

Notch signaling is reliant on γ-secretase–mediated processing, although the subcellular location where it cleaves Notch to initiate signaling remains unresolved. Findings here support a model in which Notch signaling in mammalian systems is initiated from either the plasma membrane or lysosome, but not the early endosome.
Abstract
Notch signaling is reliant on γ-secretase–mediated processing, although the subcellular location where γ-secretase cleaves Notch to initiate signaling remains unresolved. Accumulating evidence demonstrates that Notch signaling is modulated by endocytosis and endosomal transport. In this study, we investigated the relationship between Notch transport itinerary and signaling capacity. In doing so, we discovered a highly conserved dileucine sorting signal encoded within the cytoplasmic tail that directs Notch to the limiting membrane of the lysosome for signaling. Mutating the dileucine motif led to receptor accumulation in cation-dependent mannose-phosphate receptor–positive tubular early endosomes and a reduction in Notch signaling capacity. Moreover, truncated receptor forms that mimic activated Notch were readily cleaved by γ-secretase within the endosome; however, the cleavage product was proteasome-sensitive and failed to contribute to robust signaling. Collectively these results indicate that Notch signaling from the lysosome limiting membrane is conserved and that receptor targeting to this compartment is an active process. Moreover, the data support a model in which Notch signaling in mammalian systems is initiated from either the plasma membrane or lysosome, but not the early endosome.
INTRODUCTION
Notch signaling is essential for development in metazoans, in which it influences processes ranging from cell viability to cell fate specification (Kopan and Ilagan, 2009; Tien et al., 2009). In adults, elevated Notch signaling can lead to diseases such as leukemia or mammary carcinoma, while insufficient signal has been linked to malignancies in the brain and lung (Allenspach et al., 2002). Therefore it is critical that we understand the mechanisms that both promote and down-regulate the Notch signaling pathway. Ligand-dependent Notch signaling is initiated when one of several transmembrane ligands belonging to the Delta, Serrate, and Lag2 family binds Notch on the surface of a neighboring cell (Kopan and Ilagan, 2009; Pratt et al., 2010). Ligand binding is then thought to promote a conformational change within the Notch extracellular region (Gordon et al., 2007; Nichols et al., 2007), allowing cleavage by extracellular metalloproteases of the ADAM (a disintegrin and metalloprotease) family (Brou et al., 2000; van Tetering et al., 2009). This releases the Notch ectodomain from the cell surface, leaving a membrane-tethered, Notch extracellular truncation fragment (NEXT; Kopan et al., 1996; Mumm et al., 2000). In turn, NEXT undergoes a γ-secretase–dependent intramembrane cleavage event that releases the Notch intracellular domain (NICD) from the membrane (De Strooper et al., 1999). NICD then targets to the nucleus to coordinate gene expression (Kitagawa et al., 2001).
In addition to signaling that results from Notch interaction with ligand at the cell surface, genetic analysis in Drosophila reveals that Notch can also signal from the lysosome, independent of ligand (Wilkin et al., 2008). In this particular case, Notch is not targeted to the lysosome lumen for degradation. Instead, it remains on the lysosome outer/limiting membrane, where γ-secretase–mediated NICD release and subsequent signaling can occur. Moreover, when ESCRT (endosomal sorting complex required for transport)-mediated receptor transport toward the degradative pathway is disrupted in Drosophila, Notch accumulates in enlarged endosomes and Notch gain-of-function phenotypes are observed (Moberg et al., 2005; Thompson et al., 2005; Vaccari and Bilder, 2005; Childress et al., 2006; Gallagher and Knoblich, 2006; Herz et al., 2006; McGill et al., 2009). Although the latter studies failed to identify the endosomal signaling compartment, they underscore the importance of understanding the mechanisms that govern Notch transport through the endosome. In this study, we employ a mammalian cell culture system to investigate the endosomal transport itinerary of Notch and determine how Notch subcellular localization influences its processing by γ-secretase and subsequent signaling capacity.
RESULTS
Immunolocalization analysis and live-cell imaging studies reveal that Notch associates with cytoplasmic vesicles in a variety of experimental systems ranging from Caenorhabditis elegans to Drosophila melanogaster (Kooh et al., 1993; Shaye and Greenwald, 2002; Coumailleau et al., 2009). In terminally differentiated HeLa cells, endogenous mammalian Notch (mNotch1) also localizes to vesicles throughout the cytoplasm, although it is enriched in a perinuclear region (Figure 1A). Similar localization patterns were observed following low-level (two- to threefold above endogenous) expression of a recombinant full-length mammalian Notch1 chimera (CD8-mNotch1) under control of a tetracycline-regulatable promotor (pTREL; Figure 1C).
FIGURE 1:
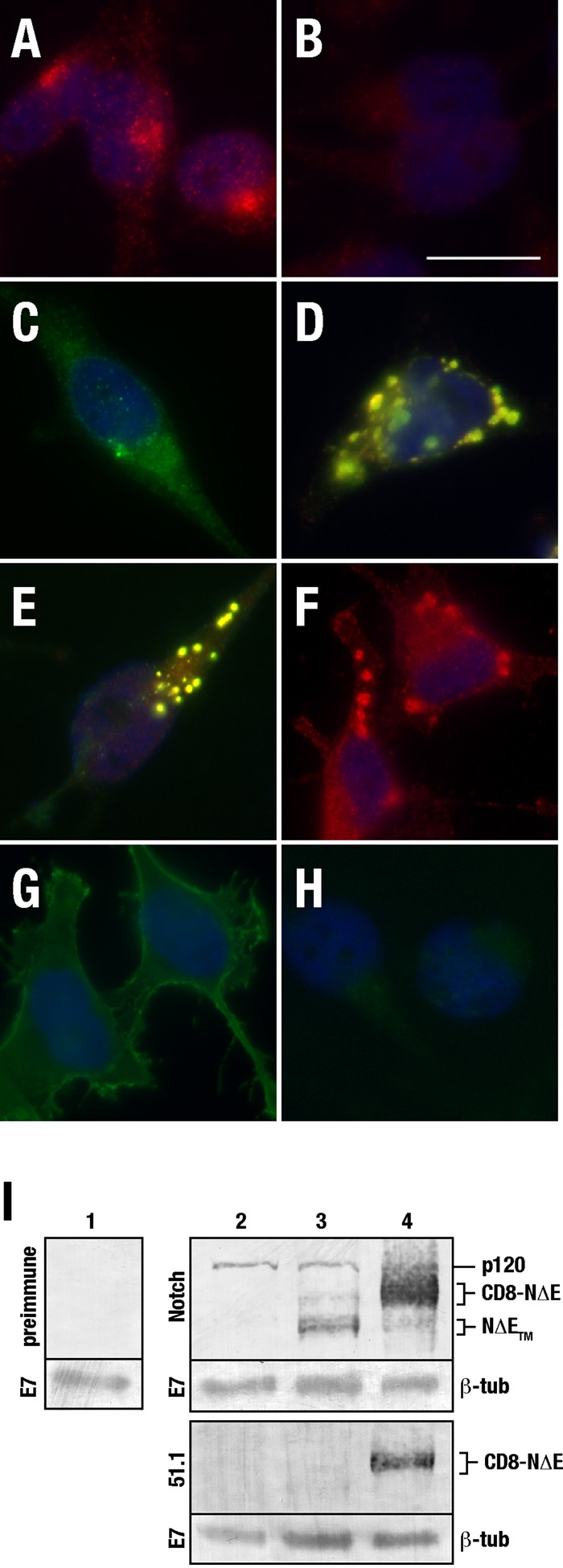
Notch immunolocalization analysis. tTA HeLa cells were fixed and probed with antisera against NICD (A) or preimmune sera (B). tTA HeLa cells were transfected with plasmid encoding CD8-mNotch under control of the tetracycline-regulatable (C, pTREL , low expression levels) or CMV promotor (D, pCMV, high expression levels), fixed, and probed for the CD8 epitope using mAb 51.1 (green) or NICD (red). tTA HeLa cells were infected with adenovirus encoding CD8-NΔE (E), NΔE-6myc (F), or CD8-LDLR (G), and probed with 51.1 (green) and/or NICD (red). (H) 51.1 antibody background. Yellow indicates overlap of the red and green channels. (I) Immunoblot analysis of control cells (lanes 1 and 2) or those expressing pTREL-CD8-NΔE (lane 3) or pCMV-CD8-NΔE (lane 4). Blots were probed using NICD antibody or 51.1. The p120 band reveals endogenous Notch levels. β-Tubulin (mAb E7) was used as a loading control.
Given that published reports suggest that endosomal sorting decisions of activated Notch are critical for signaling (Vaccari et al., 2008; Windler and Bilder, 2010), we next investigated the localization of a truncated Notch form that mimics activated receptor, CD8-NΔE (Sorensen and Conner, 2010). However, we were unable to distinguish between endogenous Notch localization and that of CD8-NΔE when expressed at low levels, since the extracellular CD8 tag was readily lost (Figure 1I). By contrast, using a strong cytomegalovirus (CMV) promotor (pCMV) to drive high levels of expression (>10-fold above endogenous), Notch chimeras localized to enlarged endosomes following transient transfection of either CD8-mNotch1 (Figure 1D) or adenovirus-mediated CD8-NΔE (Figure 1E). Although enlarged endosome formation following mNotch overexpression is consistent with published reports (Jarriault et al., 1995), we postulated that the extracellular CD8 tag or possibly adenovirus infection might artificially generate these structures. This was not the case, however, since enlarged endosomes were readily observed following overexpression of an activated Notch form lacking an extracellular tag (NΔE-6myc; Figure 1F; Schroeter et al., 1998). Similarly, adenovirus-mediated overexpression of a low-density lipoprotein receptor chimera did not result in enlarged endosome formation (CD8-LDLR; Figure 1G; Motley et al., 2003).
The aberrant and enlarged endosomes appeared similar to those that result following overexpression of late endosome or lysosomal markers, such as mannose-phosphate receptor, the major histocompatibility complex (MHC) class II invariant chain (Ii), the T-cell antigen receptor CD3γ, and the lysosomal integral membrane protein type 2 (Lotteau et al., 1990; Letourneur and Klausner, 1992; Pond et al., 1995; Lampson et al., 2001; Tiwari et al., 2011). This suggested that Notch might use a similar mechanism to directs its transport toward the late endosome or lysosome. To test this idea, we first performed colocalization analysis between endogenous Notch and recombinant cation-dependent mannose-phosphate receptor (CD-MPR), a receptor that delivers newly synthesized acid hydrolases from the trans-Golgi network (TGN) to endosomes for their transfer to lysosomes (Ghosh et al., 2003). Following transient transfection of plasmid encoding CD-MPR fused to green fluorescent protein (CD-MPR-GFP), we did not detect significant colocalization with endogenous Notch in cells expressing CD-MPR-GFP at low levels or levels at which enlarged endosomes are observed (Supplemental Figure S1, C and F). However, significant colocalization was observed between CD-MPR-GFP and CD8-NΔE when the Notch chimera was expressed at high levels (Figure S1I). To better resolve the nature of the enlarged endosome compartment, we performed immunoelectron microscopy. In doing so, we discovered that Notch chimeras are present in multivesicular endosomes and also become concentrated on multilamellar structures, which appear to result from a collapse of tubular endosomes around a central vacuole and are not observed in control cells (Figure S2).
CD-MPR can be transported directly from the TGN to the late endosome or through the endocytic pathway (Ghosh et al., 2003). Given the significant colocalization between CD-MPR and overexpressed Notch chimeras, we next attempted to determine the transport pathway by which Notch populates the enlarged endosomes. To do so, we incubated Notch chimera-expressing cells with antibody directed against the CD8 extracellular epitope (mAb 51.1) to allow endocytosis. For both CD8-mNotch and CD8-NΔE–expressing cells, significant colocalization was observed with internalized antibody (Figure S3), indicating the enlarged endosomes are populated, at least in part, by endocytosis. We interpret these results to indicate that Notch encodes a robust sorting signal that serves to package the receptor for transport between endosomal compartments following endocytosis.
Notch encodes a highly conserved dileucine sorting signal
To identify the potential sorting signal encoded with Notch, we progressively truncated the cytoplasmic receptor tail (Figure 2A), reasoning that once a sorting signal is lost, overexpressed Notch would no longer form enlarged, aberrant endosomes. For our truncation analyses, we used CD8-NΔE, because >75% of chimera-overexpressing cells form enlarged endosomes following adenovirus-mediated overexpression (Figure 2D). Following truncation of the carboxy-terminal 338 amino acids (CD8-NΔE Δ2194), we observed a similar number of enlarged endosome-forming cells relative to controls (Figure 2, C and D). By contrast, deletion of an additional 114 amino acids (CD8-NΔE Δ2080) almost completely abrogated aberrant endosome formation. Instead, CD8-NΔE Δ2080 localized to small punctate endosomes connected to an extensive tubular network (Figure 2C). These results indicated the presence a sorting signal between amino acids 2080 and 2194.
FIGURE 2:

Notch encodes an endosomal sorting signal. (A) Diagram illustrating Notch domain structure of the cytoplasmic tail and CD8-NΔE truncation mutants used for analysis. (B) Immunoblot analysis indicating expression of each CD8-NΔE form in tTA HeLa cells. (C) Immunolocalization analysis of the Notch cytoplasmic tail following infection with adenovirus encoding the indicated CD8-NΔE form (CD-NΔE, CD8-NΔ2194, CD8-NΔ2080). (D) Quantitation of cells forming enlarged endosomes (EE) following overexpression of the indicated Notch form. Cells were infected with adenovirus encoding the indicated CD8-NΔE control or truncation mutant. For each condition, more than 200 cells were counted. Boxed region indicates area of higher magnification. Error bars represent ± SEM. Scale bar: 10 μm.
Sequence analysis of this region revealed a dileucine sorting signal following the [DE]XXXL[LI] pattern (Figure 3A), similar to that found within the cytoplasmic tails of LIMPII, CD3γ, and Ii, which direct targeting to the late endosome and/or lysosome (Letourneur and Klausner, 1992; Pond et al., 1995). The motif is highly conserved between vertebrates and invertebrates, nematodes being the exception, suggesting it may be critical for Notch sorting between endosomal compartments. To test this, we next mutated the conserved leucines to alanine (DIVRAA2103, CD8-NΔE LLAA), since these amino acids are critical for motif recognition by adaptor proteins (Bonifacino and Traub, 2003). In doing so, we found that CD8-NΔE LLAA overexpression failed to generate enlarged endosomes. Instead, immunolocalization analysis of permeabilized cells revealed that CD8-NΔE LLAA is localized to an extensive interconnected tubular network (Figure 3, C–G), similar to that observed for truncation analyses (Figure 2C).
FIGURE 3:

A conserved dileucine motif directs Notch endosomal sorting. (A) Amino acid alignment of the Notch cytoplasmic tail spanning the dileucine motif. Underlined region indicates the conserved dileucine sorting motif that follows the [D/E]XXX[L][L/I] pattern. tTA HeLa cells were infected with adenovirus encoding either CD8-NΔE (B, D, F, and H) or CD8-NΔE LLAA (C, E, G, and I) and processed for immunolocalization. Permeabilized cells (B–G) were probed with antibodies to the NICD, while nonpermeabilized cells (H and I) were probed with the mAb 51.1 to detect the CD8 epitope at the cell surface. Boxed regions indicate areas of higher magnification. Scale bar: 20 μm (B,C); 10 μm (D–I).
We interpret these observations to suggest that the dileucine motif is critical for directing Notch toward the late endosome and/or lysosome. However, dileucine motifs function at multiple transport steps to promote receptor packaging into transport vesicles for delivery to the plasma membrane or internalization (Letourneur and Klausner, 1992; Pond et al., 1995). To explore these possibilities, we measured CD8-NΔE LLAA cell-surface levels by flow cytometry using mAb 51.1. We reasoned that a biosynthetic defect would lead to reduced plasma membrane targeting. However, we failed to detect a significant difference in plasma membrane-localized Notch between cells overexpressing CD8-NΔE or CD8-NΔE LLAA (Figure 4A). Consistently, we failed to detect a difference by immunolocalization analysis of nonpermeabilized cells (Figure 3, H and I). However, when Notch internalization was impaired by small interfering RNA (siRNA)-mediated clathrin heavy-chain depletion, an anticipated increase in cell-surface levels was observed for both CD8-NΔE and CD8-NΔE LLAA (Figure 4A).
FIGURE 4:
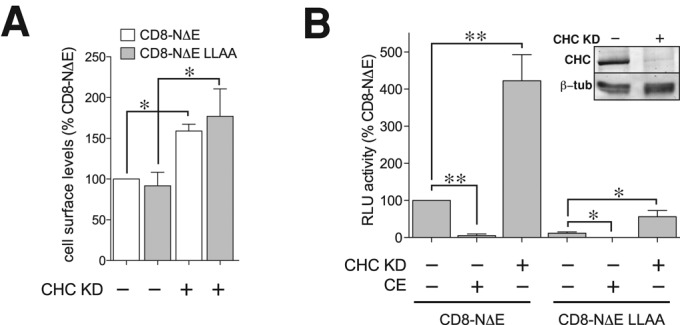
Notch internalization is independent of the dileucine motif. (A) Notch cell-surface levels were measured by flow cytometry (see Materials and Methods) in CD8-NΔE– or CD8-NΔE LLAA–overexpressing tTA HeLa cells, which were pretreated with control siRNA or siRNA against clathrin heavy chain (CHC). (B) The corresponding signaling for each Notch chimera was measured by luciferase assay in the presence or absence of 1 μM CE. Error bars represent ± SEM; p values by t test: *, p < 0.05; **, p < 0.005.
We next investigated the possibility that the dileucine motif directed Notch internalization. Our previous findings indicate that Notch signaling is elevated for CD8-NΔE when endocytosis is disrupted (Sorensen and Conner, 2010). Therefore we tested CD8-NΔE LLAA signaling capacity with the anticipation that, if the dileucine motif served to promote Notch endocytosis, CD8-NΔE LLAA signaling should be more robust relative to CD8-NΔE. On the contrary, we discovered that CD8-NΔE LLAA signaling was markedly reduced relative to CD8-NΔE (Figure 4B). This signaling was Notch-specific, given that signaling for both CD8-NΔE and CD8-NΔE LLAA was nearly eliminated by pretreating cells with Compound E (CE), a highly specific γ-secretase inhibitor (Kornilova et al., 2003) that prevents cleavage and release of the Notch cytoplasmic tail (Figure 4B). Combined, these observations lend support for the conclusion that Notch transport along the biosynthetic pathway, delivery to the plasma membrane, and subsequent internalization occur independently of the dileucine motif. Moreover, the reduction in CD8-NΔE LLAA signaling relative to controls suggests the mutant fails to target to endosomes at which signaling can occur, possibly the lysosome.
The Notch dileucine motif directs sorting from early endosomes
To more clearly resolve the endosomal transport step that relies on the conserved dileucine motif, we pursued a live-cell imaging approach. To do so, we transiently transfected PS1/2−/- mouse embryonic fibroblasts (MEFs PS1/2−/−), which lack γ-secretase activity (Herreman et al., 2000), to eliminate Notch processing, with plasmid encoding CD8-NΔE chimeras fused to either GFP or mCherry (CD8-NΔE-GFP, CD8-NΔE-mChr). Expression time was limited to avoid overexpression artifacts and enlarged endosome formation. At low expression levels, CD8-NΔE localized to punctate endosomes throughout the cytoplasm (unpublished data), similar to endogenous Notch (Figure 1). By comparison, the dileucine mutant also localized to punctate endosomes. However, CD8-NΔE LLAA was also found associated with interconnected tubular endosomes (unpublished data) similar to those observed in fixed tTA (tetracycline trans activator) HeLa cells (Figure 3). To determine the identity of the tubular endosome compartment, we next used a colocalization strategy with early and late endosomal markers.
To mark early endosomes, we tested colocalization with two early endosomal markers; smad2 activator for receptor activation (SARA; Hu et al., 2002) and wild-type rab5 (Stenmark et al., 1994). Live-cell imaging failed to reveal significant colocalization between CD8-NΔE or CD8-NΔE LLAA and either early endosome marker (Figure 5, C and F; unpublished data). However, electron microscopy (EM) analysis revealed that the early endosome also forms an extended tubular network in a variety of cell types, with particular abundance in HeLa (Hopkins et al., 1990; Tooze and Hollinshead, 1991), similar to that observed for CD8-NΔE LLAA (Figure 3). Given that CD-MPR transits the early endosome (Klumperman et al., 1993), we next evaluated its colocalization with each recombinant Notch chimera. Following transient expression in MEF Pen1/2−/− cells, GFP-CD-MPR localized to a faint interconnected tubular network at the cell periphery. In these cells, CD8-NΔE-mChr was found on punctate endosomes located between or adjacent to the GFP-CD-MPR–positive tubular network (Figure 5I). By contrast, extensive colocalization was observed between GFP-CD-MPR and CD8-NΔE LLAA-mChr (Figure 5L). The extensive colocalization and tubular pattern is consistent with the notion that CD8-NΔE LLAA transport from early endosomes is defective; however, CD-MPR is also equally present on late endosomes (Klumperman et al., 1993). To distinguish between early and late endosomal compartments, we coexpressed rab5Q79L, an activated form of rab5 that promotes early endosome fusion (Stenmark et al., 1994). We reasoned that, if the tubular network was indeed early endosome, rab5Q79L expression should promote the collapse of the tubular network into enlarged early endosomes. Indeed, GFP-rab5Q79L coexpression with CD8-NΔE LLA-mChr resulted in a loss of the tubular network and colocalization between rab5Q79L and CD8-NΔE LLAA, similar to that observed for CD8-NΔE (Figure 6). Consistently, the CD8-NΔE LLAA–positive tubular network did not colocalize with the late endosome/lysosome marker LAMP-1 (Figure 6I). On the basis of these observations, we conclude that the dileucine motif is critical for Notch sorting within the early endosome.
FIGURE 5:

CD8-NΔE LLAA accumulates in CD-MPR–positive tubular endosomes. PEN1/2 −/− MEFs were cotransfected with plasmid encoding SARA-mChr (A and D) and CD8-NΔE-GFP (B) or CD8-NΔE LLAA-GFP (E). Alternatively, cells were cotransfected with CD-MPR-GFP (G and J) and CD8-NΔE-mChr (H) or CD8-NΔE LLAA-mChr (K). Live cells were imaged after a 10-h incubation to limit recombinant protein expression levels. Merged images are shown in (C), (F), (I), and (L). Scale bar: 10 μm.
FIGURE 6:

CD8-NΔE LLAA–positive endosomes fuse into rab5 early endosomes following expression of rab5Q79L. PEN1/2 −/− MEFs were cotransfected with plasmid encoding CD8-NΔE-mChr (A) or CD8-NΔE LLAA-mChr (D) and GFP-rab5Q79L (B and E). Alternatively, cells were cotransfected with plasmid encoding LAMP-1-mRFP (monomeric red fluorescent protein) (G) and CD8-NΔE LLAA-GFP (H). Live cells were imaged after a 10-h incubation to limit recombinant protein expression levels. Merged images are shown in (C), (F), and (I). Scale bar: 10 μm
From the early endosome, Notch can be incorporated into intralumenal vesicles of the multivesicular late endosomes, which leads to receptor degradation following fusion with the lysosome. Alternatively, Notch can be directed to the lysosome limiting membrane, where ligand-independent signaling can occur (Wilkin et al., 2008). Given that colocalization analysis indicates that CD8-NΔE LLAA transport from early endosomes is altered, we next asked which sorting step might be impaired. To test this, we pursued a live-cell imaging approach with the late endosome marker rab7, expression of which promotes receptor targeting for degradation (Meresse et al., 1995; Bucci et al., 2000). We postulated that, if the dileucine motif was critical for directing Notch to the lysosome limiting membrane and not for degradation, CD8-NΔE LLAA would be readily observed inside the lumen of rab7-positive endosomes. Similar to CD8-NΔE-mChr, CD8-NΔE LLAA-mChr was observed within the lumen of rab7-positive endosomes in MEFs PS1/2−/− cells (Figure S4, C and F), indicating that the dileucine motif is not critical for Notch degradation within the lysosome. Instead, the dileucine motif likely functions in targeting Notch to the lysosome limiting membrane, consistent with decreased CD8-NΔE LLAA signaling capacity (Figure 4).
Observations in mammalian cell culture indicate that γ-secretase–mediated Notch cleavage within the endosome can lead to a proteasome-sensitive cleavage product that is rapidly degraded (Tagami et al., 2008). This raised the possibility that the dileucine mutant might be more rapidly degraded than the control. To determine the fate of CD8-NΔE LLAA, we infected HeLa cells with adenovirus expressing high levels of each Notch chimera in the presence of cycloheximide to measure protein stability over time by immunoblot analysis. In contrast to CD8-NΔE, CD8-NΔE LLAA stability was markedly reduced (Figure 7A). Quantitative analysis by densitometry revealed that CD8-NΔE LLAA is approximately threefold less stable than the CD8-NΔE control (Figure 7B). Pretreating cells with CE rescued the stability of the membrane-tethered dileucine mutant, indicating that the observed instability resulted from γ-secretase activity and not increased degradation within the lysosome lumen.
FIGURE 7:
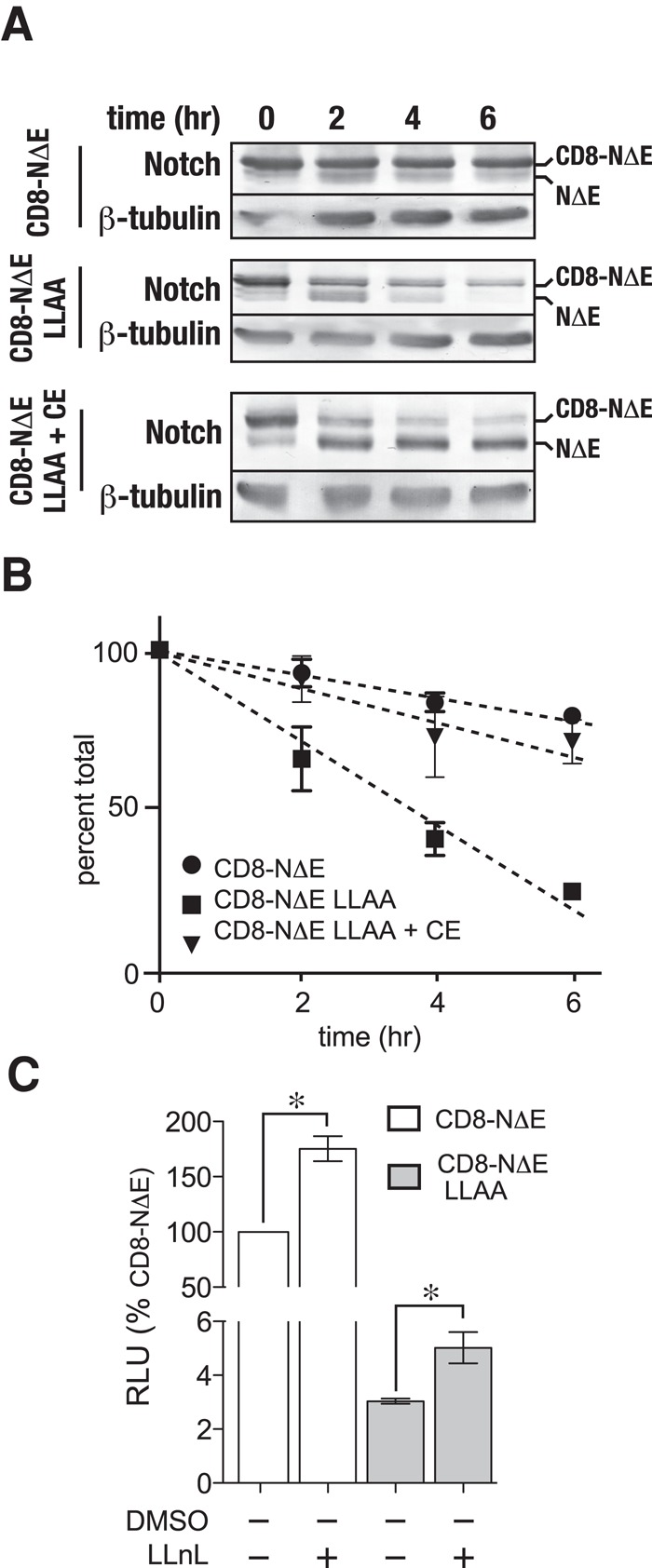
γ-secretase cleaves activated Notch forms to generate proteosome-sensitive cleavage products within the endosome. (A) tTA HeLa cells were infected with adenovirus encoding either pCMV-CD8-NΔE or pCMV-CD8-NΔE LLAA in the presence or absence of 1 μM CE. Following a 16-h incubation, protein synthesis was inhibited by treating cells with cycloheximide, and Notch chimera stability was evaluated by immunoblotting at the indicated times; a representative blot is shown. (B) Densitometric analysis of Notch chimera stability from three independent experiments. In the plot, total Notch was determined by combining densitometric values of the upper and lower Notch bands. (C) CD8-NΔE– or CD8-NΔE LLAA–expressing tTA HeLa cells were treated with either dimethyl sulfoxide (DMSO) or LLnL for 6 h to disrupt the proteosome. Notch signaling was then evaluated using the luciferase assay. Error bars indicate ± SEM of three independent experiments; p values by t test: *, p < 0.05.
On the basis of the observed protein stability differences, we next evaluated Notch signaling following proteosome inhibition with N-acetyl-l-leucyl-l-leucyl-leucyl-l-norleucinal (LLnL) to determine whether γ-secretase–dependent cleavage was generating a proteosome-sensitive product. Following a 5-h preincubation of Notch chimera–expressing cells with LLnL, a nearly twofold signaling increase was observed for the dileucine mutant (Figure 7C). Likewise, a similar increase was observed for CD8-NΔE, consistent with published observations (Tagami et al., 2008). However, CD8-NΔE LLAA signaling was not elevated to control levels, indicating that signaling differences between Notch chimeras do not result from increased exposure to endosome-localized γ-secretase. These observations indicate that both CD8-NΔE and the dileucine mutant are readily cleaved at a proteosome-sensitive site and that the increased stability of CD8-NΔE relative to the mutant likely reflects a stabilization of the control Notch chimera following incorporation into enlarged endosomes.
A variety of biochemical and mammalian cell-based studies indicate that AP-3, an endosomal adaptor protein complex, directly engages [DE]XXXL[LI] motifs to direct protein transport to the lysosome (Letourneur and Klausner, 1992; Pond et al., 1995; Reusch et al., 2002; Rodionov et al., 2002; Bonifacino and Traub, 2003; Craige et al., 2008; Sitaram et al., 2012). Moreover, genetic studies using Drosophila indicate that Notch signaling from the lysosome requires both AP-3 and the HOPS (homotypic fusion and vacuole protein sorting) tethering complex (Wilkin et al., 2008). Importantly, EM analysis also revealed that AP-3 serves to package cargo from tubular early endosomes (Peden et al., 2004). Thus we reasoned that, if the dileucine motif was critical for Notch targeting to and signaling from the lysosome, AP-3 or HOPS depletion should depress CD8-NΔE signaling to levels comparable with those observed for the dileucine mutant. To test this, we siRNA-depleted the δ-adaptin or vps39 subunits of AP-3 and HOPS, respectively, and measured signaling over a broad range of CD8-NΔE expression levels from below endogenous to 15-fold overexpression (Figure 8, A and B). Over the range of expression levels, CD8-NΔE signaling was reduced following δ-adaptin or vps39 depletion to levels comparable with those observed for CD8-NΔE LLAA (Figure 8). Consistent with the role of the dileucine sorting motif in mediating this transport step, siRNA-mediated vps39 depletion does not lead to additional decreases in CD8-NΔE LLAA signaling (Figure S5).
FIGURE 8:

Endosomal transport defects resulting from AP-3 and HOPS depletion impair Notch signaling. tTA HeLa cells were infected with increasing concentrations of adenovirus encoding pTREL-CD8-NΔE (A), pCMV-CD8-NΔE (B), or pCMV-CD8-NΔE LLAA (C). Following siRNA-mediated depletion of the HOPS subunit vps39 or the AP-3 subunit δ-adaptin, tTA HeLa cells were infected with increasing concentrations of the indicated adenovirus. Each bar within a group (left to right) corresponds to increasing adenovirus concentrations (0, 1, 5, 10, and 25 μl/ml) documented in (A–C), and Notch signaling was evaluated by luciferase assay. p values by t test: *, p < 0.05.
In Drosophila AP-3– and HOPS-dependent Notch signaling from the lysosome is stimulated by overexpression of the E3 ligase, deltex (Hori et al., 2004; Wilkin et al., 2008), whose activity in Notch signaling is tissue-specific in flies (Fuwa et al., 2006). Given that mammalian deltex (DTX1) is conserved (Matsuno et al., 1998; Kishi et al., 2001), we next tested the potential role of DTX1 in coordinating Notch function. To do so, we used an siRNA-mediated knockdown strategy and evaluated Notch localization and signaling activity following DTX1 depletion. In CD8-NΔE–expressing HeLa cells, DTX1 depletion resulted in a minor accumulation of CD8-NΔE within the endosome relative to control cells (Figure 9, A and B). By contrast, when γ-secretase activity was disrupted by treating cells with CE, CD8-NΔE also accumulated at the plasma membrane (Figure 9D). The latter observation is consistent with published findings in Drosophila, in which Notch accumulates at the cell surface in deltex mutant cells (Yamada et al., 2011), and overexpression promotes Notch redistribution from the apical membrane to intracellular vesicles (Hori et al., 2004). Moreover, it reinforces our previous findings that γ-secretase robustly cleaves activated Notch forms at the plasma membrane (Sorensen and Conner, 2010). These results suggest that DTX1 is important for Notch endocytosis. If this interpretation is correct, DTX1 depletion should promote Notch signaling for both CD8-NΔE and the dileucine mutant, as observed following clathrin depletion (Figure 4B). Indeed, DTX1 knockdown elevated signaling for CD8-NΔE and CD8-NΔE LLAA over a range of Notch chimera expression levels (Figure 9E). Given the significant signaling increases observed following DTX1 knockdown, we interpret these findings to indicate that DTX1 is not required for Notch targeting to and signaling from the lysosome via an AP-3– and HOPS-dependent pathway, as has been shown in Drosophila (Hori et al., 2004; Fuwa et al., 2006), but instead is critical for Notch removal from the plasma membrane.
FIGURE 9:
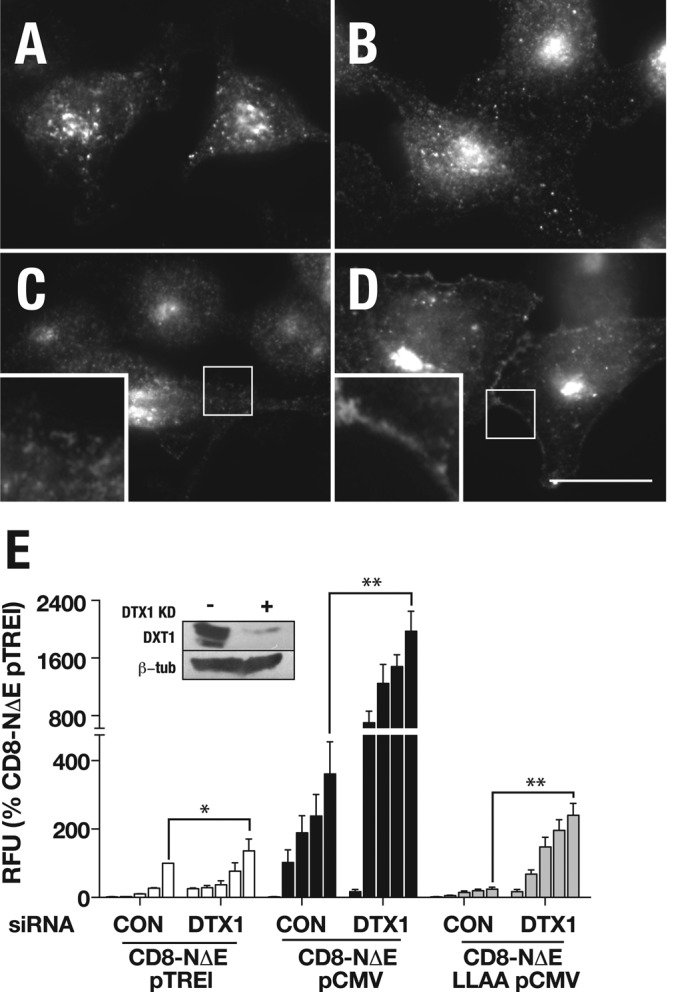
DTX1 depletion promotes signaling of activated Notch forms. tTA HeLa cells were treated with control siRNA (A and C) or siRNA directed against DTX1 (B and D), infected with 25 μl/ml pTREL-CD8-NΔE (see Figure 8), treated with either DMSO (A and B) or CE (C and D). Cells were then fixed and probed with antibody against NICD. Boxed regions indicates area of the plasma membrane at higher magnification. Scale bar: 20 μm. (E) tTA HeLa cells treated with control or DTX1 siRNA were infected with increasing concentrations of adenovirus encoding the indicated Notch chimera, and signaling was measured by luciferase assay. Each bar within a group (left to right) corresponds to increasing adenovirus concentrations (0, 1, 5, 10, and 25 μl/ml) documented in Figure 8. * and ** indicate p values by t test: *, p < 0.05; **, p < 0.005.
DISCUSSION
Results presented here provide evidence indicating that Notch sorting to the lysosome limiting membrane for signaling is directed by a highly conserved dileucine motif encoded within the cytoplasmic tail. Mutating the dileucine motif leads to receptor accumulation in CD-MPR positive early endosomes and a marked decrease in Notch signaling capacity. These findings agree well with observations in Drosophila, in which deltex-stimulated, ligand-independent Notch signaling from the lysosome occurs via an AP-3– and HOPS-dependent pathway (Wilkin et al., 2008). While our AP-3 and HOPS loss-of-function analyses reinforce observations in Drosophila, our DTX1 depletion studies reveal that Notch signaling in mammalian cell systems is not dependent on DTX1. Accumulation of activated Notch forms at the plasma membrane following DTX1 knockdown is consistent with Notch redistribution to the cell cortex in Drosophila cells mutant for deltex (Yamada et al., 2011). We interpret these observations to indicate that DTX1 likely functions to promote receptor internalization. Consistent with this notion, we find that DTX1 depletion leads to elevated signaling for activated Notch forms, similar to our previous findings (Sorensen and Conner, 2010).
Our protein stability studies comparing CD8-NΔE and the dileucine mutant indicate that activated Notch forms are readily processed by γ-secretase within an endosomal compartment upstream of the lysosome. This is particularly evident with the dileucine mutant, which accumulates in an early endosome tubular network. However, the cleavage product is proteosome-sensitive and readily degraded and does not contribute to robust signaling. The latter observation contrasts genetic studies in Drosophila, in which internalization and delivery to endosome-localized γ-secretase (upstream of the lysosome) is proposed to be a prerequisite for ligand-dependent Notch signaling. For example, clathrin loss or disrupting shibire (the Drosophila homologue of dynamin) activity prevents Notch signaling in the fly (Vaccari et al., 2008; Windler and Bilder, 2010). By contrast, loss of AP-2, a selective endocytic adaptor (McMahon and Boucrot, 2011), potently disrupts Notch internalization, yet signaling remains unaffected (Windler and Bilder, 2010). The latter observation argues that receptor internalization is not a prerequisite for signaling and that ligand-dependent Notch signaling is initiated from the plasma membrane. Consistent with this, expression of an activated Notch form bypasses the Notch signaling defect caused by expression of shibirets mutants that disrupt endocytosis (Struhl and Adachi, 2000; Vaccari et al., 2008). Likewise, internalization-defective LIN-12/Notch rescues lethality and nonvulval phenotypes that arise from loss of endogenous lin-12/Notch in C. elegans (Shaye and Greenwald, 2002, 2005). Moreover, in mammalian cells, we and others found that γ-secretase–dependent Notch signaling is either unaffected or elevated when Notch internalization is impaired (Kaether et al., 2006; Tagami et al., 2008; Sorensen and Conner, 2010).
How might these apparent discrepancies be reconciled? Our previous findings indicate that clathrin is critical for Notch transport through the trans-Golgi (Sorensen and Conner, 2010). Additionally, dynamin, the mammalian homologue of shibire, functions at multiple endosomal transport steps (van Dam and Stoorvogel, 2002). Based on these observations, it is possible that the Notch signaling defects, which arise in the absence of clathrin or dynamin in Drosophila (Windler and Bilder, 2010), might not result from impaired receptor internalization. Instead, signaling defects could result from limited receptor delivery to the plasma membrane. If clathrin and dynamin function were selective for endocytosis, one would predict that dynamin- or clathrin-deficient cells would have comparable Notch cell-surface levels relative to those lacking AP-2. However, immunolocalization analysis reveals more plasma membrane–associated Notch in AP-2–deficient cells than those lacking clathrin or dynamin (Windler and Bilder, 2010). This suggests that Notch signaling defects that arise from clathrin or dynamin loss are independent of alterations in receptor internalization. Thus we favor the previously proposed model, in which endocytosis serves to down-regulate the Notch signaling pathway, and ligand-dependent signaling is initiated from the plasma membrane (Shaye and Greenwald, 2002).
Given the high degree of conservation for the dileucine motif and its critical role in delivering Notch to the lysosome limiting membrane for signaling, our findings support the idea that Notch targeting to and signaling from the lysosome is an active process. On the basis of our findings, we propose that the Notch dileucine motif is actively recognized by AP-3 on tubular early endosomes (Peden et al., 2004), which in concert with HOPS (Angers and Merz, 2009; Salazar et al., 2009; Zlatic et al., 2011a, b), delivers Notch to the lysosome limiting membrane for signaling. This raises several interesting questions: What role does Notch signaling from the lysosome play in development and/or homeostasis? What are the regulatory steps that distinguish between Notch incorporation into intralumenal vesicles for degradation within the lysosome and receptor maintenance on the lysosome limiting membrane to allow for signaling? Given the range of human malignancies linked to Notch signaling defects (Allenspach et al., 2002), our future efforts will be directed at addressing these questions.
MATERIALS AND METHODS
Reagents
The mAbs E7, 51.1, and TD.1 were used to identify β-tubulin, CD8α, and clathrin heavy chain, respectively. Antisera to the following epitopes were purchased: CD8α (H-160; Santa Cruz Biotechnology, Santa Cruz, CA); δ-adaptin (600-101-290; Rockland, Gilbertsville, PA), vps39 (ab107570; Abcam, Cambridge, MA), and Tsg101 (H00007251-M01; Novus Biologicals, Littleton, CO). CD8-LDLR receptor construct, which was used as a backbone for CD8-mNotch1 constructs was a generous gift of Margaret Robinson (Cambridge University, UK). Plasmid encoding GFP-CD-MPR was a generous gift from Juan Bonifacino (National Institutes of Health [NIH], Bethesda, MD). Rab5 fusion constructs were a gift from Marino Zerial (Max Planck Institute, Dresden, Germany). LAMP-1-mChr was a gift from Diane Ward (University of Utah, Salt Lake City, UT). Rabbit antisera against Notch was raised against the cytoplasmic tail. γ-Secretase inhibitor XXI (CE) was purchased from Calbiochem (565790; San Diego, CA).The proteasome inhibitor LLnL (A6185) and cycloheximide (C4859) were obtained from Sigma-Aldrich (St. Louis, MO). GFP-Rab7 and PS1/2−/− MEFs were generous gifts from Marino Zerial (Max Planck Institute, Dresden, Germany) and Bart De Strooper (Institute for Biotechnology [VIB4], Leuven, Belgium), respectively.
CD8 Notch chimera cloning
CD8α signal sequence and extracellular domain were subcloned upstream of full-length mNotch1, NΔE, and truncated receptor forms, as previously reported (Sorensen and Conner, 2010). Point mutations were generated by PCR. All constructs were sequence-verified. Adenovirus production was performed as previously described (Damke et al., 1995). NΔE in pCeMM-CTAP(SG)-6W and full-length Notch in pCS2, which were used for cloning, were generous gifts from Alain Israel (Pasteur Institute, Paris, France).
siRNA-mediated depletion
siRNA depletions were performed essentially as previously described (Motley et al., 2003). In short, two siRNA transfections were performed, one on day 1 and another on day 2. After a 48-h incubation, cells were infected with adenovirus encoding the indicated Notch chimera and processed for immunolocalization 16 h later. siRNAs: clathrin heavy-chain target sequence, UAAUCCAAUUCGAAGACCAAU; AP-3D1, GCCUCCAAGUUCACCUUCAAGCGAA; vps39, GGUAAAGAAGCUGAAUGACUCUGAU; DTX1, CACAUUCCUUUAACGGAGGUCUCUA were obtained from Invitrogen (Carlsbad, CA) and Shanghai GenePharma (Shanghai, China). Silencer negative control #1 siRNA was obtained from Ambion (Austin, TX). Expression knockdowns were validated by immunoblot.
Immunolocalization
tTA HeLa cells were grown on coverslips and infected with adenovirus or transfected with plasmid encoding the indicated construct. Cells were fixed with ice-cold acetone for 5 min and extracted with methanol. Cells were then washed with phosphate-buffered saline (PBS) containing 0.1% Triton-X 100. For cell-surface labeling, cells were fixed with 4% paraformaldehyde without permeabilization. Cells were then incubated with primary antibody for 1 h at room temperature. Cells were washed and incubated for 1 h at room temperature with the appropriate secondary antibody conjugated to either Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen). Samples were then visualized by epifluorescence using a Zeiss Axio Imager M1 (Zeiss, Thornwood, NY) and captured with a monochrome Jenoptik CCD camera (Jena, Germany). Images were then imported, cropped, and assembled into panels using Photoshop CS4 and Illustrator CS4 (Adobe Systems, San Jose, CA).
51.1. uptake assay
tTA HeLa cells grown on coverslips were infected with the appropriate adenovirus in the continuous present of 51.1 mAb in DMEM containing 10% fetal bovine serum for 16 h at 37°C. Cells were then processed for immunolocalization as indicated above.
Transmission electron microscopy
Samples were fixed in 2% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) at room temperature for 1 h, rinsed in buffer, and washed with fresh buffered sodium borohydride solution (2 mg/ml). Cells were then rinsed with buffer, dehydrated in an ethanol series, and embedded in LR White resin (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections (80 to 100 nm thick) were cut on a Leica (Buffalo Grove, IL) Ultracut UCT microtome using a diamond knife and collected on Formvar carbon-coated nickel grids. Immunogold labeling was performed following published protocols (Lonsdale et al., 1999). The primary antibody Notch was diluted 1:25,000, and the secondary antibody (goat anti–rabbit immunoglobulin G preconjugated to 20-nm diameter colloidal gold; BB International, Cardiff, UK) was diluted 1:100. After labeling, grids were stained with 3% uranyl acetate followed by triple-lead stain (Sato, 1968). Sections were examined with an FEI (Phillips, Eindhoven, The Netherlands) CM 12 transmission electron microscope operating at 60 kV. Images were recorded with a Maxim DL digital capture system (Diffraction Limited, Ottawa, Canada).
Notch chimera stability assay
tTA HeLa cells grown in a 12-well dish were infected with adenovirus encoding either CD8-N∆E or CD8-N∆E LLAA for 16 h at 37°C. The media was then replaced with fresh media supplemented with 100 μg/ml cycloheximide to stop protein synthesis and 1 μM CE, where indicated. Cells were then returned to 37°C. At 2-h intervals, cell samples were removed, solubilized with protein sample buffer, and processed for immunoblotting to evaluate total Notch chimera stability. Densitometric analysis of immunoblots was performed using the ImageJ software package (NIH).
Notch signaling reporter assay
Signaling was evaluated using a dual-luciferase RBP-Jκ reporter assay (SA Biosciences, Valencia, CA) according to the manufacturer’s published protocols (Promega, Madison, WI). Relative luciferase units represent signaling expressed as a ratio of Notch-promoted firefly luciferase activity over constitutively expressed Renilla luciferase.
Flow cytometry
Cell surface levels of CD8-∆E and CD8-∆E-LLAA were measured by flow cytometry. In short, Notch chimera–expressing cells were first washed in PBS and detached from dishes using PBS supplemented with 5 mM EDTA. Cells were then gently pelleted, resuspended in ice-cold PBS containing 4% paraformaldehyde, and fixed for 20 min. Following a PBS wash, cells were incubated in PBS containing the mAb 51.1 for 1 h at room temperature. Cells were washed again and incubated with Alexa Fluor 488–labeled goat anti–mouse secondary antibody (Invitrogen) for 1 h. Surface antibody levels were quantified, gating on intact cells, and the median fluorescence intensity was determined from 10,000 cells.
Supplementary Material
Acknowledgments
We thank Gail Celio for technical support in performing immunoelectron microscopy and M. B. Rock for nontechnical assistance. We also thank Gary Struhl for helpful suggestions. This work was supported in part by an NIH grant (GM085029) to S.D.C. and a Developmental Biology Training grant (2T32-HD007480-11A1) to E.B.S.
Abbreviations used:
- CD-MPR
cation-dependent mannose-phosphate receptor
- CE
Compound E
- CMV
cytomegalovirus
- DMSO
dimethyl sulfoxide
- DTX1
mammalian deltex
- EM
electron microscopy
- GFP
green fluorescent protein
- HOPS
homotypic fusion and vacuole protein sorting
- LLnL
N-acetyl-l-leucyl-l-leucyl-leucyl-l-norleucinal
- MEFs
mouse embryonic fibroblasts
- mNotch1
endogenous mammalian Notch
- NEXT
Notch extracellular truncation fragment
- NICD
Notch intracellular domain
- PBS
phosphate-buffered saline
- pCMV
CMV promotor
- pTREL
tetracycline-regulatable promotor
- SARA
smad2 activator for receptor activation
- siRNA
small interfering RNA
- TGN
trans-Golgi network
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0081) on November 21, 2012.
REFERENCES
- Allenspach EJ, Maillard I, Aster JC, Pear WS. Notch signaling in cancer. Cancer Biol Ther. 2002;5:466–476. doi: 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- Angers CG, Merz AJ. HOPS interacts with Apl5 at the vacuole membrane and is required for consumption of AP-3 transport vesicles. Mol Biol Cell. 2009;20:4563–4574. doi: 10.1091/mbc.E09-04-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress JL, Acar M, Tao C, Halder G. Lethal giant discs, a novel C2-domain protein, restricts notch activation during endocytosis. Curr Biol. 2006;16:2228–2233. doi: 10.1016/j.cub.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumailleau F, Furthauer M, Knoblich JA, Gonzalez-Gaitan M. Directional Delta and Notch trafficking in Sara endosomes during asymmetric cell division. Nature. 2009;458:1051–1055. doi: 10.1038/nature07854. [DOI] [PubMed] [Google Scholar]
- Craige B, Salazar G, Faundez V. Phosphatidylinositol-4-kinase type II alpha contains an AP-3-sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell. 2008;19:1415–1426. doi: 10.1091/mbc.E07-12-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Gossen M, Freundlieb S, Bujard H, Schmid SL. Tightly regulated and inducible expression of dominant interfering dynamin mutant in stably transformed HeLa cells. Methods Enzymol. 1995;257:209–220. doi: 10.1016/s0076-6879(95)57026-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B, et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Fuwa TJ, Hori K, Sasamura T, Higgs J, Baron M, Matsuno K. The first deltex null mutant indicates tissue-specific deltex-dependent Notch signaling in Drosophila. Mol Genet Genomics. 2006;275:251–263. doi: 10.1007/s00438-005-0087-3. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Knoblich JA. The conserved c2 domain protein lethal (2) giant discs regulates protein trafficking in Drosophila. Dev Cell. 2006;11:641–653. doi: 10.1016/j.devcel.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007;14:295–300. doi: 10.1038/nsmb1227. [DOI] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- Herz HM, Chen Z, Scherr H, Lackey M, Bolduc C, Bergmann A. vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development. 2006;133:1871–1880. doi: 10.1242/dev.02356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR, Gibson A, Shipman M, Miller K. Movement of internalized ligand-receptor complexes along a continuous endosomal reticulum. Nature. 1990;346:335–339. doi: 10.1038/346335a0. [DOI] [PubMed] [Google Scholar]
- Hori K, Fostier M, Ito M, Fuwa TJ, Go MJ, Okano H, Baron M, Matsuno K. Drosophila deltex mediates suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development. 2004;131:5527–5537. doi: 10.1242/dev.01448. [DOI] [PubMed] [Google Scholar]
- Hu Y, Chuang JZ, Xu K, McGraw TG, Sung CH. SARA, a FYVE domain protein, affects Rab5-mediated endocytosis. J Cell Sci. 2002;115:4755–4763. doi: 10.1242/jcs.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006;7:408–415. doi: 10.1111/j.1600-0854.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- Kishi N, et al. Murine homologs of deltex define a novel gene family involved in vertebrate Notch signaling and neurogenesis. Int J Dev Neurosci. 2001;19:21–35. doi: 10.1016/s0736-5748(00)00071-x. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Oyama T, Kawashima T, Yedvobnick B, Kumar A, Matsuno K, Harigaya K. A human protein with sequence similarity to Drosophila mastermind coordinates the nuclear form of notch and a CSL protein to build a transcriptional activator complex on target promoters. Mol Cell Biol. 2001;21:4337–4346. doi: 10.1128/MCB.21.13.4337-4346.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J, Hille A, Veenendaal T, Oorschot V, Stoorvogel W, von Figura K, Geuze HJ. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J Cell Biol. 1993;121:997–1010. doi: 10.1083/jcb.121.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooh PJ, Fehon RG, Muskavitch MA. Implications of dynamic patterns of Delta and Notch expression for cellular interactions during Drosophila development. Development. 1993;117:493–507. doi: 10.1242/dev.117.2.493. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornilova AY, Das C, Wolfe MS. Differential effects of inhibitors on the γ-secretase complex. Mechanistic implications. J Biol Chem. 2003;278:16470–16473. doi: 10.1074/jbc.C300019200. [DOI] [PubMed] [Google Scholar]
- Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol Biol Cell. 2001;12:3489–3501. doi: 10.1091/mbc.12.11.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F, Klausner RD. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell. 1992;69:1143–1157. doi: 10.1016/0092-8674(92)90636-q. [DOI] [PubMed] [Google Scholar]
- Lonsdale JE, McDonald KL, Jones RL. High pressure freezing and freeze substitution reveal new aspects of fine structure and maintain protein antigenicity in barley aleurone cells. Plant J. 1999;17:221–229. [Google Scholar]
- Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, Quaranta V, Peterson PA. Intracellular transport of class II MHC molecules directed by invariant chain. Nature. 1990;348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Eastman D, Mitsiades T, Quinn AM, Carcanciu ML, Ordentlich P, Kadesch T, Artavanis-Tsakonas S. Human deltex is a conserved regulator of Notch signalling. Nat Genet. 1998;19:74–78. doi: 10.1038/ng0598-74. [DOI] [PubMed] [Google Scholar]
- McGill MA, Dho SE, Weinmaster G, McGlade CJ. Numb regulates post-endocytic trafficking and degradation of notch1. J Biol Chem. 2009;284:26427–26438. doi: 10.1074/jbc.M109.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Meresse S, Gorvel JP, Chavrier P. The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J Cell Sci. 1995;108:3349–3358. doi: 10.1242/jcs.108.11.3349. [DOI] [PubMed] [Google Scholar]
- Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- Nichols JT, Miyamoto A, Olsen SL, D’Souza B, Yao C, Weinmaster G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J Cell Biol. 2007;176:445–458. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004;164:1065–1076. doi: 10.1083/jcb.200311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond L, Kuhn LA, Teyton L, Schutze MP, Tainer JA, Jackson MR, Peterson PA. A role for acidic residues in di-leucine motif-based targeting to the endocytic pathway. J Biol Chem. 1995;270:19989–19997. doi: 10.1074/jbc.270.34.19989. [DOI] [PubMed] [Google Scholar]
- Pratt EB, Wentzell JS, Maxson JE, Courter L, Hazelett D, Christian JL. The cell giveth and the cell taketh away: an overview of Notch pathway activation by endocytic trafficking of ligands and receptors. Acta Histochem. 2010;113:248–255. doi: 10.1016/j.acthis.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch U, Bernhard O, Koszinowski U, Schu P. AP-1A and AP-3A lysosomal sorting functions. Traffic. 2002;3:752–761. doi: 10.1034/j.1600-0854.2002.31007.x. [DOI] [PubMed] [Google Scholar]
- Rodionov DG, Honing S, Silye A, Kongsvik TL, von Figura K, Bakke O. Structural requirements for interactions between leucine-sorting signals and clathrin-associated adaptor protein complex AP3. J Biol Chem. 2002;277:47436–47443. doi: 10.1074/jbc.M207149200. [DOI] [PubMed] [Google Scholar]
- Salazar G, Zlatic S, Craige B, Peden AA, Pohl J, Faundez V. Hermansky-Pudlak syndrome protein complexes associate with phosphatidylinositol 4-kinase type II α in neuronal and non-neuronal cells. J Biol Chem. 2009;284:1790–1802. doi: 10.1074/jbc.M805991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T. A modified method for lead staining of thin sections. J Electron Microsc (Tokyo) 1968;17:158–159. [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Shaye DD, Greenwald I. Endocytosis-mediated downregulation of LIN-12/Notch upon Ras activation in Caenorhabditis elegans. Nature. 2002;420:686–690. doi: 10.1038/nature01234. [DOI] [PubMed] [Google Scholar]
- Shaye DD, Greenwald I. LIN-12/Notch trafficking and regulation of DSL ligand activity during vulval induction in Caenorhabditis elegans. Development. 2005;132:5081–5092. doi: 10.1242/dev.02076. [DOI] [PubMed] [Google Scholar]
- Sitaram A, et al. Differential recognition of a dileucine-based sorting signal by AP-1 and AP-3 reveals a requirement for both BLOC-1 and AP-3 in delivery of OCA2 to melanosomes. Mol Biol Cell. 2012;23:3178–3192. doi: 10.1091/mbc.E11-06-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen EB, Conner SD. γ secretase-dependent cleavage initiates Notch signaling from the plasma membrane. Traffic. 2010;11:1234–1245. doi: 10.1111/j.1600-0854.2010.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000;6:625–636. doi: 10.1016/s1097-2765(00)00061-7. [DOI] [PubMed] [Google Scholar]
- Tagami S, et al. Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol Cell Biol. 2008;28:165–176. doi: 10.1128/MCB.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621–629. doi: 10.1083/jcb.200811141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Jung JJ, Inamdar SM, Brown CO, Goel A, Choudhury A. Endothelial cell migration on fibronectin is regulated by syntaxin 6-mediated α5β1 integrin recycling. J Biol Chem. 2011;286:36749–36761. doi: 10.1074/jbc.M111.260828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M. Tubular early endosomal networks in AtT20 and other cells. J Cell Biol. 1991;115:635–653. doi: 10.1083/jcb.115.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell. 2005;9:687–698. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Vaccari T, Lu H, Kanwar R, Fortini ME, Bilder D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J Cell Biol. 2008;180:755–762. doi: 10.1083/jcb.200708127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam EM, Stoorvogel W. Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol Biol Cell. 2002;13:169–182. doi: 10.1091/mbc.01-07-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. The metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284:31018–31027. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin M, et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev Cell. 2008;15:762–772. doi: 10.1016/j.devcel.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Windler SL, Bilder D. Endocytic internalization routes required for Delta/Notch signaling. Curr Biol. 2010;20:538–543. doi: 10.1016/j.cub.2010.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fuwa TJ, Ayukawa T, Tanaka T, Nakamura A, Wilkin MB, Baron M, Matsuno K. Roles of Drosophila deltex in Notch receptor endocytic trafficking and activation. Genes Cells. 2011;16:261–272. doi: 10.1111/j.1365-2443.2011.01488.x. [DOI] [PubMed] [Google Scholar]
- Zlatic SA, Tornieri K, L’hernault SW, Faundez V. Clathrin-dependent mechanisms modulate the subcellular distribution of class C Vps/HOPS tether subunits in polarized and non-polarized cells. Mol Biol Cell. 2011a;22:1699–1715. doi: 10.1091/mbc.E10-10-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic SA, Tornieri K, L’hernault SW, Faundez V. Metazoan cell biology of the HOPS tethering complex. Cell Logist. 2011b;1:111–117. doi: 10.4161/cl.1.3.17279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.