandem affinity purification (TAP) and mass spectrometry were previously used to identify gp78-associated proteins (Jo et al., 2011b). These studies utilized CHO-7 cells, designated CHO/gp78-TAP, that stably overexpress gp78 fused to a TAP tag consisting of three T7 epitopes and protein A separated by a cleavage site for the tobacco etch virus (TEV) protease. In the current study, we subjected detergent lysates of CHO/gp78-TAP cells to affinity chromatography using immunoglobulin G (IgG) beads; this was followed by elution of gp78 and associated proteins with TEV protease. Eluted proteins were subjected to SDS–PAGE and colloidal blue staining; visible bands were excised and digested with trypsin, and proteins were identified by mass spectrometry (Supplemental Figure S1). One of these proteins, Aup1, was chosen for further study owing to its localization to lipid droplets and its previously described role in ERAD (Mueller et al., 2008; Spandl et al., 2009; 2011; Klemm et al., 2011).
In Figure 1A, detergent lysates of SV-589 cells treated in the absence or presence of the oxysterol 25-hydroxycholesterol (25-HC) plus mevalonate and the proteasome inhibitor MG-132 were immunoprecipitated with preimmune or anti-gp78 polyclonal antibodies. Immunoblot analysis of precipitated material revealed specific pulldown of gp78 (Figure 1A, panel 1, compare lanes 1, 2, 5, and 6 with lanes 3, 4, 7, and 8). Aup1 coprecipitated with gp78 (Figure 1A, panel 2, lanes 3 and 4) and MG-132 significantly increased the association (Figure 1A, panel 2, lanes 7 and 8). Treatment with 25-HC plus mevalonate caused reductase to disappear from the immunoprecipitation supernatant (Figure 1A, panel 3, lanes 10 and 12) and this was blocked by MG-132 (Figure 1A, panel 3, lanes 14 and 16). Reductase coimmunoprecipitated with gp78 in lysates from MG-132–treated cells (Figure 1A, panel 3, lane 7); this coprecipitation was enhanced by 25-HC plus mevalonate treatment (Figure 1A, panel 3, lane 8).
FIGURE 1:
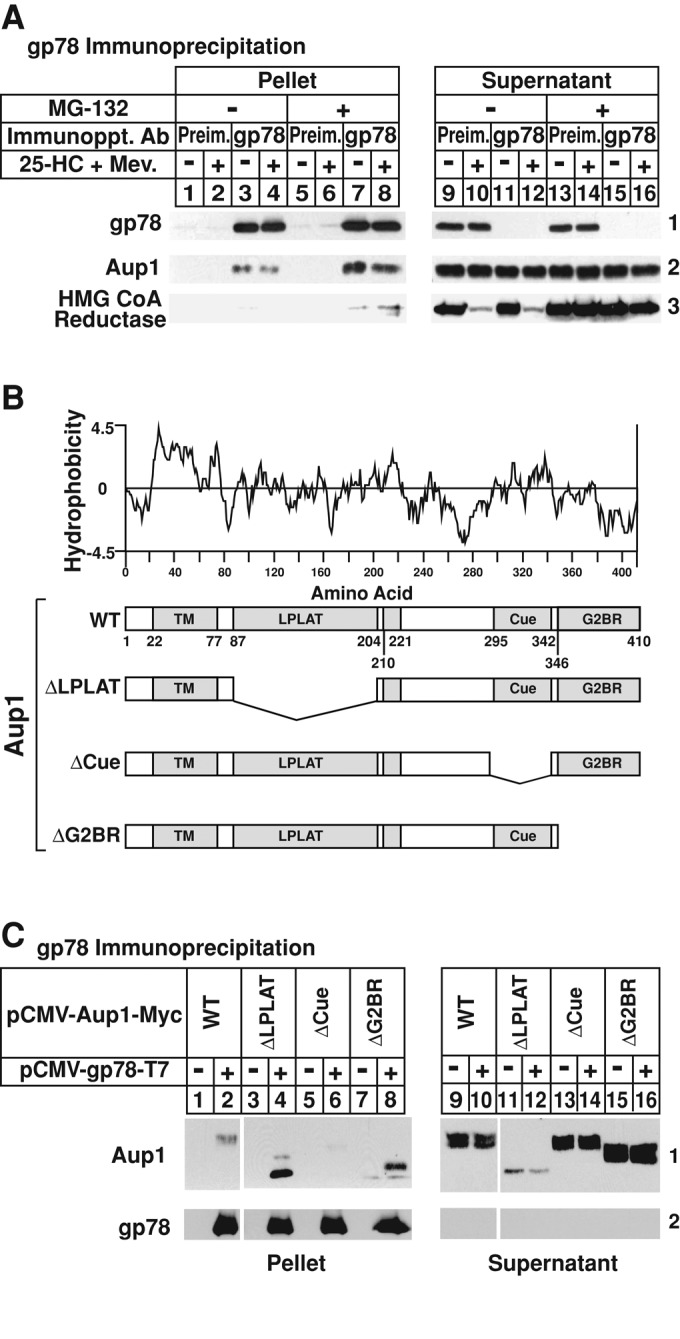
Association of Aup1 with the membrane-bound ubiquitin ligase gp78. (A) SV-589 cells were set up on day 0 at 6 × 105 cells/100-mm dish in medium B supplemented with 10% FCS. On day 2, the cells were depleted of sterols through incubation for 16 h at 37°C in medium B containing 10% LPDS, 50 μM sodium compactin, and 50 μM sodium mevalonate. The cells were subsequently re-fed sterol-depleting medium containing 1 μg/ml 25-HC, 10 mM mevalonate, and 10 μM MG-132, as indicated. Following incubation for 2 h at 37°C, the cells were harvested, lysed, and immunoprecipitated with preimmune polyclonal IgG or IgG-740F against gp78 as described in Materials and Methods. Pellet and supernatant fractions of the immunoprecipitations were subjected to SDS–PAGE, which was followed by immunoblot analysis with IgG-740F, anti-Aup1 IgG, and IgG-A9 (against reductase). (B) Hydrophobicity of Aup1 and schematic representation of wild-type and mutant versions of the protein analyzed in (C). (C) CHO-7 cells were set up for experiments on day 0 at 5 × 105cells/60-mm dish in medium A containing 5% LPDS. On day 1, the cells were transfected with 0.1 μg/dish pCMV-gp78-T7 and 0.1 μg/dish wild-type or mutant pCMV-Aup1-Myc in medium A containing 5% LPDS as indicated and described in Materials and Methods. Following incubation for 3–6 h, the cells were depleted of sterols by the direct addition of medium A containing 5% LPDS, 10 μM compactin, and 50 μM mevalonate (final concentration). After 16 h at 37°C, cells were subjected to treatment with sterol-depleting medium containing 10 μM MG-132 for 2 h, after which they were harvested, lysed, and immunoprecipitated with anti-T7–coupled agarose beads. Resulting pellet and supernatant fractions of the immunoprecipitations were subjected to SDS–PAGE and immunoblot analysis with anti-T7 IgG (against gp78) and IgG-9E10 (against Aup1).
Detergent lysates of CHO-7 cells transfected with expression plasmids encoding T7-tagged gp78 and wild-type or various deletion mutants of Myc-tagged Aup1 (Figure 1B) were immunoprecipitated with anti-T7–coupled agarose beads. Subsequent immunoblot analysis showed that immunoprecipitation of T7-tagged gp78 brought down wild-type Aup1 (Figure 1C, panel 1, lane 2), as well as mutant Aup1 lacking either the lysophospholipid acyltransferase (LPLAT) domain or the G2BR (Figure 1C, panel 1, lanes 4 and 8, respectively). However, deletion of the Cue domain completely abolished Aup1-gp78 coprecipitation (Figure 1C, panel 1, lane 6).
The requirement of Aup1 for sterol-induced ubiquitination of reductase was examined in Figure 2A. Cells were transfected with small interfering RNAs (siRNAs) targeting either the VSV-G mRNA, a viral gene that is not expressed in the cells, or the Aup1 mRNA. Following sterol depletion, cells were treated with MG-132 in the absence or presence of 25-HC plus mevalonate, and subsequently harvested for antireductase immunoprecipitation. Antiubiquitin immunoblot revealed sterol-induced ubiquitination of reductase in control-transfected cells treated with 25-HC plus mevalonate (Figure 2A, panel 1, lane 2). This ubiquitination was inhibited in cells that received the Aup1 siRNA (Figure 2A, panel 1, lane 4). A similar experiment was carried out in the absence of MG-132 to monitor sterol-accelerated ERAD of reductase in Aup1 knockdown cells. As expected, reductase ERAD was stimulated by 25-HC plus mevalonate in control-transfected cells (Figure 2B, panel 1, lane 2). This degradation was blunted in cells transfected with the Aup1 siRNA (Figure 2B, panel 1, lane 4), but not in those that received mutant Aup1 siRNA (Figure 2B, panel 1, lane 6). Aup1 knockdown was specific, as revealed by a marked reduction in the level of the protein in knockdown cells compared with that in controls (Figure 2B, panel 2, compare lanes 1, 2, 5, and 6 with lanes 3 and 4). Levels of Bat3, a gp78-associated ERAD factor involved in cytosolic dislocation of some substrates (see Figure S1; Claessen and Ploegh, 2011), were constant in the control and Aup1 knockdown cells, regardless of treatment conditions (panel 3).
FIGURE 2:
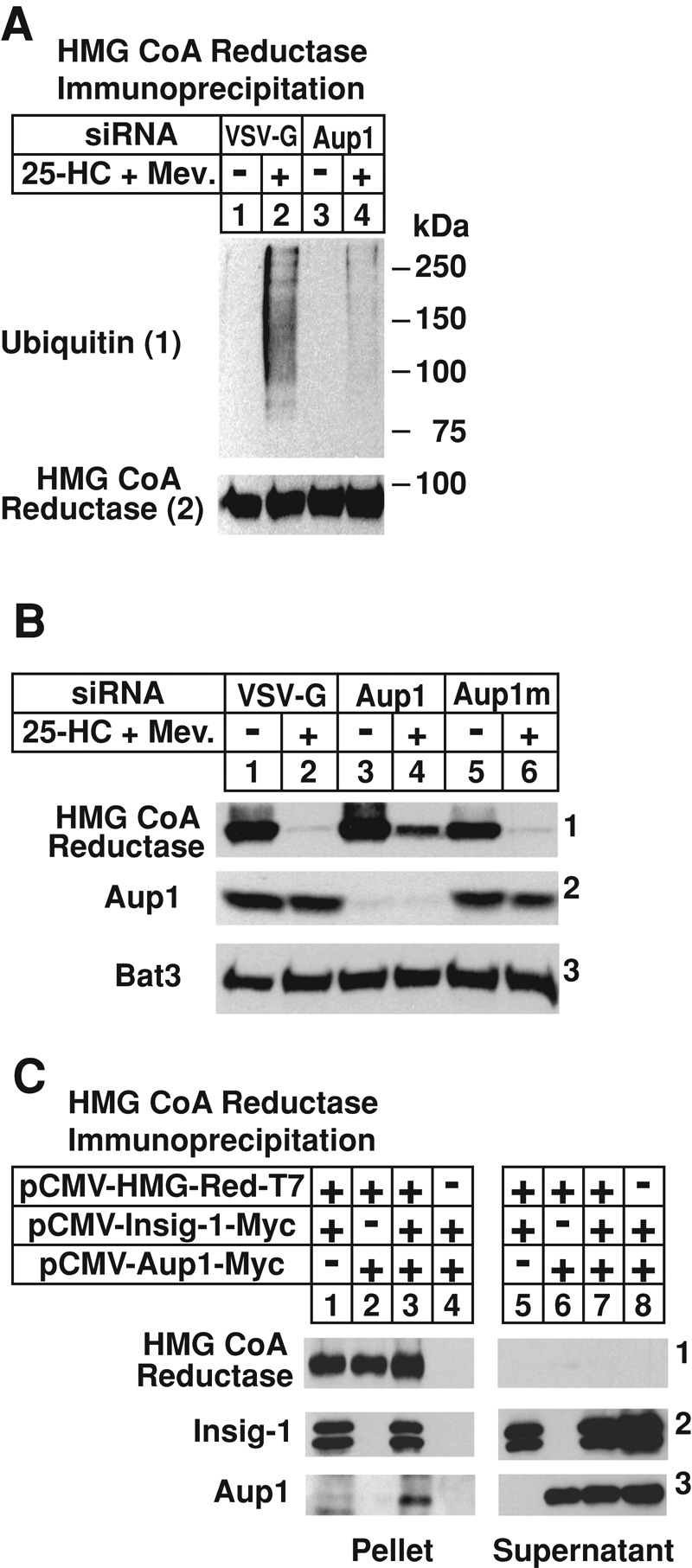
RNA-interference–mediated knockdown of gp78-associated Aup1 blunts sterol-accelerated ubiquitination and degradation of endogenous HMG CoA reductase. (A and B) SV-589 cells were set up on day 0 at 2 × 105 cells/60-mm dish in medium B containing 10% FCS. On days 1 and 2, the cells were transfected with control VSV-G or Aup1 siRNAs as indicated and described in Materials and Methods. (B) Some of the dishes received a mutant version of the Aup1 siRNA designated Aup1m. Four hours following siRNA transfections on day 2, cells were depleted of sterols for 16 h at 37°C. (A) Sterol-depleted cells were re-fed medium B supplemented with 10% LPDS, 50 μM compactin, and 10 μM MG-132 in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate. Following incubation for 0.5 h at 37°C, the cells were harvested, lysed, and subjected to immunoprecipitation with polyclonal antibodies against reductase. Aliquots of the immunoprecipitates were subjected to SDS–PAGE, and immunoblot analysis was carried out with IgG-A9 (against reductase) and IgG-P4D1 (against ubiquitin). (B) Sterol-depleted cells were incubated for 2.5 h at 37°C in medium B supplemented with 10% LPDS, 50 μM compactin, and 1 μg/ml 25-HC and 10 mM mevalonate, as indicated. The cells were subsequently harvested for subcellular fractionation and aliquots of the membrane fractions (normalized for equal protein loaded/lane) were subjected to SDS–PAGE, which was followed by immunoblot analysis with IgG-A9 (against reductase), anti-Aup1 IgG, and anti-Bat3 IgG. (C) CHO-7 cells were set up on day 0, transfected on day 1 with 1 μg/dish of pCMV-HMG-Red-T7, 0.1 μg/dish of pCMV-Insig-1-Myc, and 0.1 μg/dish of pCMV-Aup1-Myc, as indicated, and depleted of sterols as described in the legend to Figure 1. Cells were subsequently re-fed sterol-depleting medium containing 10 μM MG-132 and 1 μg/ml 25-HC plus 10 mM mevalonate and further incubated for 2 h at 37°C. Cells were subsequently harvested, lysed, and immunoprecipitated with anti-T7. Immunoblot analysis of the resulting pellet and supernatant fractions of the immunoprecipitations was carried out with anti-T7 IgG (against reductase) and IgG-9E10 (against Insig-1 and Aup1).
To determine whether one of the Insig proteins bridge Aup1 to reductase, we transfected CHO-7 cells with various combinations of expression plasmids encoding T7-tagged reductase, Myc-tagged Insig-1, and Myc-tagged Aup1. Detergent lysates were immunoprecipitated with anti-T7 to pull down transfected reductase. Immunoblotting revealed that, when transfected together, reductase and Insig-1 coprecipitated in the cells treated with 25-HC plus (Lee et al. 2006) mevalonate (Figure 2C, panels 1 and 2, lane 1). Immunoprecipitation of reductase failed to pull down Aup1 when the proteins were expressed together in the absence of Insig-1 (Figure 2C, panel 3, lane 2); however, reductase-Aup1 coprecipitation was observed upon Insig-1 coexpression (Figure 2C, panel 3, lane 3).
The mammalian Insig proteins consist of six membrane-spanning helices separated by short hydrophilic loops (Feramisco et al., 2004). Despite the high degree of homology within this region (85% amino-acid identity), Insig-1, but not Insig-2, is subjected to gp78-mediated ubiquitination and proteasomal degradation (Lee et al., 2006). A role for gp78-associated Aup1 in reductase degradation having been established, an RNAi experiment was next designed to determine whether Aup1 is required for Insig-1 degradation. In the experiment of Figure 3A, control and Aup1 knockdown cells were subjected to treatment with the protein synthesis inhibitor cycloheximide for various periods of time. Immunoblot analysis of membrane fractions from control-transfected cells revealed a marked reduction in the amount of Insig-1 protein after 1 h of cycloheximide treatment (Figure 3A, panel 1, compare lanes 1 and 3); reduction of the protein was nearly complete after 2 h of treatment (Figure 3A, panel 1, lane 4). In contrast, Insig-1 protein persisted throughout the cycloheximide chase in Aup1 knockdown cells (Figure 3A, panel 1, lanes 6–8). Reductase was similarly stabilized by the RNAi-mediated knockdown of Aup1 (Figure 3A, panel 2, compare lanes 1–4 with lanes 5–8).
FIGURE 3:

Aup1 mediates ER-associated degradation of key modulators of the cholesterol biosynthetic pathway. SV-589 cells were set up on day 0 and transfected on days 1 and 2 with VSV-G or Aup1 siRNAs as described in the legend to Figure 2. Four hours following transfection on day 2, the cells received a direct addition of medium B supplemented with 10% FCS (final concentration). (A) On day 3, cells were re-fed medium B containing 10% FCS and 50 μM cycloheximide (Cyclohex.) and incubated at 37°C for the indicated period of time. Cells were then harvested for the preparation of membrane fractions that were subjected to immunoblot analysis with IgG-A9 (against reductase), IgG-13F5 (against Insig-1), anti-Aup1 IgG, anti-calnexin IgG, IgG-2179 (against SREBP-1), IgG-22D5 (against SREBP-2), and IgG-R139 (against Scap). These results are representative of four independent experiments conducted under similar conditions. (B) Cells were re-fed on day 3 with medium B containing 10% FCS and 15 μCi/ml [14C]acetate; cold acetate was added to achieve a final concentration of 0.5 mM. Following incubation for the indicated period of time, cell lysates were prepared and lipids were extracted as described in Materials and Methods. Aliquots of the resulting lipid extracts were then subjected to TLC in parallel with authentic standards for squalene, lanosterol, and cholesterol. Incorporation of [14C]acetate into squalene, lanosterol, and cholesterol was determined by scintillation counting. Data are presented as mean values from triplicate dishes.
In addition to reductase, sterols promote binding of Insigs to the cholesterol-sensing protein Scap (Yabe et al., 2002; Yang et al., 2002). In sterol-deprived cells, Scap escorts the membrane-bound transcription factors sterol-regulatory, element-binding proteins SREBP-1 and SREBP-2 from the ER to the Golgi for proteolytic activation (Goldstein et al., 2006). This activation results in the release of transcriptionally active fragments of SREBP-1 and SREBP-2 that migrate to the nucleus, where they activate genes encoding enzymes required for the synthesis of cholesterol and other lipids. The full-length, membrane-bound SREBP-1 and SREBP-2 precursors are known to be substrates of the ERAD pathway, especially in the absence of Scap (Rawson et al., 1999). Consistent with this, SREBP-1 and SREBP-2 precursors were stabilized significantly in Aup1 knockdown cells (Figure 3A, panels 5 and 7, compare lanes 9–12 with lanes 13–16). A corresponding increase in the processed nuclear form of SREBPs was also observed (Figure 3A, panels 6 and 8). Aup1 knockdown also led to the slight stabilization of Scap (Figure 3A, panel 9). Finally, the metabolic labeling experiment of Figure 3B shows that knockdown of Aup1 resulted in the increased incorporation of [14C]acetate into cholesterol synthesis intermediates squalene and lanosterol, as well as cholesterol itself.
The presence of the G2BR domain in Aup1 prompted us to next compare its binding to Ubc7 and two other ubiquitin-conjugating enzymes, cytosolic Ubc1 and membrane-bound Ubc6. Detergent lysates of cells transfected with Myc-tagged Ubc1, Ubc6, or Ubc7 together with T7-tagged Aup1 were immunoprecipitated with anti-Myc. The results show that Aup1 failed to coprecipitate with Ubc1 (Figure 4A, panel 1, lanes 1–4). A small but measurable amount of Aup1 coprecipitated with Ubc6 (Figure 4A, panel 1, lanes 5 and 6). However, a significant fraction of Aup1 coprecipitated with Ubc7 (Figure 4A, panel 1, compare lanes 7 and 8 with lanes 17 and 18). Figure 4B compares the association of Ubc7 with wild-type Aup1 and deletion mutants of the protein shown in Figure 1B. Immunoprecipitation of Ubc7 brought down wild-type Aup1 (Figure 4B, panel 1, lane 2), as well as deletion mutants of Aup1 lacking either the LPLAT or Cue domains (Figure 4B, lanes 4 and 6, respectively). In contrast, truncated Aup1 lacking the G2BR did not coprecipitate with Ubc7 (Figure 4B, panel 1, lane 8). Comparative sequence analysis of the G2BR from Aup1, gp78, and Cue1p, which mediates recruitment of Ubc7 to ER membranes in the yeast Saccharomyces cerevisiae (Ponting, 2000; Hampton and Garza, 2009), revealed the presence of several conserved amino acids (Figure 4C). Therefore we next measured the binding of Ubc7 to a series of Aup1 mutants containing substitutions of alanine for several of these conserved residues (Figure 4, D–F). As expected, wild-type Aup1 coprecipitated with Ubc7 (Figure 4, D–F, panel 1, lane 2), whereas Aup1 lacking the G2BR did not (Figure 4, D–F, panel 1, lane 4). Aup1 continued to coprecipitate with Ubc7 when an alanine residue was substituted for Glu-384 (Figure S2), Glu-388 (Figure 4D, panel 1, lane 12), Arg-399 (Figure 4E, panel 1, lane 12), or Asp-410 (Figure 4F, panel 1, lane 12). In contrast, Aup1-Ubc7 coprecipitation was significantly reduced by alanine substitutions for Arg-382 (Figure 4D, panel 1, lane 6), Gln-383 (Figure 4D, panel 1, lane 8), Arg-389 (Figure 4D, panel 1, lane 14), Arg-398 (Figure 4E, panel 1, lane 10), or Glu-408 (Figure 4F, panel 1, lane 10). Even more complete inhibition of this coprecipitation was observed when Ala was substituted for Leu-386 (Figure 4D, panel 1, lane 10), Lys-390 (Figure 4E, panel 1, lane 6), Leu-393 (Figure 4E, panel 1, lane 8), Phe-401 (Figure 4E, panel 1, lane 14), Arg-400 (Figure 4F, panel 1, lane 6), or Arg-404 (Figure 4F, panel 1, lane 8).
FIGURE 4:

Association of Aup1 with the ubiquitin-conjugating enzyme Ubc7 (UBE2G2). CHO-7 cells were set up for experiments on day 0, transfected on day 1 with 0.1 μg/dish pCMV-Aup1-T7 and either 1.0 μg or 1.9 μg/dish pCMV-Ubc1-Myc, 0.05 μg/dish pCMV-Ubc6-Myc, or 0.05 μg/dish pCMV-Ubc7-Myc (A); 0.5 μg/dish pCMV-Ubc7-T7 (B); or 1.0 μg/dish pCMV-Ubc7-Myc (D–F) together with 0.1 μg/dish wild-type or mutant pCMV-Aup1-Myc (B) and pCMV-Aup1-T7 (D–F), and depleted of sterols as described in the legend to Figure 1. After 16 h at 37°C, cells were re-fed medium A containing 5% LPDS, 10 μM compactin, and 10 μM MG-132 in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate and incubated for 2 h at 37°C. The cells were subsequently harvested, lysed, and immunoprecipitated with anti-Myc (A and D–F) or anti-T7 (B). Resulting pellet and supernatant fractions were subjected to immunoblot analysis with anti-T7 IgG (against Aup1 in A and D–F; Ubc7 in B) and IgG-9E10 (against Ubc1 and Ubc6 in A; Ubc7 in A and D–F; Aup1 in B). (C) Comparison of the amino acid residues in the G2BR domain from human Aup1, human gp78, and yeast Cue1p. Residues identical in all three proteins are highlighted in red and residues present in two of three proteins are highlighted in green. One asterisk denotes amino acids whose mutation to alanine have no effect on Aup1-Ubc7 binding; two asterisks denote alanine substitutions that significantly inhibit Aup1-Ubc7 binding; and three asterisks denote those that completely abolish binding.
The intracellular localization of Aup1 fused to red fluorescent protein (RFP) was next examined in Figure 5. Soluble RFP localized to the cytosol of transfected cells, as determined by immunoblot analysis (Figure 5A, lane 2) and confocal microscopy (Figure 5, B–D). In contrast, a fusion protein consisting of wild-type Aup1 and RFP was present in both the membrane and lipid droplet fractions (Figure 5E, lanes 1 and 3). This result is consistent with microscopic images that revealed the Aup1-RFP fusion protein localized to reticular structures indicative of ER membranes (Figure 5F) and to punctate structures representative of lipid droplets, as indicated by their staining with the lipophilic dye Bodipy (Figure 5, G and H). The region in Aup1 responsible for lipid droplet localization was identified by studying the localization of RFP fused to amino acids 1–49, 1–86, or 210–221 of Aup1, as each of these sequences exhibits significant hydrophobicity (Figure 1B). Subcellular fractionation (Figure 5, I and Q, lane 1) and confocal microscopy (Figure 5, J–L and R–T) showed that RFP fused to amino acids 1–49 or 210–221 of Aup1 was primarily localized to membranes, whereas Aup1 (1–86)-RFP localized to both membranes and lipid droplets (Figure 5, M, lanes 1 and 3, and N–P).
FIGURE 5:

Localization of Aup1 to lipid droplets. CHO-7 cells were set up for subcellular fractionation (A, E, I, M, and Q) on day 0 at 6 × 105 cells/60-mm dish in medium B containing 5% LPDS. On day 1, the cells were transfected with 2 μg/dish pCMV-RFP, -Aup1-RFP, -Aup1(1–49)-RFP, -Aup1(1–86)-RFP, or -Aup1(210–221)-RFP, as indicated and described in the legend to Figure 1, and incubated for 16 h at 37°C in medium A containing 5% dFCS and 100 μM oleate–bovine serum albumin. The cells were then incubated for 2 h at 37°C in sterol-depleting medium containing 10 μM MG-132, after which they were harvested and subjected to subcellular fractionation. Aliquots of the resulting membrane, cytosol, and lipid droplet fractions were analyzed by immunoblot with anti-RFP IgG. For confocal microscopy, SV-589 cells were set up on day 0 at 1.5 × 105 cells/well of six-well plates with glass coverslips in medium B containing 10% FCS. On day 1, cells were transfected with 2 μg/dish pCMV-RFP, -Aup1-RFP, -Aup1(1-49)-RFP, -Aup1(1-86)-RFP, or -Aup1(210-221)-RFP, as indicated, and subsequently incubated at 37°C in medium B containing 10% dFCS. The cells were then subjected to MG-132 treatment in medium B containing 10% dFCS as described above. Cells were subsequently fixed, permeabilized, and stained with 5 μg/ml DAPI to visualize nuclei (blue) and 10 μg/ml Bodipy 493/503 to visualize lipid droplets (green). Images were taken using a Zeiss LSM510 laser-scanning confocal microscope with a META spectral detector, a Chameleon XR NIR laser (Bitplan, St. Paul, MN), and a Zeiss 63×, oil-immersion objective.
Correlative fluorescence and electron microscopy (CFEM) was next used to study the colocalization of Aup1 and Ubc7 fused to green fluorescent protein (GFP) and RFP, respectively. Confocal microscopy revealed that Aup1-GFP and Ubc7-RFP localized to discrete intracellular puncta (Figure 6, A and E, respectively, white arrows), with a fraction of both proteins present in reticular structures. Analysis of the identical cells by transmission electron microscopy revealed the presence of lipid droplets scattered throughout the cells (Figure 6B, black arrows). Magnification of the image obtained from one of the serial sections revealed a close association between these lipid droplets and ER membranes (Figure 6, B–D; see Figure S3). Overlay of the confocal image and the electron micrograph revealed colocalization of Aup1-GFP, Ubc7-RFP, and lipid droplets (Figure 6F, black arrows).
FIGURE 6:

Aup1 mediates recruitment of Ubc7 to lipid droplets. (A–F) SV-589 cells were set up on day 0 at 1.5 × 105 cells/well of six-well plates with glass coverslips in medium B containing 10% FCS. On day 1, the cells were transfected with 1 μg/dish pCMV-Aup1-GFP and pCMV-Ubc7-RFP as described in the legend to Figure 1. On day 2, the cells were fixed for confocal and transmission electron microscopy as described in Materials and Methods. The white boxes shown in (B) and (C) were magnified in (C) and (D), respectively. (G–L) SV-589 cells were set up for confocal microscopy on day 0 as described in the legend to Figure 4. On days 1 and 2, the cells were transfected with control VSV-G or Aup1 siRNAs as described in legend to Figure 2. Four hours following siRNA transfections on day 2, cells were transfected with 0.3 μg pCMV-Ubc7-RFP and subsequently depleted of sterols for 16 h at 37°C. The cells were then incubated for 2 h at 37°C in the sterol-depleting medium containing 10 μM MG-132 and subsequently fixed for confocal microscopy as described in the legend to Figure 4. (M) SV-589/Ubc7-Myc cells were set up on day 0, transfected with control VSV-G or Aup1 siRNA on days 1 and 2, and depleted of sterols as described in the legend to Figure 2. Cells were then incubated in sterol-depleting medium containing 10% LPDS and 50 μM compactin in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate. Following incubation for 2.5 h at 37°C, the cells were harvested and subjected to subcellular fractionation. Aliquots of resulting membrane and cytosol fractions were fractionated by SDS–PAGE, which was followed by immunoblot analysis with IgG-9E10 (against Ubc7), IgG-A9 (against reductase), anti-Aup1, anti-actin, and anti-calnexin IgG.
A role for Aup1 in localization of Ubc7 to lipid droplets was next examined using RNAi. SV-589 cells transfected with control VSV-G or Aup1 siRNAs and the Ubc7-RFP expression plasmid were analyzed by confocal microscopy. Several lipid droplets were observed scattered throughout cells transfected with the control siRNA, as determined by Bodipy staining (Figure 6G). Knockdown of Aup1 had little effect on the number and morphology of these lipid droplets (Figure 6J). The majority of Ubc7-RFP colocalized with lipid droplets in control-transfected cells (Figure 6, H and I). This colocalization was completely abrogated in Aup1 knockdown cells; Ubc7-RFP was redistributed to the cytosol and to reticular structures consistent with the ER (Figure 6, K and L). Immunoblot analysis of subcellular fractions obtained from SV-589 cells stably overexpressing Ubc7-Myc (designated SV-589/Ubc7-Myc) showed that RNAi-mediated knockdown of Aup1 led to the significant recovery of Ubc7 in the cytosol (Figure 6M, panel 1, compare lanes 3 and 4 with lanes 7 and 8) and blunted sterol-accelerated reductase ERAD (Figure 6M, panel 3, compare lanes 1 and 2 with lanes 5 and 6).
We previously showed that ubiquitination is required for extraction of reductase from lipid droplet–associated ER membranes into the cytosol (Hartman et al., 2010). To determine whether ubiquitination is also required for sterol-induced translocation of reductase to lipid droplet–associated ER, we examined the subcellular localization of a mutant form of the enzyme harboring substitutions of arginine for Lys-89 and Lys-248 in the membrane domain. These substitutions are known to prevent sterol-mediated ubiquitination, as well as the subsequent cytosolic dislocation and proteasomal degradation of reductase (Sever et al., 2003a; Hartman et al., 2010). Figure 7A shows that the membrane domain of reductase became degraded from membranes when the cells were treated with 25-HC plus mevalonate (Figure 7A, panel 1, lane 2). This degradation was blocked by MG-132 (Figure 7A, panel 1, lane 4), which caused the appearance of reductase in the cytosol and lipid droplet fractions isolated from cells treated with 25-HC plus mevalonate (Figure 7A, panels 2 and 3, respectively, lane 4). The ubiquitination-resistant K89R/K248R mutant of reductase resisted sterol-induced degradation (Figure 7A, panel 1, lane 6) and cytosolic dislocation (panel 2, lane 8). However, the protein continued to be recovered from the lipid droplet fraction of cells in a sterol-regulated manner (Figure 7A, panel 3, lane 8). The subcellular fractions were also immunoblotted for endogenous reductase and, as expected, 25-HC plus mevalonate stimulated the protein’s ERAD in the absence of MG-132 (Figure 7A, panel 4, lanes 2 and 6). MG-132 blocked this ERAD (Figure 7A, panel 4, lanes 4 and 8), and reductase was observed in the cytosol and lipid droplet fractions of cells treated with 25-HC plus mevalonate (Figure 7A, panels 5 and 6, respectively, lanes 4 and 8).
FIGURE 7:

Sterol-induced ubiquitination of HMG CoA reductase occurs in lipid droplet–associated ER membranes. (A) CHO-K1 cells were set up on day 0 at 5 × 105 cells/100-mm dish in medium A containing 5% FCS, transfected on day 2 with 5.82 μg/dish of wild-type or K89R/K248R versions of pCMV-HMG-Red-T7(TM1-8) together with 0.18 μg/dish of pCMV-Insig-1-Myc, as indicated, and depleted of sterols as described in the legend to Figure 1. The cells were then subjected to treatment with sterol-depleting medium containing 10 μM MG-132 in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate as indicated. After 5 h at 37°C, the cells were harvested and subjected to subcellular fractionation. Aliquots of membrane (Memb.), cytosol (Cyt.), and lipid droplet (LD) fractions were subjected to SDS–PAGE, which was followed by immunoblot analysis with anti-T7 IgG (against transfected reductase), IgG-A9 (against endogenous reductase), anti-actin IgG, and anti-ADRP IgG. (B–D) SV-589/pUbc7-Myc cells were set up on day 0 at 8 × 106 cells/100-mm dish in medium B containing 10% FCS. On day 2, the cells were depleted of sterols through incubation for 16 h at 37°C in medium B containing 10% dFCS, 50 μM compactin, and 50 μM mevalonate. Cells were subsequently treated with the identical medium containing 10 μM triacsin C for 3–4 h at 37°C, after which they received 10 μM MG-132 and were further incubated for an additional 3–3.5 h. Cells were then harvested and subjected to either subcellular fractionation (B and C) or anti-Aup1 immunoprecipitation (D). Aliquots of resulting membrane, cytosol, lipid droplet, immunoprecipitation pellet, and supernatant fractions were subjected to SDS–PAGE and immunoblot analysis with IgG-A9 (against reductase), anti-Aup1 IgG, IgG-9E10 (against Ubc7), anti-calnexin IgG, IgG-740F (against gp78), anti-ADRP IgG, anti-Ubxd8 IgG, and anti-actin IgG. (E–J) SV-589/pUbc7-Myc cells were set up for experiments on day 0 and depleted of sterols on day 2 as described above. Following sterol depletion, the cells were re-fed medium B containing 10% LPDS, 50 μM compactin, and 10 μM triacsin C. After 4 h at 37°C, cells received 10 μM MG-132 and were incubated for an additional 2 h. The cells were subsequently fixed, permeabilized, and stained with 5 μg/ml DAPI to visualize nuclei (blue) and 2–5 μg/ml anti-Aup1 IgG together with 15 μg/ml Alexa Fluor 594 donkey anti–rabbit IgG to visualize Aup1 (red) or 15 μg/ml IgG-9E10 together with 15 μg/ml Alexa Fluor 488 donkey anti–mouse IgG to visualize Ubc7-Myc (green), and analyzed by confocal microscopy.
To determine whether lipid droplets are required for Aup1-Ubc7 binding, we treated SV-589/pUbc7-Myc cells with triacsin C, an inhibitor of fatty acyl–CoA synthetase that blocks incorporation of free fatty acids into triglycerides and prevents lipid droplet formation in lipid-loaded or MG-132–treated cells (Omura et al., 1986; Hartman et al., 2010). The results show that triacsin C treatment had no effect on levels of Aup1, Ubc7-Myc, or the calnexin loading control (Figure 7B, panels 1–3, compare lanes 1, 2, 5, and 6 with lanes 3, 4, 7, and 8). Consistent with our previous findings (Hartman et al., 2010), triacsin C blunted sterol-accelerated ERAD of reductase (panel 4, lane 4) and resulted in reduced recovery of Aup1, Ubc7, ADRP, Ubxd8, and VCP/p97 from the lipid droplet fraction (Figure 7C, panels 1–5, lane 6). Despite this, triacsin C treatment did not prevent the interaction between Aup1 and Ubc7 or gp78 in SV-589/pUbc7-Myc cells (Figure 7D, panels 1 and 2, lanes 1 and 2) or in transfected CHO-7 cells (Supplemental Figure S4). These results are consistent with confocal images that show that Aup1 and Ubc7 colocalized to lipid droplets in the absence of triacsin C (Figure 7, E–G). Multiple Z-stacks at a fixed interval were acquired from randomly chosen cells and used to generate three-dimensional images for each cell. Quantitative analysis of these images revealed that 54% of Aup1 colocalized with lipid droplets, whereas 90% of signal corresponding to lipid droplets colocalized with Aup1 (Pearson’s coefficient = 0.66). In the same cells, roughly 60% of Aup1 colocalized with Ubc7 and 84% of Ubc7 colocalized with Aup1 (Pearson’s coefficient = 0.72). Triacsin C treatment caused redistribution of Aup1 and Ubc7 to a more reticular-type staining pattern (Figure 7, H–J). Quantitative analysis revealed that triacsin C treatment reduced the amount of Aup1 that colocalized with lipid droplets from 54% to 12%.
Considering our finding that Aup1 binds to Ubc7 and gp78 through distinct domains (Figures 1 and 4), and that Ubc7 has been identified as a gp78-associated protein (Chen et al., 2006), we next sought to determine whether Aup1 facilitates the well-documented association between gp78 and Ubc7. SV-589 cells were transfected sequentially with VSV-G or Aup1 siRNAs and Myc-tagged Ubc7 in the absence or presence of T7-tagged gp78 (Figure 8A). Immunoprecipitation of gp78 brought down Ubc7 in cells transfected with the control siRNA (Figure 8A, panel 1, lane 2); this was marginally reduced in Aup1 knockdown cells (Figure 8A, lane 4). MG-132 treatment increased gp78-Ubc7 coprecipitation in control cells (Figure 8A, lane 6); however, this coprecipitation was significantly reduced in Aup1 knockdown cells (Figure 8A, lane 8). In Figure 8B, we measured the effect of Aup1 overexpression on gp78-Ubc7 coprecipitation. The results show that gp78 immunoprecipitation brought down a small fraction of Ubc7 when the proteins were overexpressed together in MG-132–treated cells (Figure 8B, panel 2, lanes 3 and 4). This coprecipitation was significantly enhanced by Aup1 coexpression (Figure 8B, compare lanes 3 and 4 with lanes 5 and 6). In contrast, coexpression of Ubc7 had little effect on coprecipitation of Aup1 and gp78 (Figure 8B, panel 1, lanes 1, 2, 5, and 6).
FIGURE 8:

Aup1 facilitates the association of Ubc7 with gp78 and Trc8. (A) SV-589 cells were set up on day 0 and transfected on days 1 and 2 with control VSV-G or Aup1 siRNAs as described in the legend to Figure 2. Four hours after siRNA transfections on day 2, the cells were transfected with 1 μg/dish pCMV-Ubc7-Myc in the absence or presence of 0.1 μg/dish pCMV-gp78-T7 and depleted of sterols as described in the legend to Figure 1. The cells were then switched to sterol-depleting medium containing 10 μM MG-132 as indicated, incubated for 2 h at 37°C, after which they were harvested for anti-T7 immunoprecipitation. Pellet and supernatant fractions were subjected to immunoblot analysis with anti-T7 IgG (against gp78), IgG-9E10 (against Ubc7), and anti-Aup1 IgG. (B–D) CHO-7 cells were set up for experiments on day 0, transfected on day 1 with 0.01 μg/dish pCMV-gp78-T7, 0.1 μg/dish pCMV-Aup1-Myc, and 1 μg/dish pCMV-Ubc7-Myc (B); 0.1 μg/dish pCMV-Aup1-T7 together with 0.1 μg/dish pCMV-gp78-Myc or 1.8 μg/dish pCMV-Trc8-Myc (C); 0.1 μg/dish pCMV-Aup1-Myc together with 0.5 μg/dish pCMV-Ubc7-Myc and 1.4 μg/dish pCMV-Trc8-T7 (D) as indicated, and depleted of sterols as described in the legend to Figure 1. Sterol-depleted cells were then re-fed medium A containing 5% LPDS, 10 μM compactin, and 10 μM MG-132 in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate. After 2 h at 37°C, the cells were harvested for anti-T7 (B and D) or anti-Myc (C) immunoprecipitation; resulting pellet and supernatant fractions were subjected to immunoblot analysis with anti-T7 IgG (against gp78 in B; Aup1 in C; Trc8 in D) and IgG-9E10 (against Ubc7 in B, and D; Aup1 in B and D; gp78 and Trc8 in C).
We recently discovered that another membrane-bound ubiquitin ligase, called Trc8, contributes to sterol-induced ubiquitination and degradation of reductase (Jo et al., 2011a). Figure 8C shows that Aup1 coprecipitated with Trc8 in transfected cells (Figure 8C, panel 1, lanes 3 and 4) with an efficiency equivalent to that observed for gp78-Aup1 coprecipitation (Figure 8C, lanes 1 and 2). Similar to the results seen in Figure 8B, coexpression of Aup1 markedly enhanced coprecipitation of Ubc7 with Trc8 (Figure 8D, panel 2, compare lanes 3 and 4 with lanes 5 and 6), whereas Ubc7 did not enhance Aup1-Trc8 coprecipitation (Figure 8D, panel 1, compare lanes 1 and 2 with lanes 5 and 6).
DISCUSSION
Aup1, a highly conserved protein in multicellular organisms, appears to play a key role in two seemingly disparate cellular functions: storage of neutral lipids and quality control of misfolded ER proteins (Fujimoto and Parton, 2011). Its connection to lipid droplets is provided by proteomic studies that identify Aup1 as a lipid droplet–associated protein (Brasaemle et al., 2004; Liu et al., 2004; Sato et al., 2006; Bartz et al., 2007b; Wan et al., 2007). Subsequent studies confirmed this observation by showing that endogenous, as well as overexpressed, Aup1 localizes to the surface of lipid droplets, and its knockdown blunts lipid droplet accumulation in lipid-laden cells (Spandl et al., 2009, 2011; Klemm et al., 2011). The first connection of Aup1 to ERAD was provided by its identification as a component of the Hrd1 ubiquitin ligase quality control complex (Mueller et al., 2008). This connection is further implicated by the domain structure of Aup1, which includes a Cue domain and a G2BR (see Figure 1B). The Cue domain binds ubiquitin and mediates the association of Aup1 with ubiquitinated ERAD substrates and with various components of the ERAD pathway (Klemm et al., 2011; Spandl et al., 2011). The G2BR of Aup1 binds to the ubiquitin-conjugating enzyme Ubc7 (Klemm et al., 2011). A functional role for Aup1 in ERAD is revealed by RNAi studies showing that its knockdown impairs cytosolic dislocation of several ERAD substrates (Klemm et al., 2011).
The role of Aup1 in ERAD is further established through its identification as an associated protein of gp78 (Figure S1). This interaction was confirmed in subsequent studies that showed coprecipitation between either endogenous or overexpressed gp78 and Aup1 (Figure 1, A and C). These studies also revealed that the Cue domain of Aup1 mediates its association with gp78 (Figure 1C). This result is consistent with the finding of an earlier study (Klemm et al., 2011), which showed that deletion of the Cue domain of Aup1 slightly reduced its binding to the Hrd1 ubiquitin ligase. Although it cannot be determined whether Cue-mediated binding of Aup1 to gp78 is mediated by indirect or direct mechanisms, it should be noted that the proteasome inhibitor MG-132 enhances the interaction (Figure 1A). This suggests that proteasome inhibition 1) stabilizes an unknown ubiquitinated protein that mediates Aup1-gp78 binding; 2) increases levels of ubiquitinated forms of Aup1 that bind to gp78; or 3) stabilizes a transient interaction that can only be observed when ERAD is disrupted (see below).
Consistent with two previous reports (Klemm et al., 2011; Spandl et al., 2011), we found that Aup1 binds to the ubiquitin-conjugating enzyme Ubc7 (Figure 4A) and localizes to lipid droplets, as well as to ER membranes (Figure 5). Importantly, the hydrophobic N-terminus of Aup1 (amino acids 1–86) is capable of directing a significant fraction of the normally cytosolic RFP to lipid droplets (Figure 5, M–P). Studies employing RNAi show that binding of Ubc7 to Aup1 mediated by the Aup1 G2BR (Figure 4, B and D–F) results in recruitment of Ubc7 to lipid droplets (Figure 6). However, lipid droplet formation does not appear to be a requirement for Aup1-Ubc7 or Aup1-gp78 association, as indicated by resistance of these interactions to disruption by the inhibitor triacsin C (Figure 7).
Coimmunoprecipitation studies showed that gp78 associated with Ubc7 when the two proteins were overexpressed together in cells (Figure 8A). The RNAi-mediated knockdown of Aup1 had no effect on this interaction. Similar to results for Aup1-gp78 binding (Figure 1A), proteasome inhibition caused a marked stabilization of Ubc7-gp78 binding, which was reduced by Aup1 knockdown. This finding is supported by the observation that overexpression of Aup1 enhanced gp78-Ubc7 coprecipitation in MG-132–treated cells (Figure 8B). Similarly, Aup1 enhances the association of Ubc7 with another membrane-bound ubiquitin ligase, Trc8 (Figure 8D). These results suggest that one mechanism through which Aup1 contributes to ERAD involves its bridging of Ubc7 to at least two ubiquitin ligases, gp78 and Trc8. However, we can only observe this role when proteasomes are inhibited, which stimulates lipid droplet formation in addition to blocking ERAD (Hartman et al., 2010). We reason that, under these conditions, factors involved in substrate ubiquitination, membrane extraction, and cytosolic dislocation become concentrated in an ER subdomain that is contiguous with lipid droplets. This concentration leads to the stabilization of normally transient interactions, such as the Aup1-mediated binding of Ubc7 to either gp78 or Trc8, allowing their detection by coimmunoprecipitation.
A functional role for Aup1 in the metabolically regulated ERAD of the cholesterol biosynthetic enzyme HMG CoA reductase is demonstrated in Figure 2. RNAi-mediated knockdown of Aup1 significantly inhibited sterol-accelerated ubiquitination of reductase (Figure 2A), which is mediated by gp78 and Trc8 (Jo et al., 2011a), and blunted the enzyme’s sterol-accelerated ERAD (Figure 2B). Moreover, Aup1 associated with reductase in sterol-treated cells through a mechanism that required the polytopic ER membrane protein Insig-1 (Figure 2C), which bridges reductase to gp78 and Trc8 for ubiquitination (Jo et al., 2011a). These results are consistent with a model in which sterols cause reductase to become incorporated into a multiprotein complex that at least contains Insig-1, gp78 or Trc8, Aup1, and Ubc7. Reductase’s incorporation into this complex results in its ubiquitination, which appears to occur in ER membranes closely associated with lipid droplets. This is indicated by the observation in Figure 7A that sterols triggered translocation of a ubiquitination-resistant form of reductase to the lipid droplet fraction, but not into the cytosol. It is notable that lipid droplets are relatively protein poor, and the amount of reductase detected in the lipid droplet fraction relative to that in other fractions may be overrepresented in this study and those we have previously reported (Hartman et al., 2010). However, the appearance of reductase in the lipid droplet fraction was only observed in the presence of 25-HC, implicating the physiological relevance of the reaction.
The RNAi-mediated knockdown of Aup1 also blunted the ERAD of Insig-1, the precursors of SREBP-1 and SREBP-2, and, to a lesser extent, Scap (Figure 3A). Although Insig-1 ERAD was slowed, nuclear forms of SREBPs were increased, and the net effect of Aup1 knockdown was an increase in de novo cholesterol synthesis of cholesterol (Figure 3B). This unexpected result was likely due to enhanced stability of SREBP-1 and SREBP-2 precursors, Scap, and reductase, which is consistent with a prominent role for these proteins in lipid metabolism. An important goal for future studies is to determine whether the mechanism for Aup1-mediated ERAD of Insig-1, SREBP-1/-2, and Scap is similar to that shown for reductase in the current study.
In addition to being a locus for preubiquitination steps of reductase ERAD, the lipid droplet–associated ER also appears to be a locus for postubiquitination steps of the reaction. We previously found that, in VCP/p97 knockdown cells, endogenous reductase continued to translocate to lipid droplet–associated ER in the presence of sterols, even though it was not dislocated into the cytosol (Hartman et al., 2010). Importantly, these findings with reductase are consistent with those recently reported for another ERAD substrate, apolipoprotein B-100 (Suzuki et al., 2012). Knockdown of Ubxd8, the lipid droplet–associated VCP/p97 recruitment factor (Liu et al., 2004; Bartz et al., 2007b; Schuberth and Buchberger, 2008), resulted in the accumulation of apolipoprotein B-100 on the surface of lipid droplets. The study also found that the putative retrotranslocon component Derlin-1 localized to lipid droplet–associated ER and mediated a step in apolipoprotein B-100 ERAD prior to the action of Ubxd8 (and presumably VCP/p97). Considered together, these studies argue that ERAD components converge at the juncture between lipid droplets and ER membranes to mediate ubiquitination, membrane extraction, and cytosolic dislocation. Future challenges include further dissection of lipid droplet–ER membrane association and determination of whether lipid droplet–mediated ERAD is specific for a subset of substrates or general for all ERAD substrates.
MATERIALS AND METHODS
Materials
We purchased 25-HC from Steraloids (Wilton, NH) and MG-132 from Peptides International (Osaka, Japan); [1-2-14C]acetic acid, sodium salt, and [3-3H]lanosterol were obtained from American Radiolabeled Chemicals (St. Louis, MO); silica gel TLC plates were from Macherey-Nagel (Düren, Germany); digitonin was from Calbiochem (San Diego, CA); horseradish peroxidase–conjugated donkey anti-mouse, anti-rabbit, anti-goat, and anti–guinea pig were from Jackson ImmunoResearch Laboratories (West Grove, PA). Lipoprotein-deficient serum (LPDS; d > 1.215 g/ml) and delipidated fetal calf serum (dFCS) were prepared from newborn calf serum by ultracentrifugation, as described previously (Goldstein et al., 1983; Hannah et al., 2001). All other reagents were obtained from previously described sources (DeBose-Boyd et al., 1999).
Expression plasmids
The following plasmids have been described in the indicated references: pCMV-HMG-Red-T7 encoding hamster HMG CoA reductase followed by three tandem copies of the T7 epitope tag (MASMTGGQQMG) under transcriptional control of the cytomegalovirus (CMV) promoter (Sever et al., 2003a); pCMV-HMG-Red-T7 (TM1-8) encoding the membrane domain of hamster reductase (amino acids 1–346) followed by three copies of the T7 epitope (Sever et al., 2003b); pCMV-HMG-Red-T7 (TM1-8) K89R/K248R encodes the membrane domain of hamster reductase containing substitutions of arginine for Lys-89 and Lys-248 (Sever et al., 2003a); pCMV-Insig-1-Myc encoding human Insig-1 followed by six tandem copies of an epitope derived from c-Myc (EQKLISEEDL; Yang et al., 2002); pCMV-gp78-Myc, which encodes human gp78 followed by five tandem copies of the Myc epitope (Song et al., 2005b); pCMV-Trc8-Myc encoding human Trc8 followed by five copies of the c-Myc epitope (Jo et al., 2011b). The pCMV-Trc8-T7 expression plasmid encodes human Trc8 fused to six copies of the T7 epitope. The cDNAs encoding Ubc6 and Ubc7 were kind gifts from Alan Weissman (National Cancer Institute, Bethesda, MD); human Ubc1 (UbcH1, E2-25K) and Aup1 cDNAs were purchased from Thermo Fisher Scientific (Huntsville, AL) and cloned into the pcDNA3.1(+) vector using standard PCR methods. The expression plasmids designated pCMV-Ubc1-Myc, pCMV-Ubc1-T7, pCMV-Ubc6-Myc, pCMV-Ubc6-T7, pCMV-Ubc7 (Ube2g2)-Myc, and pCMV-Ubc7-T7 encode the respective human proteins followed by either five copies of the c-Myc epitope or six copies of the T7 epitope; pCMV-Aup1-T7 and pCMV-Aup1-Myc encode human Aup1 fused to three copies of the T7 or Myc epitope. The expression plasmids pCMV-Aup1-Myc (ΔLPLAT), (ΔCue), and (ΔG2BR) contain deletions of the LPLAT, Cue, and G2BR domains of Aup1, respectively. The expression plasmid pCMV-Aup1–enhanced green fluorescent protein (eGFP) was generated by fusing the cDNA encoding eGFP to the C-terminus of pCMV-Aup1 and pCMV-Ubc7-RFP was generated by fusing RFP to the C-terminus of Ubc7 in pCMV-Ubc7. The plasmid pCMV-Aup1-RFP was generated by fusing the RFP cDNA to the C-terminus of Aup1; pCMV-Aup1(1-49)- and Aup1(1-86)-RFP were generated by fusing amino acids 1–49 or 1–86 of Aup1 to the N-terminus of RFP; pCMV-RFP-Aup1(210–221) is a fusion protein between the C-terminus of RFP and amino acids 210–221 of Aup1.
Cell culture
CHO-7 cells are a line of CHO-K1 cells selected for growth in LPDS (Metherall et al., 1989). Monolayers of CHO-7 and CHO-K1 cells were maintained in medium A (1:1 mixture of Ham’s F-12 medium and DMEM containing 100 U/ml penicillin and 100 mg/ml streptomycin sulfate) supplemented with 5% (vol/vol) LPDS at 37°C, 8–9% CO2. Stock cultures of SV-589 cells, a line of immortalized human fibroblasts expressing the SV40 large T antigen (Yamamoto et al., 1984), were grown in monolayer at 37°C, 5% CO2 in medium B (DMEM containing 1000 mg glucose/l, 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate) supplemented with 10% fetal calf serum (FCS).
SV-589/pUbc7-Myc cells, a line of SV-589 cells that stably express Myc-tagged Ubc7 were generated as follows: On day 0, SV-589 cells were set up at a density of 2 × 105 cells per 100-mm dish in medium B supplemented with 10% FCS. On day 1, cells were transfected with 0.1, 0.3, and 1.0 μg/dish of pCMV-Ubc7-Myc using the FuGENE6 transfection reagent, as described above. Following incubation for 16 h at 37°C, the cells were switched to medium B supplemented with 10% FCS and 700 μg/ml of G418. Fresh medium was added every 2–3 d until colonies formed after ∼2 wk. Individual colonies were isolated using cloning cylinders, and expression of Ubc7-Myc was determined by immunoblot analysis. Cells from a single colony of cells expressing a moderate level of Ubc7-Myc were selected, cloned by limiting dilution, and maintained in medium B supplemented with 10% FCS and 700 μg/ml G418 at 37°C, 5% CO2.
Transfection, cell fractionation, and immunoblot analysis
Transfection of CHO-7 and SV-589 cells with FuGENE6 transfection reagent (Roche, Indianapolis, IN) was performed as previously described (Sever et al., 2003b; Jo et al., 2011b). Conditions of subsequent incubations are described in the figure legends. Following incubations, triplicate dishes of cells for each variable were harvested and pooled for analysis. Immunoprecipitation with anti-T7 agarose (Novagen, Darmstadt, Germany), anti-Myc agarose (Sigma-Aldrich, St. Louis, MO), or anti-Aup1 IgG and subcellular fractionation by differential centrifugation was performed as previously described (Sever et al., 2003b; Jo et al., 2011b). Aliquots of resulting supernatant and pellet fractions from immunoprecipitations and cytosol, membrane, or lipid droplet fractions (normalized for equal protein loaded per lane) obtained from subcellular fractionations were subjected to SDS–PAGE and immunoblot analysis. Primary antibodies used for immunoblot analysis included: mouse monoclonal anti-T7 Tag IgG (Novagen); IgG-9E10, a mouse monoclonal antibody against c-Myc purified from the culture medium of hybridoma clone 9E10 (American Type Culture Collection, Manassas, VA); IgG-A9, a mouse monoclonal antibody against the catalytic domain of reductase (Liscum et al., 1983); IgG-P4D1, a mouse monoclonal antibody against bovine ubiquitin (Santa Cruz Biotechnology, Santa Cruz, CA); mouse polyclonal Ubxd8 IgG (Abnova, Taipei City, Taiwan); IgG-13F5, a mouse monoclonal antibody against human Insig-1; IgG-2179 and IgG-22D5, rabbit polyclonal antibodies against human SREBP-1 and SREBP-2, respectively (Lee et al., 2008); IgG-R139, a rabbit polyclonal antibody against hamster Scap (Sakai et al., 1997); rabbit polyclonal anti-calnexin IgG (Novus Biologicals); rabbit polyclonal anti-Aup1 and anti-actin IgG (Sigma); IgG-740F, a rabbit polyclonal antibody against human gp78 (Lee et al. 2006); guinea pig polyclonal anti-ADRP IgG (Fitzgerald, Acton, MA); chicken polyclonal anti-BAT3 IgG (Abcam, Cambridge, MA); and mouse monoclonal anti-VCP/p97 IgG (BD Transduction Laboratories, San Jose, CA).
Immunocytochemistry and CFEM
SV-589 and SV-589/pUbc7-Myc cells were set up for experiments on day 0 as described in the figure legends. Following incubations described in the figure legends, cells were washed with PBS, fixed with 3% paraformaldehyde, and permeabilized in PBS containing 0.1% Triton X-100. Following incubation in 10% normal goat serum (Invitrogen, Carlsbad, CA), coverslips were incubated for 1 h at room temperature with primary antibodies (2–5 μg/ml) diluted in 10% normal goat serum. Bound antibodies were visualized with anti–mouse IgG conjugated to Alexa Fluor 488 or Alexa Fluor 647 and anti–rabbit IgG conjugated to Alexa Fluor 594 or Alexa Fluor 568 (Invitrogen) as described in the figure legends. In addition, some of the coverslips were stained with 50 ng/ml of 4′,6-diamidino-2-phenylindole (DAPI) and 5 μg/ml of Bodipy to visualize nuclei and lipid droplets, respectively. Images were taken using a Chameleon XR NIR laser confocal microscope with a Zeiss LSM510 META spectral detector (Jena, Germany).
For CFEM experiments, SV-589 cells were set up on day 0 at a density of 8 × 104 cells per 35-mm glass-bottom dish (MatTek, Ashland, MA) in medium B supplemented with 10% FCS. On day 1, the cells were transfected with 1 μg/dish of pCMV-Aup1-GFP and pCMV-Ubc7-RFP using FuGENE6, as described above. Following incubations described in the figure legends, cells were fixed with 3% paraformaldehyde, and images were taken using a Zeiss L5M510 laser-scanning confocal microscope. Cells were fixed again in 0.1 M cacodylate buffer containing 2.5% glutaraldehyde and subsequently processed for electron microscopy; images were taken using a FEI Tecnai G2 Spirit BioTWIN transmission electron microscope (Hillsboro, OR).
Ubiquitination of HMG CoA reductase
Conditions of the incubations prior to harvest are described in the figure legends. Following treatments, triplicate dishes of cells were harvested, lysed, and subjected to immunoprecipitation with polyclonal antireductase; this was followed by immunoblot analysis with mouse monoclonal IgG-A9 (against reductase) and IgG-P4D1 (against ubiquitin) as previously described (Sever et al., 2003a).
RNAi and real-time PCR
RNAi was performed as previously described with minor modifications (Liang et al., 2002). Duplexes of siRNAs were designed and synthesized by Dharmacon/Thermo Fisher Scientific. SV-589 and SV-589/pUbc7-Myc cells were set up for experiments on day 0 as described in the figure legends. On day 1, the cells were incubated with 600 pmol of siRNA duplexes (VSV-G, GGCUAUUCAAGCAGACGGU; Aup1, GCACAUGAAGCGACAAAGA; Aup1m, GCACAUGggGCGgCAAAGA) mixed with Oligofectamine (Invitrogen, Grand Island, NY) diluted in Opti-MEM I reduced serum medium (Life Technologies, Grand Island, NY) according to the manufacturer’s protocol. Following incubation for 5 h at 37°C, the cells received a direct addition of FCS to achieve a final concentration of 10%. On day 2, the RNAi procedure was repeated as described above, except that, after 5 h, the cells were switched to medium B containing 10% LPDS, 50 μM compactin, and 50 μM mevalonate and incubated for 16 h at 37°C. The cells were subsequently treated and analyzed as described in the figure legends.
Metabolic labeling studies
Incorporation of [14C]acetate into sterols was determined as previously described (Nguyen et al., 2007), with minor modifications. SV-589 cells were set up and transfected with siRNAs as indicated in the figure legends. On day 3, the cells were re-fed medium B containing 10% FCS and 15 μCi/ml (30 μCi/dish) of [14C]acetate; cold acetate was added to achieve a final acetate concentration of 0.5 mM. Following incubation at 37°C for the times indicated in the figure legends, cell monolayers were washed with PBS, harvested, and dissolved in 0.1 N NaOH at room temperature for 30 min. Cholesterol, lanosterol, and squalene dissolved in chloroform/methanol (2:1) were added to each sample as carrier sterols, and 5 nCi of [3H]lanosterol was added as an internal standard. Lipids were extracted three times with 3.5 ml of petroleum ether, and the organic phase was evaporated under pressurized air. Lipids were resuspended in heptane and separated by TLC on plastic-backed silica gel TLC plates developed in benzene:ethyl acetate (4:1). Values were corrected for recovery efficiency of the internal standard. An aliquot of each sample was taken prior to saponification for protein determination. The mean values from triplicate dishes of cells are presented as picomoles of [14C]acetate incorporation per milligram of protein.
Supplementary Material
Acknowledgments
We thank Michael S. Brown and Joseph L. Goldstein for their continued encouragement and insightful advice. We also thank Tammy Dinh for excellent technical assistance and Lisa Beatty, Angela Caroll, Muleya Kapaale, Shomanike Head, and Ijeoma Onwuneme for help with tissue culture. This work was supported in whole or in part by National Institutes of Health grants HL20948 and GM090216. R.A.D.-B. is an Early Career Scientist of the Howard Hughes Medical Institute.
Abbreviations used:
- 25-HC
25-hydroxycholesterol
- Aup1
ancient ubiquitous protein-1
- CFEM
correlative fluorescence and electron microscopy
- CMV
cytomegalovirus
- DAPI
4′,6-diamidino-2-phenylindole
- dFCS
delipidated fetal calf serum
- eGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- FCS
fetal calf serum
- G2BR
UBE2G2-binding region
- GFP
green fluorescent protein
- HMG CoA
3-hydroxy-3-methylglutaryl CoA
- IgG
immunoglobulin G
- LPDS
lipoprotein-deficient serum
- LPLAT
lysophospholipid acyltransferase
- RFP
red fluorescent protein
- RNAi
RNA interference
- siRNA
small interfering RNA
- SREBP
sterol-regulatory, element-binding protein
- TAP
tandem affinity purification
- TEV
tobacco etch virus
- Ubxd
ubiquitin regulatory x domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-07-0564) on December 5, 2012.
REFERENCES
- Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RG, Liu P, Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007a;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- Bartz R, Zehmer JK, Zhu M, Chen Y, Serrero G, Zhao Y, Liu P. Dynamic activity of lipid droplets: protein phosphorylation and GTP-mediated protein translocation 2. J Proteome Res. 2007b;6:3256–3265. doi: 10.1021/pr070158j. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci USA. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Ploegh HL. BAT3 guides misfolded proteins out of the endoplasmic reticulum. PLoS One. 2011;6:e28542. doi: 10.1371/journal.pone.0028542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBose-Boyd RA, Brown MS, Li WP, Nohturfft A, Goldstein JL, Espenshade PJ. Transport-dependent proteolysis of SREBP: relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell. 1999;99:703–712. doi: 10.1016/s0092-8674(00)81668-2. [DOI] [PubMed] [Google Scholar]
- Farese RV, Jr., Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feramisco JD, Goldstein JL, Brown MS. Membrane topology of human insig-1, a protein regulator of lipid synthesis. J Biol Chem. 2004;279:8487–8496. doi: 10.1074/jbc.M312623200. [DOI] [PubMed] [Google Scholar]
- Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J Biol Chem. 2002;277:17377–17380. doi: 10.1074/jbc.R100068200. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Parton RG. Not just fat: the structure and function of the lipid droplet. Cold Spring Harb Perspect Biol. 2011;3:a004838. doi: 10.1101/cshperspect.a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 2001;276:4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- Hartman IZ, Liu P, Zehmer JK, Luby-Phelps K, Jo Y, Anderson RG, DeBose-Boyd RA. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 2010;285:19288–19298. doi: 10.1074/jbc.M110.134213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, DeBose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45:185–198. doi: 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc Natl Acad Sci USA. 2011a;108:20503–20508. doi: 10.1073/pnas.1112831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, Sguigna PV, DeBose-Boyd RA. Membrane-associated ubiquitin ligase complex containing gp78 mediates sterol-accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2011b;286:15022–15031. doi: 10.1074/jbc.M110.211326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang RS, Daniels CM, Francis SA, Shih SC, Salerno WJ, Hicke L, Radhakrishnan I. Solution structure of a CUE-ubiquitin complex reveals a conserved mode of ubiquitin binding. Cell. 2003;113:621–630. doi: 10.1016/s0092-8674(03)00362-3. [DOI] [PubMed] [Google Scholar]
- Klemm EJ, Spooner E, Ploegh HL. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J Biol Chem. 2011;286:37602–37614. doi: 10.1074/jbc.M111.284794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol Chem. 2006;281:39308–39315. doi: 10.1074/jbc.M608999200. [DOI] [PubMed] [Google Scholar]
- Lee JN, Zhang X, Feramisco JD, Gong Y, Ye J. Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J Biol Chem. 2008;283:33772–33783. doi: 10.1074/jbc.M806108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- Liscum L, Finer-Moore J, Stroud RM, Luskey KL, Brown MS, Goldstein JL. Domain structure of 3-hydroxy-3-methylglutaryl coenzyme A reductase, a glycoprotein of the endoplasmic reticulum. J Biol Chem. 1985;260:522–530. [PubMed] [Google Scholar]
- Liscum L, Luskey KL, Chin DJ, Ho YK, Goldstein JL, Brown MS. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J Biol Chem. 1983;258:8450–8455. [PubMed] [Google Scholar]
- Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279:3787–3792. doi: 10.1074/jbc.M311945200. [DOI] [PubMed] [Google Scholar]
- Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- Metherall JE, Goldstein JL, Luskey KL, Brown MS. Loss of transcriptional repression of three sterol-regulated genes in mutant hamster cells. J Biol Chem. 1989;264:15634–15641. [PubMed] [Google Scholar]
- Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol Chem. 2007;282:27436–27446. doi: 10.1074/jbc.M704976200. [DOI] [PubMed] [Google Scholar]
- Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell. 2006;17:2674–2683. doi: 10.1091/mbc.E05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR. Lipid droplet formation is dispensable for endoplasmic reticulum-associated degradation. J Biol Chem. 2011;286:27872–27874. doi: 10.1074/jbc.C111.266452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S, Tomoda H, Xu QM, Takahashi Y, Iwai Y. Triacsins, new inhibitors of acyl-CoA synthetase produced by Streptomyces sp. J Antibiot (Tokyo) 1986;39:1211–1218. doi: 10.7164/antibiotics.39.1211. [DOI] [PubMed] [Google Scholar]
- Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- Ponting CP. Proteins of the endoplasmic-reticulum-associated degradation pathway: domain detection and function prediction. Biochem J. 2000;351:527–535. [PMC free article] [PubMed] [Google Scholar]
- Prag G, Misra S, Jones EA, Ghirlando R, Davies BA, Horazdovsky BF, Hurley JH. Mechanism of ubiquitin recognition by the CUE domain of Vps9p. Cell. 2003;113:609–620. doi: 10.1016/s0092-8674(03)00364-7. [DOI] [PubMed] [Google Scholar]
- Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem. 1999;274:28549–28556. doi: 10.1074/jbc.274.40.28549. [DOI] [PubMed] [Google Scholar]
- Roitelman J, Olender EH, Bar-Nun S, Dunn WA, Jr., Simoni RD. Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J Cell Biol. 1992;117:959–973. doi: 10.1083/jcb.117.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Nohturfft A, Cheng D, Ho YK, Brown MS, Goldstein JL. Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J Biol Chem. 1997;272:20213–20221. doi: 10.1074/jbc.272.32.20213. [DOI] [PubMed] [Google Scholar]
- Sato S, Fukasawa M, Yamakawa Y, Natsume T, Suzuki T, Shoji I, Aizaki H, Miyamura T, Nishijima M. Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J Biochem. 2006;139:921–930. doi: 10.1093/jb/mvj104. [DOI] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell Mol Life Sci. 2008;65:2360–2371. doi: 10.1007/s00018-008-8072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003a;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003b;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- Song BL, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005a;1:179–189. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005b;19:829–840. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Spandl J, Lohmann D, Kuerschner L, Moessinger C, Thiele C. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J Biol Chem. 2011;286:5599–5606. doi: 10.1074/jbc.M110.190785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandl J, White DJ, Peychl J, Thiele C. Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic. 2009;10:1579–1584. doi: 10.1111/j.1600-0854.2009.00980.x. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, Fujimoto T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HC, Melo RC, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB J. 2007;21:167–178. doi: 10.1096/fj.06-6711com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Proteins under new management: lipid droplets deliver. Trends Cell Biol. 2007;17:363–369. doi: 10.1016/j.tcb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.