

Abstract
The efficient synthesis of an Fmoc-Gly-Ile phosphinic pseudodipeptide was desired as an eventual building block for construction of matrix metalloproteinase inhibitors. A Michael-type addition reaction of bis(tri-methylsilyl) phosphonite with the appropriate acrylate generated the pseudodipeptide bond. Additional of adamantyl (Ad) protection by our prior route (reaction of in situ generated phosphinic acid chloride with the sodium salt of adamantanol) was surprisingly inefficient. Adamantyl protection was achieved in high yield by refluxing the phosphinic acid, Ag2O, and 1-AdBr in chloroform. Subsequently a concise one-pot three-step reaction comprising a double deprotection of the N- and C-termini under catalytic hydrogenation conditions followed by selective protection of the N-terminus with an Fmoc group yielded Fmoc-NHCH2PO(OAd)CH2CH(2-butyl)CO2H in 41 % overall yield. These results indicate that, as the diversity of phosphinic pseudodipeptides is increased to create selective matrix metalloproteinase inhibitors, different synthetic pathways may be required for efficient building block preparation.
Keywords: Matrix metalloproteinase, Peptidomimetic, Phosphinate, Protease, Protease inhibitor, Transition state analog
Introduction
Collagen catabolism (collagenolysis) is an important physiological process essential to tissue and organ development, morphogenesis, and wound healing (Song et al. 2006). However, abnormal collagen degradation may result in severe pathological conditions such as primary and meta-static tumor growth, arthritis, arteriosclerosis, and periodontitis (Egeblad and Werb 2002; Fingleton 2007; Overall and Kleifeld 2006). Amongst the different protease families, members of the matrix metalloproteinases (MMPs) have been described as exhibiting efficient collagenolytic activity (Lauer-Fields et al. 2002; Overall and Kleifeld 2006).
A well-established strategy to develop a protease inhibitor is the backbone modification of a peptide substrate (Lauer-Fields et al. 2007, 2008). This is often achieved by replacing a hydrolytically susceptible trigonal planar amide or ester bond within the peptide sequence with a hydrolytically-stable functional group possessing tetrahedral geometry to mimic the intermediate formed during enzymatic hydrolysis (Schramm 2011; Wolfenden 1976). The use of phosphinic pseudodipeptides (PPDs) is a very effective approach to develop highly selective and potent inhibitors of a variety of Zn2+ metalloproteases, as PPDs contain a hydrolytically stable tetrahedral phosphinate functional group with a pair of electronically equivalent anionic oxygens which mimics the tetrahedral intermediate formed during enzyme catalyzed hydrolysis (Buchardt et al. 1999, 2000; Lauer-Fields et al. 2007, 2008; Vassiliou et al. 1999; Yiotakis et al. 1996). Our laboratory has utilized PPDs to create triple-helical peptide inhibitors for collagenolytic MMPs (Lauer-Fields et al. 2007, 2008, 2009).
Phosphinic pseudodipeptides are of growing interest as a building block in medicinal chemistry approaches, but their practical application in peptide modification is limited due to lack of accessibility. Several groups (Buchardt et al. 1999; Yiotakis et al. 1996, and Yamagishi et al. 2008) have reported the synthesis of different phosphinate dipeptide building blocks. However, applying these previously reported procedures for the synthesis of Fmoc-NHCH2PO(OAd)-CH2 CH(Pri)CO2H 1a (Fig. 1) required a low yielding (35 %) and problematic final step involving a Ru-catalyzed deprotection of an allyl ester (Lauer-Fields et al. 2007). We recently reported an efficient PPD synthetic method by developing a one-pot reaction procedure utilizing a single step bis-deprotection followed by Fmoc-protection leading to the phosphinate dipeptide 1a and its Gly-Leu analog 1b (Fig. 1) (Bhowmick et al. 2011). Surprisingly, synthesis of the Gly-Ile analog (1c, Fig. 1) utilizing the same methodology did not provide the desired product. Herein we report the synthesis of PPD Gly-Ile analog 1c via α′-substitution dependent adamantyl (Ad) protection followed by our previously reported bis-deprotection strategy, and discuss the implication of these results for assembly of diverse PPDs.
Fig. 1.
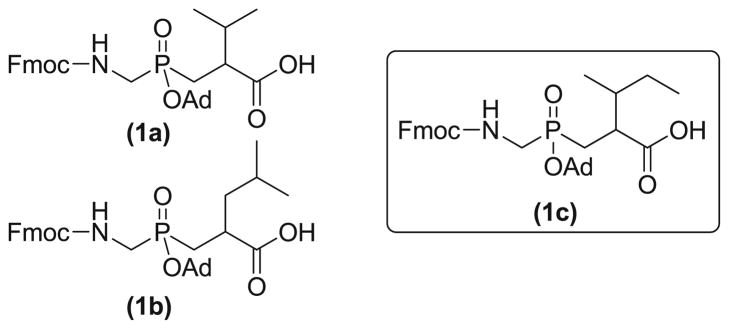
Structures of PPDs. The pseudodipeptides are analogs of Gly-Val (1a), Gly-Leu (1b), and Gly-Ile (1c)
Materials and Methods
Materials
All the reactions were carried out under an atmosphere of nitrogen or argon in oven-dried glassware with magnetic stirring. Purification of reaction products was carried out by flash column chromatography using Flash Silica gel (40–63 μm). Analytical thin layer chromatography was performed on 0.25 mm precoated (silica gel 60 F-254) plates. TLC visualization was accomplished with UV light (λ = 254 nm) or aqueous potassium permanganate solution staining followed by air heating.
1H NMR spectra were recorded on a Bruker 400 (400 MHz) spectrometer and chemical shifts are reported in parts per million (ppm, δ) relative to CHCl3 (δ = 7.26) in CDCl3. Data for 1H NMR are reported as follows: multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), integration and coupling constant (Hz). 13C NMR spectra were recorded on a Bruker 400 (100 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to the central CDCl3 resonance (δ = 77.0). Multiplicities in 13C NMR spectra due to phosphorous coupling are reported as d = doublet, PC = phosphorous coupling.
Methods
General Procedure for Michael-type Addition (Bhowmick et al. 2011)
In a dry round-bottom flask, a mixture of 2 (Scheme 1) (1 equiv) and lithium hexamethyldisilazide (HMDS) (5 equiv) was refluxed neat at 110–120 °C under argon atmosphere. After 2 h the temperature was lowered to 90–95 °C and a racemic mixture of acrylate 3 (Lee et al. 2003) (Scheme 1) (1.2 equiv) was added to the reaction mixture within approximately 30 min. Refluxing was continued for 4 h at 95 °C. The temperature was lowered to 70 °C and absolute EtOH (3 mL/mmol of 2) was added dropwise (to avoid vigorous reaction). The reaction mixture was cooled to room temperature and evaporated in vacuo to afford a gummy compound. The crude mixture was redissolved in EtOAc and washed with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with another portion of EtOAc. The combined EtOAc extracts were washed with water and brine and dried over Na2SO4. The solvent was removed under reduced pressure to provide a gummy crude product. The crude product was washed with dry hexane (to remove the unreacted acrylate) to produce the diastereoisomeric mixture of P-(benzyloxycarbonylaminomethyl)-P-{benzyl-2-(2-butyl)propionate-3-yl}-phosphinic acid (4, Scheme 1) as a white solid, which was then dried in high vacuo and found to be sufficiently pure for the next step. 1H NMR (400 MHz, CDCl3, mixture of diastereomers): δ 0.75–0.95 (m, 6H), 1.00–1.20 (m, 1H), 1.20–1.45 (m, 1H), 1.50–2.00 (m, 2H), 2.10–2.45 (m, 1H), 2.70–2.95 (m, 1H), 3.40–3.75 (m, 2H), 5.00–5.25 (m, 4H), 5.90 (brs, 1H), 7.20–7.40 (m, 10H), 9.60 (brs, 1H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers): δ 11.6, 11.7, 15.4, 16.0, 26.1, 26.4, 26.8, 26.99, 37.85 (d, J = 12.5 Hz, PC), 38.5 (d, J = 12.5 Hz, PC), 39.5 (d, J = 5.0 Hz, PC), 40.5 (d, J = 6.0 Hz, PC), 43.5 (d, J = 3.2 Hz, PC), 43.7 (d, J = 3.4 Hz, PC), 66.7, 66.8, 67.2, 128.1 (2C), 128.13, 128.2, 128.3 (2C), 128.4 (2C), 128.5 (2C), 135.8, 136.3, 156.6, 174.1, 174.6.
Scheme 1.
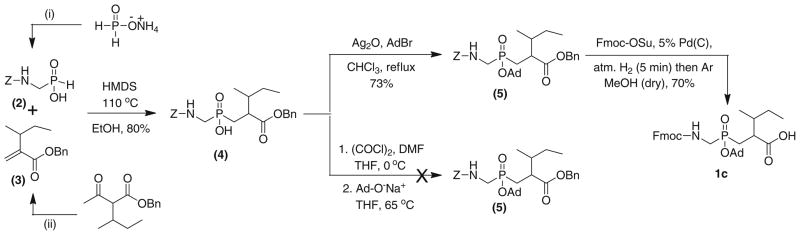
Synthesis of Fmoc-protected PPD 1c. The synthesis of 2 via (i) is as described previously (Li et al. 2007), as is the synthesis of 3 via (ii) (Lee et al. 2003)
Procedure for Adamantyl Protection (Buchardt et al. 1999)
A mixture of 4 (1.0 g, 2.236 mmol) and Ag2O (1.032 g, 4.472 mmol) was refluxed in dry chloroform (8 mL) under argon atmosphere. After 30 min a solution of AdBr (0.528 g, 2.46 mmol) in 8 mL dry chloroform was added dropwise to the refluxing reaction mixture over a 45 min period. Refluxing was continued for another 2 h and the mixture was allowed to cool at room temperature. After overnight stirring, the crude reaction mixture was filtered through Celite pad, concentrated, and purified by flash chromatography using 30 % EtOAc in toluene as eluent to provide pure diastereoisomeric mixture of O-adamantyl-P-(benzyloxycarbonylaminomethyl)-P-{benzyl-2-(2-butyl)propionate-3-yl}-phosphinate (5, Scheme 1) (0.944 g) in 73 % yield as a white solid. 1H NMR (400 MHz, CDCl3, mixture of diastereomers): δ 0.85–0.95 (m, 6H), 1.0–1.25 (m, 1H), 1.25–1.45 (m, 1H), 1.45–1.62 (m, 6H), 1.62–1.90 (m, 2H), 2.00 (brs, 3H), 2.05 (brs, 3H), 2.13 (brs, 3H), 2.20–2.45 (m, 1H), 2.75–2.95 (m, 1H), 3.25–3.55 (m, 1H), 3.55–3.75 (m, 1H), 4.95–5.25 (m, 4H), 5.25–5.45 (m, 1H), 7.25–7.45 (m, 10H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers): δ 11.6, 11.77, 15.49, 16.0, 26.55 (d, J = 6.9 Hz, PC), 26.88 (d, J = 4.0 Hz, PC), 31.1 and 31.2 (3C each), 35.6 and 35.7 (3C each), 38.0, 38.8, 43.5, 43.7, 44.3 (3C), 66.6, 67.1, 83.3, 83.4 (d, J = 9.4 Hz, PC), 128.1, 128.2, 128.38, 128.4, 128.44, 128.5 (3C), 135.8, 136.4, 156.3, 174.1, 174.57.
Procedure for Tandem Bis-deprotection and Fmoc-amine Formation (Bhowmick et al. 2011)
In a 50 mL round-bottom flask 0.400 g (0.688 mmol) of 5 and 0.278 g (0.826 mmol) of Fmoc-OSu were dissolved in 35 mL of dry MeOH under Ar atmosphere. 0.145 g (0.0688 mmol) of 5 % Pd on activated charcoal was added at room temperature and hydrogen gas was bubbled through the reaction mixture. After approximately 5 min (when the starting material was fully consumed, as confirmed by TLC), the hydrogen gas source was removed and argon was bubbled through the reaction mixture for approximately 15 min (to ensure that all H2 was removed from the reaction vessel). The reaction mixture was stirred for an additional 3 h. The catalyst was removed by filtration through a bed of Celite and washed with another 50 mL of MeOH. The solvent was removed under reduced pressure to afford the gummy crude reaction mixture. Flash column chromatography using 3 % MeOH in CHCl3 as eluent provided 0.279 g (70 %) of the diastereoisomeric mixture of the pure compound O-adamantyl-P-(9-fluorenylmethyloxycarbonyl-aminomethyl)-P-{2-(2-butyl)propionic acid-3-yl}-phosphinate (1c) as a white solid. 1H NMR (400 MHz, CDCl3, mixture of diastereomers): δ 0.80–1.05 (m, 6H), 1.10–1.48 (m, 2H), 1.48–1.64 (m, 6H), 1.64–1.78 (m, 1H), 1.78–1.94 (m, 1H), 1.94–2.25 (m, 9H), 2.25–2.60 (m, 1H), 2.60–3.00 (m, 1H), 3.45–3.68 (m, 1H), 3.68–3.95 (m, 1H), 4.10–4.55 (m, 3H), 7.28–7.36 (m, 2H), 7.36–7.5 (m, 2H), 7.60–7.74 (m, 2H), 7.74–7.82 (m, 2H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers): δ 11.7, 11.9, 16.1, 16.5, 26.2 (d, J = 25.4 Hz, PC), 27.22 (d, J = 30.5 Hz, PC), 31.1 and 31.2 (3C each), 35.6 (3C), 37.7, 38.4, 44.1, 44.3 (d, J = 3.1 Hz, PC) and 44.4 (d, J = 3.4 Hz, PC, 3C each), 47.1, 67.5, 83.7 (d, J = 10.3 Hz, PC) and 84.3 (d, J = 9.9 Hz, PC), 119.8 and 119.9 (2C each), 125.4 and 125.7 (2C each), 127.0 and 127.1 (2C each), 127.6 and 127.7 (2C each), 141.2 and 141.3 (2C each), 143.9 and 144.2 (2C each), 156.7 (d, J = 6.7 Hz, PC) and 157.0 (d, J = 5.6 Hz, PC), 176.5 and 176.9.
Results and Discussion
Our general strategy for the synthesis of 1c (Fig. 1) involved a Michael-type addition reaction for the formation of a new phosphorous-carbon bond followed by Ad protection and finally Pd(C)/H2 reduction (Scheme 1) (Bhowmick et al. 2011). We successfully synthesized Cbz-protected aminomethyl phosphinic acid 2 starting from commercially available ammonium hypophosphite using previously described literature procedures (Bhowmick et al. 2011; Li et al. 2007). Z-protected aminomethyl phosphinic acid 2 was then treated with HMDS and converted to its active trivalent state (Georgiadis et al. 2001). Michael-type addition of activated 2 with an α-(2-butyl)-α,β-unsaturated ester 3 afforded Z-protected phosphinic pseudodipeptide benzyl ester 4 in 80 % yield. The acrylate 3 was prepared from commercially available benzyl acetoacetate ester by following a previously reported procedure (Lee et al. 2003).
Adamantyl protection of 4, applying the procedure of Buchardt et al. (1999) subsequently used for our prior building blocks (Bhowmick et al. 2011), in which in situ generated phosphinic acid chloride is reacted with the sodium salt of adamantanol, failed to provide protected dipeptide 5 (Scheme 1). It was possible that a steric effect of the bulky 2-(2-butyl) and benzyl ester groups prevented the nucleophilic attack of the anionic oxygen of adamantanol to form the Ad ester. The esterification was achieved by treating 4 with Ag2O and 1-AdBr under refluxing conditions in chloroform in an inert atmosphere (Buchardt et al. 1999; Georgiadis et al. 2001). We believe that, in the presence of Ag2O, the Ag(I) chelates the double-bonded phosphorous oxygen and the N-terminal carbonyl oxygen (from the Fmoc group). This chelation results in the bulky 2-(2-butyl) and benzyl ester groups being sterically distanced from the phosphorus OH group. The oxygen lone pair from OH can now easily attack the adamantane carbocation formed in the reaction between Ag2O and 1-AdBr. This alternate esterification procedure provided a fully protected PPD 5 in very good yield (73 %). Finally, catalytic hydrogenation of 5 with 5 % Pd/C under hydrogen (atmospheric pressure) in the presence of Fmoc-OSu afforded the desired pseudodipeptide 1c as a 1:1 mixture of diastereomers in 70 % yield (Scheme 1). Although a diastereomeric building block was produced, MMPs are often well inhibited by peptides containing a mixture of PPD diastereomers (Devel et al. 2006; Gall et al. 2001; Lauer-Fields et al. 2008, 2009). Once effective inhibitors are initially identified, diastereomeric peptides can then be separated and studied (Lauer-Fields et al. 2007). Interestingly, depending upon the MMP and peptide, the nature of the diastereomer may (Buchardt et al. 1999; Makaritis et al. 2003) or may not (Gall et al. 2001; Lauer-Fields et al. 2007) have a significant effect on inhibitory activity.
Conclusion
We have successfully developed an easy and scalable method for the synthesis of Fmoc-protected Gly-Ile PPD (Fig. 1, 1c) in 41 % overall yield. This modified synthetic route consists of an alternate Ad protection strategy compared with Gly-Val and Gly-Leu PPDs (Fig. 1, 1a and 1b), followed by a concise protecting group manipulation procedure. As diversity in both the substrate P1 and subsites can be exploited for selectivity of MMP inhibitors (Rao 2005; Verma and Hansch 2007), the synthesis of diverse PPDs is highly desirable. The present study indicates that even subtle alterations in side chain composition (such as changing from Leu to Ile in the subsite) can have significant effects on the efficiency of PPD synthetic pathways. Thus, effective large-scale preparation of PPD building blocks, allowing the synthesis of a variety of transition state analog triple-helical MMP inhibitors for in vitro and in vivo testing, requires careful exploration of experimental conditions.
Supplementary Material
Acknowledgments
This work was supported by NIH grant CA98799, NIH contract 268201000036C, and the Multiple Sclerosis National Research Institute (to GBF). We thank Dr. Deboprosad Mondal, The Scripps Research Institute/Scripps Florida, Jupiter, FL and The University of Texas Health Science Center at San Antonio, TX for the acquisition of NMR spectra.
Abbreviations
- Ad
Adamantyl
- Bn
Benzyl
- Z
N-Benzyloxycarbonyl
- Fmoc
N-(9-Fluorenyl)methoxycarbonyl
- HMDS
Lithium hexamethyldisilazide
- MMP
Matrix metalloproteinase
- PPD
Phosphinic pseudodipeptide
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10989-012-9307-y) contains supplementary material, which is available to authorized users.
References
- Bhowmick M, Sappidi RR, Fields GB, Lepore SD. Efficient synthesis of Fmoc-protected phosphinic pseuedodipeptides: building blocks for the synthesis of matrix metalloproteinase inhibitors (MMPIs) Biopolymers (Pept Sci) 2011;96:1–3. doi: 10.1002/bip.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchardt J, Ferreras M, Krog-Jensen C, Delaisse J-M, Foged NT, Meldal M. Phosphinic peptide matrix metalloproteinase-9 inhibitors by solid-phase synthesis using a building block approach. Chem Eur J. 1999;5:2877–2884. [Google Scholar]
- Buchardt J, Schiodt CB, Krog-Jensen C, Delaissé J-M, Foged NT, Meldal M. Solid phase combinatorial library of phosphinic peptides for discovery of matrix metalloproteinase inhibitors. J Comb Chem. 2000;2:624–638. doi: 10.1021/cc000031q. [DOI] [PubMed] [Google Scholar]
- Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr Pharm Des. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- Gall AL, Ruff M, Kannan R, Cuniasse P, Yiotakis A, Dive V, Rio MC, Basset P, Moras D. Crystal structure of the stromelysin-3 (MMP-11) catalytic domain complexed with a phosphinic inhibitor mimicking the transition-state. J Mol Biol. 2001;307:577–586. doi: 10.1006/jmbi.2001.4493. [DOI] [PubMed] [Google Scholar]
- Georgiadis D, Matziari M, Yiotakis A. A highly efficient method for the preparation of phosphinic pseudodipeptidic blocks suitably protected for solid-phase peptide synthesis. Tetrahedron. 2001;57:3471–3478. [Google Scholar]
- Lauer-Fields JL, Juska D, Fields GB. Matrix metalloproteinases and collagen catabolism. Biopolymers (Pept Sci) 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]
- Lauer-Fields JL, Brew K, Whitehead JK, Li S, Hammer RP, Fields GB. Triple-helical transition-state analogs: a new class of selective matrix metalloproteinase inhibitors. J Am Chem Soc. 2007;129:10408–10417. doi: 10.1021/ja0715849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J Biol Chem. 2008;283:20087–20095. doi: 10.1074/jbc.M801438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer-Fields JL, Chalmers MJ, Busby SA, Minond D, Griffin PR, Fields GB. Identification of specific hemopexin-like domain residues that facilitate matrix metalloproteinase collagenolytic activity. J Biol Chem. 2009;284:24017–24024. doi: 10.1074/jbc.M109.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-S, Park J-S, Kim BM, Gellman SH. Efficient synthesis of enantiomerically pure β2-amino acids via chiral isoxazolidinones. J Org Chem. 2003;68:1575–1578. doi: 10.1021/jo026738b. [DOI] [PubMed] [Google Scholar]
- Li S, Whitehead JK, Hammer RP. Application of in situ silylation for improved, convenient preparation of fluorenylmethoxycarbonyl (Fmoc)-protected phosphinate amino acids. J Org Chem. 2007;72:3116–3118. doi: 10.1021/jo070266p. [DOI] [PubMed] [Google Scholar]
- Makaritis A, Georgiadis D, Dive V, Yiotakis A. Diastereoselective solution and multipin-based combinatorial array synthesis of a novel class of potent phosphinic metalloprotease inhibitors. Chem Eur J. 2003;9:2079–2094. doi: 10.1002/chem.200204456. [DOI] [PubMed] [Google Scholar]
- Overall CM, Kleifeld O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- Rao BG. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr Pharm Des. 2005;11:295–322. doi: 10.2174/1381612053382115. [DOI] [PubMed] [Google Scholar]
- Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Wisithphrom K, Zhou J, Windsor LJ. Matrix metalloproteinase dependent and independent collagen degradation. Front Biosci. 2006;11:3100–3120. doi: 10.2741/2036. [DOI] [PubMed] [Google Scholar]
- Vassiliou S, Mucha A, Cuniasse P, Georgiadis D, Lucet-Levannier K, Beau F, Kannan R, Murphy G, Knauper V, Rio MC, et al. Phosphinic pseudotripeptides as potent inhibitors of matrix metalloproteinases: a structure-activity study. J Med Chem. 1999;42:2610–2620. doi: 10.1021/jm9900164. [DOI] [PubMed] [Google Scholar]
- Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007;15:2223–2268. doi: 10.1016/j.bmc.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Wolfenden R. Transition state analog inhibitors and enzyme catalysis. Annu Rev Biophys Bioeng. 1976;5:271–306. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, Ichikawa H, Haruki T, Yokomatsu T. Org Lett. 2008;10:4347–4350. doi: 10.1021/ol801743d. [DOI] [PubMed] [Google Scholar]
- Yiotakis A, Vassiliou S, Jiracek J, Dive V. Protection of the hydroxyphosphinyl function of phosphinic dipeptides by adamantyl: application to the solid-phase synthesis of phosphinic peptides. J Org Chem. 1996;61:6601–6605. doi: 10.1021/jo9603439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.