

Abstract
Bacterial toxins damage the host at the site of bacterial infection or distanced from the site of infections. Bacterial toxins can be single proteins or organized as oligomeric protein complexes and are organized with distinct AB structure-function properties. The A domain encodes a catalytic activity; ADP-ribosylation of host proteins is the earliest post-translational modification determine to be performed by bacterial toxin, and now include glucosylation and proteolysis among other s. Bacterial toxins also catalyze the non-covalent modification of host protein function or can modify host cell properties through direct protein-protein interactions. The B domain includes two functional domains: a receptor-binding domain, which defines the tropism of a toxin for a cell and a translocation domain that delivers A domain across a lipid bilayer, either on the plasma membrane or the endosome. Bacterial toxins are often characterized based upon the section mechanism that delivers the toxin out of the bacterium, termed type I–VII. This review will overview the major families of bacterial toxins and will also describe the specific structure-function properties of the botulinum neurotoxins.
Keywords: Toxins, ADP-ribosylation, post-translational modification, secretion systems, diphtheria toxin, cholera toxin, superantigens, botulinum neurotoxins
Introduction
Bacterial pathogens damage the host via invasive and toxic attributes. Invasion involves the ability of the bacterium to grow in the host, either in an extracellular environment or intracellular environment. Invasive bacteria injure the host through the production of extracellular enzymes that damage host tissue, or through the modulation of the host response system such as the up regulation of cytokine expression. In contrast, bacterial pathogens that damage the host through the action of toxins are often non-invasive and have limited capacity to disseminate in the host. Bacterial toxins often act distanced from the site of infection and are physically organized into distinct domains that recognize receptors on the surface of sensitive cells and possess enzymatic capacity to modulate the action of an intracellular host target, often a protein, termed A–B structure-function organization (Figure 1). The A domain, also described as an effector, is usually an enzyme or a factor that functions through protein-protein interactions within the cell. The B domain comprises the receptor-binding function, providing tropism to specific cell types through receptor binding capacity. The B domain also includes a domain that translocates the A domain across a lipid bilayer, either at the plasma membrane or within the endosomal compartment. Translocation of the A domain across the lipid bilayer is hypothesized in most cases to occur through a pore/channel formed by the B domain. The B domain can be a single subunit (B) or an oligomeric (B5) form. The A and B domains may be linked by a disulfide bond or associated by non-covalent interactions. This review will present an overview of the properties of the major families of bacterial toxin and then describe in more detail the structure-function properties of the botulinum neurotoxins.
Figure 1. AB organization of bacterial toxins.

Diphtheria toxin is an AB toxin where the N terminal A domain (black) encodes an Enzyme activity, an ADP-ribosyl trasferase activity, NAD + EF-2 → ADP-r-EF-2 + nicotinamide + H+. The C-terminal B domain encodes a receptor binding function (grey) that binds to a grwoth factor receptor and enters cells through receptor-mediated endocytosis and a translocation function (white) which undergoes a pH-dependent conformation a change where chaafed amino acids are protonated which allows a pair of hydrophobic alpha helices to insert into the endosome membrane which is responsible for the delivery of A domain in to the host cytosol. Structure: PDB 1fol
Heat Stable Enterotoxins
Heat-stable enterotoxins (ST) are a family of conserved peptides expressed by pathogenic strains of Escherichia coli. ST elicits fluid accumulation in the intestine [1, 2], which is often responsible for diarrhea in travelers, young children, and domesticated animals in developing countries. ST includes two subfamilies, STa and STb.
The STa subfamily that intoxicate humans (STah) and porcine (STap) [3, 4] comprise s~18 amino acid peptides, which includes an N-terminal α-helix, a type I β-turn in the central region, and a type II β-turn at the C terminus of the peptide [5]. STa binds the guanylate cyclase C (GC-C) receptor on the surface of epithelial cells of the small intestine and colon. STa binding to the GC-C receptor mimics guanylin, a protein ligand of GC-C, where residues between Cys5 and Cys17(termed the central region) of STa are essential for binding. These residues are conserved within the STa subfamily, and comprise three disulfide bonds within the central region that are necessary for functional binding [6]. The central β-turn (Asn11-Cys14) interacts with three residues (T389, F390, W392) of the extra-cellular domain of the GC-C receptor [7–9]. Peptides comprising residues 5-17 and 6-18 possess equivalent biological and immunological activities as native STa, showing that the central region is necessary and sufficient to elicit toxicity [10, 11]. STa-bound GC-C activates protein kinase G (PKG) and protein kinase C (PKC), increasing IP3-mediated calcium to elevate intracellular cyclic GMP (cGMP)[12]. Increased cGMP activates phosphorylation of the Cl− ion channel, cystic fibrosis transmembrane regulator, increasing the concentration of Cl− ions in the extracellular space and causing fluid accumulation within the lumen of the intestine [3, 13].
STb is a 48 amino acid peptide which causes diarrhea in porcine and humans [14, 15]. Despite the similarity of nomenclature, STb does not have primary amino acid homology with STa. STb is composed of two anti-parallel α-helices connected by a glycine-rich loop which is stabilized by two disulfide bonds that are necessary for cellular intoxication [16–18]. STb binds to sulfatide, a glycosphingolipid on epithelial cells [19] and internalization is required for intoxication. While the molecular mechanism remains to be determined, STb does not cause cGMP elevation as observed with STa [20]. STb stimulates fluid accumulation through a pertussis toxin-sensitive GTP-binding regulatory protein, which has been proposed to stimulate the elevation of intracellular calcium through an unidentified ligand-gated Ca2+ channel [21]. Recent studies observed that STb forms multimeric complexes and enhance membrane permeability, implicating a pore-forming toxin-like activity [14, 17].
While details of the structure-function properties of the ST were resolved shortly after the identification of this family of toxins, recent studies address how STs contribute to the pathology of the enterotoxgenic E. coli (ETEC). For example, Moxley and co-workers utilized isogenic strains of ETEC to address the role of STb and other virulence factors in gnotobiotic piglets [22], while Lucas et al,[23] recently addressed the role of STa in the elicitation of distanced effects on fluid absorption in the intestine of rats.
Pore forming toxins
Bacterial pore-forming toxins (PFT) are a large group of protein toxins which forms pores in the membranes of bacteria, plants, and mammals, causing membrane permeability and ion imbalance. Bacteria release PFTs as soluble subunits which form stable multimeric complexes on the membranes of various target membranes, and translocate across lipid membranes through several mechanisms. PFTs are classified into two groups based on the multimeric structure involved in membrane insertion. α-PFTs describe those PFTs which insert into membranes as an α-helix, while β-PFTs insert into the membrane as β-sheets [24, 25].
α-PFTs are represented by colicins that are produced in E. coli and share structural organization with diphtheria toxin. Colicins are cytotoxic for E. coli and other closely related species. Colicins encode α-helices that are utilized for translocation of the catalytic domains across a bacterial outer membrane, the bacterial outer membrane. The proposed mechanism of membrane insertion and translocation of the colicin catalytic domain utilized early biochemical studies, the crystal structure of colicin E (ColE) bound to the BtuR receptor [26], and the crystal structure of OmpR, the putative pore utilized for translocation of the catalytic domain across the outer membrane [27]. ColE3 is 551 amino acids with an internal receptor binding domain, an N-terminal translocation domain, and a C-terminal ~96 amino acids that function as an endoribonuclease. Entry of ColE into the bacterial cell involves the binding of the internal receptor binding domain to the But R protein, which concentrates into the outer membrane to coordinate the subsequent binding of the translocation domain into the translocator, OmpF. Translocation involves the association of residues within an α-helix of the translocation domain with internal regions of OmpF [27] with the subsequent movement of the nicked catalytic domain across the bacterial inner membrane via a TonB dependent-mechanism.
While not considered a pore forming toxin, the delivery mechanism that diphtheria toxin utilizes to translocate the catalytic subunit across the endosome membrane and into the host cytosol has properties that are analogous to the α-PFTs. Diphtheria toxin is a single chain protein that elicts a lethal phenotype in humans through the ADP-ribosylation of elongation factor-2. Diphtheria toxin is a 535-amino acid AB toxin with the N terminus encoding the ADP-ribosyltrasferase activity and the C terminus comprises a translocation domain and a C-terminal receptor binding domain. Diphtheria toxin binds to a growth factor receptor to traffic into early endosomes via receptor-mediated endocytosis where hydrophobic α-helixes of the translocation domain insert into the endosomal membrane by a pH-dependent mechanism [28].
Insertion of these helices into bilayers opens a channel, analogous to the channel formed by the α-PFTs that facilitates the translocation of an extended form of the N-terminal catalytic domain across the membrane. The catalytic domain refolds within the cytosol and ADP-ribosylates elongation factor-2, which inhibits protein synthesis. Recent studies have implicated a role for host chaperones in the A domain translocation event [29]. The crystal structure of native diphtheria toxin, along with biophysical studies, provide a model describing the molecular basis for the translocation of the catalytic domain of diphtheria toxin across the endosome membrane where hydrophilic loops, containing several charged amino acids with pH sensitive ionizable R-groups, stabilize hydrophobic α-helices within the B domain of diphtheria toxin (Figure 1).
β-PFTs are produced as soluble proteins which oligomerize into multimeric complexes on the mammalian plasma membrane, where one or two amphipathic β-hairpins on a monomeric subunit contribute to the organization of the pore [30]. These β-hairpins contain a hydrophobic outer surface which favors insertion into the membrane [31, 32]. The pores are organized in a variety of subunit numbers (7–50) and sizes (2nm–50nm) [33–35]. The largest group of β-PFTs is the cholesterol-dependent cytolysins (CDC), which are primarily produced by Gram-positive, pathogenic bacteria such as Lysteria sp., Streptococcus pneumoniae, and Bacillus anthracis, but are also found in the mammalian immune system [36–38]. CDC pore complex formation occurs before protein insertion into the membrane and is pH-independent [39]. The mechanism of pore insertion includes steps that involve soluble monomer association with the plasma membrane, lateral diffusion of monomers with the membrane, pre-pore monomer oligomerization, pore formation, and insertion of the oligomer into the membrane [39]. CDC monomers are organized into four distinct domains, (termed D1–D4), which control monomer-receptor binding, monomer to oligomer association, and membrane interactions. For most CDCs, the D4 domain is responsible for interactions with cholesterol through a tryptophan-rich area called the undecapeptide, which inserts a short distance into the membrane. Cholesterol-bound monomers concentrate and then associate through lateral diffusion on the membrane surface. A conformational change in D3, triggered by D4 domain-membrane interaction, exposes the β-strand 4 of D3 domain and hydrogen bonding between β-strand 4 of D3 domain with the β-strand 1 of D3 domain in the adjacent monomer initiate pre-pore formation [40–43]. The D3 domain also undergoes a second conformational change that causes rearrangement of three α-helices into two β-hairpins for each monomer, which represents the transmembrane component of the pore. These β-hairpins stabilize interactions with neighboring β-hairpins through π-bond stacking with tyrosine and phenylalanine residues. Once the pre-pore complex has assembled on the membrane, a conformational change occurs in which the complex inserts into the membrane due to β-barrel pore formation.
Superantigens
Superantigens (SAgs) are a group of secreted protein toxins produced by an increasing numbers of bacteria, including Staphylococcus aureus, Streptococcus sp., Mycoplasma arithiditis, and Yersinia pseudotuberculosis [44, 45]. SAgs bind major histocompatibility complex II (MHC II) and stimulate peptide-independent MHC II/T cell receptor (TCR) interaction and immune activation. SAgs is responsible for Toxic Shock Syndrome (TSS) and food poisoning [46]. A second group of Sags are the superantigen-like toxins (SSL), which possess much of the conserved domain structure of the SAgs, but do not bind MHC II or activate T cells, but do target the innate immune system [47]. SAgs and SSLs contain two conserved structures at the N-terminal and C-terminal domains. The N-terminal domain contains a conserved oligonucleotide-OB fold, whereas the C-terminal domain contains a β grasp-fold. The OB-fold and the β-grasp-fold are closely packed, which contributes to protein stability upon heating, a property of SAgs. Within the family of SAgs, the N terminus includes a groove formed between the OB-fold and the β grasp-fold that comprise TCR binding domain [47]. Members of the SAgs family possess a variety of structures that bind MHC II. Yersinia SAgs (YPM-A, YPM-B, and YPM-C) are unique and contain a single domain that comprises a jelly-roll motif consisting of 2 β-strands [48]. The SAg of M. arithritidis (MAM), creates an ‘L’ structure through the assembly of 10 α-helices. While the SSL toxins show an overall conserved structure relative to the SAgs, the surfaces for MHC II and TCR binding are altered [49–51]. In contrast to Sags, SSLs do not bind to the MHC II complex or TCR. Binding sites and function vary among SSLs, demonstrated by SSL 11 binding sialyllactosamine and SSL 7 binding C5 and IgA [51, 52]. Crystallized SSL 11 has an elongated β-strand within the β-grasp fold which allows dimerization to occur, to presumably increase affinity for cellular glycoproteins [51].
SAgs can bind MHC II by several mechanisms (Figure 2) and are classified by how each binds MHC II. TSST-1 binds the MHC II α-chain and a region of the β-chain by extending over the presented peptide (Class I) [53]. Staphylococcal and Streptococal SAgs bind the MHC II α-chain through the OB-domain and do not contact the peptide (Class II) [53]. Classes IV and V SAgs bind through the C-terminal domain to the β-chain of MHC II, utilizing a coordinated zinc site as well as the presented peptide [54, 55]. MAM binds in a different fashion by binding the MHC II α1-helix and β1-helix, causing the dimerization of the two MHC II molecules [56].
Figure 2. Binding of superantigen SEC3 to TCR β.
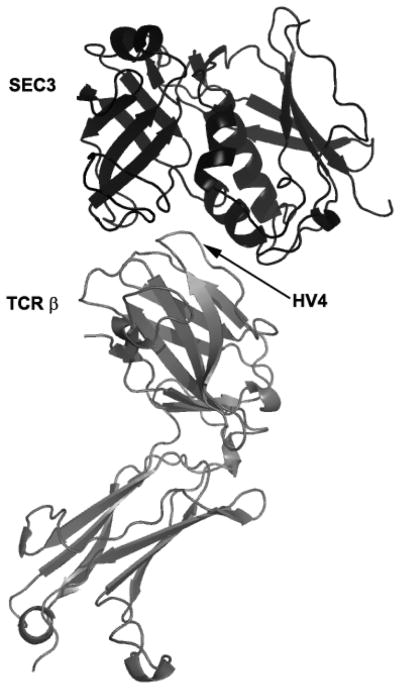
Staphylococcus aureus superantigen SEC3 binds to the β chain of TCR through the hypervariable domain 4 (HV4), acting as a wedge to encourage TCR and MHCII interaction and activation lacking foreign peptide specificity. Arrow indicates HV4 loop on TCR β. Structure: PDB 1jck.
Binding to MHC II and TCR by SAgs appears to be sequential, based on kinetic binding data [47]. SAgs affinity to MHC II is between 10- to 100-fold higher than to TCR proteins [57]. This determined in vitro affinity is most likely a low estimation, since APCs bind SAgs in a stable conformation for over 30 hours [58]. SAg binding to TCR molecules for non peptide-specific activation occurs through the β-chain, via diverse mechanisms, that include binding to the CDR2 loop of TCR Vβ [59]. This interaction is sufficient to activate up to 20% of T cells [60]. Most SAgs, however, have an increased specificity to TCR which is defined by specific interactions with the TCR hyper-variable areas surrounding the CDR2 loop [59]. Another action of SAgs is to directly block TCR interaction with the presented peptide, keeping the TCR and MHC II dimers physically separated where SAgs wedge between the two receptors (Group II), allowing only minimal contact (TCR α-chain and MHC II β-chain), physically block peptide interaction (Class 1), or bind to cause a sharp angle in between TCR and MHC II/peptide interface (class V) [57]. Current studies on SAgs involve development of structural mimics to neutralize pathology in animal models [61] and to determine the specificity of immune stimulation by SAgs of clinical bacterial isolates [62].
Secretion of Toxins from the Bacterium
Bacterial toxins are transported across the bacterial membranes through co-translational and post-translational mechanisms to reach their targets. Toxin transport occurs by multiple mechanisms, which have been characterized within Gram Negative and Gram Positive bacteria. Most secretion systems utilize active transport, requiring at least one energy requiring step. For example, types-II, -III, and -IV secretion systems contain an oligomeric ring of ATPases on the cytoplasmic side of the secretion system inner membrane complexes. While signal sequences often coordinate the secretion process, signal sequences are not universal. This review describes the general properties of the Type I–VII secretion systems as related to the secretion of bacterial toxins.
Type I Secreted Toxins
Type I secretion is observed in Gram Negative and Gram Positive bacteria and transports a variety of proteins across the bacterial inner membrane into the periplasmic space in an active, single step process. Proteins secreted via the Type I pathway contain glycine-rich repeats, usually at the C terminus, which bind calcium [63–65]. The organization of Type I secretion complex is similar to the ABC family of membrane transporters, composed of a tripartite protein complex that includes an inner membrane transporter, a membrane fusion protein within the periplasm, and an outer membrane pore protein [65]. Central to Type I secretion is the inner membrane transporter that is a transmembrane protein and encodes an ATP-binding domain for recognition of the secretion signal. The membrane fusion protein exists within the periplasmic space and also contains a region which spans the inner membrane that is exposed to the cytoplasm [66, 67] and links inner and outer membrane-spanning proteins. The outer membrane pore protein is a trimer that forms a channel for direct access the extracellular space. This trimer assembles into the secretion complex when the ATP-binding domain recognizes a signal sequence contained by the secreted protein [67]. Current studies on Type I secretion address the physical link between energy utilization (ATP) and protein transport [62].
The adenylcyclase-hemolysin (CyaA) of Bordetella pertussis is a Type I secreted toxin [68]. Entry into the host cell, via the alpha(M)beta(2) integrin (CD11b/CD18) cell receptor [69], involves a two step process with insertion into the membrane and calcium-dependent unfolding of the N terminus of CyaA [70–72]. Upon entry into the cytosol of a host cell, CyaA is activated by the cofactor, calmodulin, and catalyzes the conversion of ATP to cAMP. Elevated intracellular cAMP disrupts signaling events within the cell and inhibits the ability of phagocytes to respond to B. pertussis infections [70]. Specifically, deregulation of cell signaling by CyaA affects protein kinase A, which modulates neutrophil migration, cytokine synthesis, oxidative bursts and organization of the actin cytoskeleton [73, 74]. CyaA is a ~200 kDa protein with two functional domains, a 400 amino acid N-terminal domain that expresses adenylate cyclase activity, and a C-terminal domain that contains the hemolytic activity [75]. Current studies on CyaA involve characterization of structural organization [76] and functional properties [77] of the protein’s multiple activities.
Type II Secreted Toxins
Type II secretion is facilitated by the Sec system and comprises protein complexes that span the bacterial inner and outer membranes. Sec secretion includes three groups of proteins, a protein complex that spans the inner membrane, a periplasm-spanning protein complex, and outer membrane associated proteins. Type II secretion involves recognition of an N-terminal signal peptide on the nascent protein within the sequence recognition particle which is analogous to eukaryotic protein secretion in the endoplasmic reticulum [78]. Upon recognition and docking, the growing polypeptide undergoes co-translational secretion and folding to a mature state in the periplasm where the protein is then translocated through the outer membrane [79]. The signal sequence consists of N-terminal positive charged amino acids, internal hydrophobic amino acids, and a C-terminal domain with prolines and glycine [80, 81]. Type II secretion is an active process, requiring a hexameric ATPase associated with the cytoplasmic portion of the inner membrane protein complex and periplasmic proteins that link the inner membrane complex with the outer membrane associated proteins. One group of periplasmic proteins are homologous to pilin-like structures (known as pseudopilins) and regulatory proteins of the Type IV secretion system, suggesting that these proteins interact to make a tube spanning the periplasmic space [82] and may be involved in the transfer the secreted protein to the outer membrane complex.
Cholera toxin (CT) of Vibrio cholera is a Type II secreted toxin. CT is an AB5 toxin, where the A domain (~27.4 kDa) consists of two components, CT-A1 and CT-A2 and the B domain (~58 kDa) is a homopentameric protein complex [83]. CT-A1 ADP-ribosylates the Gα-subunit of the heterotrimeric protein, Gs. CT-A1 associates with the CT-A2 via a disulfide bond where CT-A2 inserts into the channel within the center of B5. The B5 domain binds specifically to the ganglioside, GM1a on the surface of intestinal epithelial cells [84]. Once bound, CT enters the cell through both clathrin-dependent and non-clathrin-dependent vesicle mechanisms involving lipid rafts [85]. After internalization, CT retrograde traffics to the endoplasmic reticulum (ER) via a KDEL-like sequence on the C terminus of A2 (note the KDEL sequence is not absolutely required for the intracellular trafficking of cholera toxin) where the A1 subunit utilizes a retro-translocation mechanism to cross the ER plasma membrane through a degradation pathway that recycles miss folded hosts proteins called ERAD [86]. Cytosolic CT-A1 binds A DP-ribosylation factor (ARF) and the activated CT-A1 mono-ADP-ribosylates Arg 201 of Gsα, which blocks intrinsic GTPase activity and constitutively activates Gsα [87, 88]. Gsα is a positive regulator of adenylate cyclase. Increased intracellular cAMP activates protein kinase A (PKA), which phosphorylates the cystic fibrosis transmembrane conductance regulator (CFTR), increasing active secretion of chloride ions [89]. Inhibition of the Na/K/2Cl co-transporter at the same time increases the unidirectional flow of chloride into the gut lumen, causing osmotic H20 flow into the gut lumen, the pathological outcome of cholera [90]. Current studies on cholera toxin involve the utilization of the toxin as an adjuvant in vaccine development [91] and as a tool to study intracellular protein trafficking [85].
Type III Secreted Toxins
The Type III secretion system (TTSS) is a bacterial virulence factor which injects cytotoxins (also termed effectors) in an unfolded or semi-folded state into a host cell. Like Type II secretion, TTSS comprises inner and outer membrane protein complexes, but includes a hollow, pilin-like structure that extends beyond the outer membrane ring complex. The genetic material encoding the TTSS is a gene duplication of the inner and outer membrane ring complexes of the bacterial flagella protein complex [92]. The inner membrane complex is comprised of two interacting ring structures, each composed of a different oligomeric protein [93]. Like Type II secretion, the cytoplasmic domain of the inner membrane complex associates with an ATPase, which assembles in a hexameric ring to provide the energy for the unfolding and secretion of cytotoxins. The outer membrane structure is also similar to the Type II secretion system outer membrane ring and the Type IV pilus. Extracellular components of the TTSS include a needle, needle extension, and translocation pore, which deliver cytotoxins into the cytoplasm of host cells. The needle is a single protein oligomer which forms a hollow tube which has direct interactions between head and tail of the protein subunits and have been implicated in the secretion process upon conformational changes [94]. The needle tip proteins appear to be adaptors that link the translocator proteins within the host membrane to the needle body for efficient transfer of cytotoxins. Translocator proteins form a host membrane-spanning pore for the TTSS effectors [95]. The translocon protein complex prevents cytotoxin secretion before contact with the host cell by physically blocking the hollow channel of the needle complex [96].
ExoS is a Type III secret ed cytotoxin. ExoS is a 453 amino acid protein produced by Pseudomonas aeruginosa and is a dual function toxin, containing a Rho GTPase Activating Protein (Rho GAP) activity in the N terminus (96–219) and an ADP-ribosylation domain in the C terminus (234–453) (Figure 3). ExoS Rho GAP domain increases γ-phosphate hydrolysis of GTP bound to Rho GTPases and in actives RhoA, Rac1, and Cdc42 [97, 98]. Structural studies showed that the ExoS Rho GAP domain was a molecular mimic of the host Rho GAP, which implicated a convergent mechanism of protein evolution for ExoS their mammalian counterparts [99]. The C-terminal ADP-ribosylation domain of ExoS has the capacity to ADP-ribosylate multiple substrates including Ras at Arg 41, which blocks association with the guanine nucleotide exchange factor leading to an inactivation of Ras signaling [100]. Recently, ExoS has been shown also to ADP-ribosylate Rab proteins, such as Rab5, where and the ADP-ribosylation of Rab5 uncoupled clathrin-mediate endocytosis [101] and ADP-ribosylation of ezrin/radixin/moesin, caused cytoskeletal defects [102]. ExoS binds a cellular cofactor to express ADP-ribosyltrasferase activity. The factor was termed factor activating exoenzyme S (FAS) and later identified as a 14-3-3 protein [103]. Activation of a cytotoxin upon binding to a host protein is becoming a common feature of toxin action. Current studies on ExoS involve characterization of how ExoS traffics within host cells to efficiently target intracellular substrates [104], the determination of the role of ExoS and other Type III cytotoxins in P. aeruginosa pathogenesis [105], and the development of diagnostics for clinical therapy based upon the seroconversion of CF patients to components of the TTSS upon initial infection by P. aeruginosa [106].
Figure 3. Functional orgainzation of the type III cytotoxin Pseudomaons aeruginosa.
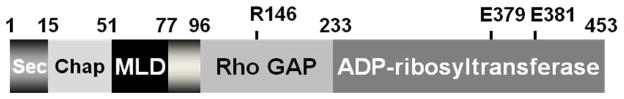
ExoS is a bi-functional toxins and is orgainzed inot discret functional doamisn (amino acids): secretion domain (1–15), chaperone binding domain (16–51), membrane localization domain (51–77), Rho GAP domain, active site residue R146 (96–243), and ADP-ribosyltransferase domain, active site residues E379, E381 (233–453).
Type IV Secreted toxins
The Type IV secretion system is a multi-functional protein complex, which transfers DNA between bacteria through conjugation (Type IVA), and transports effector proteins into host cells to regulate host responses to bacterial infection (Type IVB). Type IV transport of effectors across the inner and outer bacterial membranes can follow a single step or two step process [107]. DNA is transferred as a complex bound to transfer proteins which bind by a C-terminal signal to DNA [108]. The VirT IVA secretion system in Agrobacterium tumefaciens is the best characterized Type IV secretion system, which transfers DNA and proteins into plants to cause disease. VirB contributes to the transfer of effectors through the Type IV secretion apparatus into the host cell [109]. DNA transfer requires a protein relaxase, which binds covalently to the 5′ end of ssDNA and causes secretion specifically in a 5′ to 3′ direction. VirB11 is an ATPase which provides energy for the secretion process analogous to other secretion systems [110, 111]. Pilin homologs form an outer connecting transfer tube between the bacteria and the target cell or bacteria [108]. Recent structural studies suggest that the inner membrane protein complex spans the majority of the periplasmic space [112]. In addition, Type IV secretion contributes in the pathogenesis of several intracellular bacterial pathogens, including Legionella, Brucella and Coxiella [113, 114].
SidC is a type IV effector of Legionella pneumophila, the causative agent of Legionnaire’s disease [115]. SidC localizes to legionella containing vesicles via phosphatidylinositol-4 phosphate where deletion of SidC interferes with the recruitment of endoplasmic reticulum-derived vesicles to the legionella containing vesicles. This is required for the maturation of the legionella containing vesicles with lysosomal marker proteins.
Type V Secreted toxins
The Type V secretion system, also termed the autotransporter secretion group, includes 3 transport secretion mechanisms: termed Va, Vb, and Vc. Va group autotransporters are translated as a single protein composed of a N-terminal signal peptide sequence for transport by the Sec system, the effector domain, and a C-terminal outer membrane translocation domain [116]. The N-terminal secretion signal utilizes Sec for transport into the periplasm, where the inner membrane secretion signal is cleaved by a periplasmic peptidase. While early models predicted that transported protein folded in mature configuration, recent studies suggest that an unfolded conformation is necessary for transport of the effector across the outer membrane [117]. The C-terminal translocation domain forms a β-barrel [118, 119] that utilizes a co-secreted chaperone protein to protect against proteases and premature folding and to fold effectors upon secretion [120]. A role for ATP has not been determined. Details for effector transport through the Type V secretion system are under investigation [121–123]. The Vb two-partner secretion subgroup follows a similar mechanism as Va, involving a Sec dependent pathway as two distinct proteins [124, 125] where the translocation protein is predicted to contain β-strands and to interact with effector proteins for efficient translocation across the outer membrane [126–128].
IgA1 protease is Type V secreted protein that is produced by several genera of bacteria that cleave mammalian IgA [129]. The IgA1 protease group contains a general structure containing a protease and a β–barrel forming domain. This protease group cleaves proline-threonine and proline-serine bonds in the hinge region of human IgA1 [130]. The general structures of the IgA group of proteins comprise a zinc-like protease bound to a β-barrel.
Type VI and Type VII Secretion Systems
A type VI secretion system has been proposed in V. cholerae [131] and P. aeruginosa [132]. Type VI effectors do not possess N-terminal signal peptides and are Sec secretion-independent, implicating a unique mechanism for effector transport relative to the Types I–V secretion systems. Recently, a secretion system was identified in the mycobacteria that was classified as the Type VII secretion for Gram-Positive bacteria [133]. Current studies address the identification of mechanisms for effector protein secretion by these new secretion systems.
Botulinum neurotoxin
The botulinum neurotoxins (BoNT) are the most toxic proteins for humans and have been characterized as Category A agents by the Center for Disease Control and Prevention. BoNTs block neurotransmitter signal transduction in peripheral α-motor neurons, causing flaccid paralysis, termed botulism [134]. Botulism poisoning in humans is typified by slurred speech, dry mouth, blurred or double vision, peripheral muscle weakness and paralysis. There are seven serotypes of the BoNTs, termed A–G.
Three types of human botulism include, wound, ingestion, and infant. Wound botulism occurs upon the delivery of clostridial spores into a deep, anaerobic wound where the spores germinate into the vegetative cells that produce BoNT. BoNT circulates in the bloodstream to target α-motor neurons. Food botulism results for the ingestion of preformed toxin in improperly cooked or stored food that allows growth of clostridial and subsequent production of BoNT. Ingested BoNT transcytoses across endothelial cells of the small intestine and migrates to the α-motor neurons. Infant botulism is caused by ingestion of clostridial spores by a child with an underdeveloped intestinal microbiota system. This provides a metabolic niche for the spores to germinate to vegetative cells that produced BoNT, which transcytoses epithelial barrier to cause paralysis. One potential source of clostridial spores for infant botulism appears to be raw or pasteurized honey in children <2 years of age [135]. A medically important characteristic of botulism intoxication is the extended duration of the toxin within neuron cells. The recommended treatment for botulism poisoning is a trivalent (ABE) equine antitoxin serum, obtained through the Centers for Disease Control and Prevention (CDC).
BoNT as a clinical therapeutic agent
BoNT/A holotoxin is also a clinically useful reagent, commercially available as BoTox®. The medical importance of BoTox transcends superficial cosmetic treatments and is used for a large range of muscle-induced impairments, as well as pain management [136]. Myobloc® is the commercial version of BoNT/B, which can be utilized as an alternative to BoTox treatment [137]. Despite the low immunoreactivity there are reports of α-BoNT/A and α-BoNT/B antibodies developing in patients after long-term use of the toxin [138, 139]. Determining the antigenic epitopes in medically relevant BoNT and how these epitopes affect toxin action is an important area of research for development of non-reactive clinical BoNTs therapies in the future.
BoNT-Producing Clostridial Species
BoNT is produced by a heterogeneous group of Clostridial species. While antigenic nomenclature separates the BoNTs by antibody cross-neutralization, clostridia that produce BoNT are differentiated into four groups based on physiology, Groups I–IV [140]. Clostridium botulinum, C. butyricum, C. baratii, and C. argentinense produce BoNTs. BoNT genes are found on a variety of genetic elements with BoNT/A, B, E, and F encoded on the chromosome, plasmids, and bacteriophages in various Clostridia sp. BoNT/C and D are found primarily in bacteriophage elements [141].
BoNT Operon Organization and Secretion
BoNT is transcribed and translated as a single ~150 kDa protein with little proteolytic activity. BotR is a positive regulator of BoNT and associated protein cluster, which is often transcribed as a set of three polycistronic transcripts [142]. BoNT form complexes with non-toxinogenic proteins transcribed and translated together with the BoNT gene. These proteins include haemagglutinin-active (HA) components, which associate with BoNT as a noncovalent, pH-stable complex, but are not necessary for full toxicity [143–145]. The most basic BoNT protein complex is the M complex (~300 kDa), consisting of BoNT and a non-haemagglutinin protein, which is found in BoNT-producing strains except BoNT/G[146]. The gene encoding the non-haemagglutinin protein is located upstream of the BoNT gene [147]. Other protein complexes include the L complex (~500 kDa), which encodes a number of proteins associated with BoNT which includes a haemagglutinin activity. The genes encoding the haemagglutinin-active proteins are located upstream of the non-haemagglutinin protein, and are found in clostridia producing BoNT serotypes A, B, C, D and G[146]. The haemagglutinin-active proteins were once hypothesized to protect the BoNT against acid damage during passage through the stomach, but ingested holotoxin without accessory proteins has demonstrated equal toxicity [148]. Currently, these proteins are hypothesized to increase access to the bloodstream (discussed below).
BoNT are single-chain proteins, which are cleaved to di-chain proteins that are linked by a disulfide bond with AB structure function properties [149] (Figure 4). The N-terminal A domain (Light chain, LC) is an ~50 kDa zinc metalloprotease with the characteristic thermolysin-family zinc coordination motif (HExxH) [134]. The C-terminal B domain (Heavy Chain, HC) is ~100 kDa and is composed of two functional domains that are involved in receptor recognition (HCR) and translocation of the LC across the endosomal membrane (HCT) [149]. Antibody cross-neutralization differentiates BoNTs into seven serotypes, A–G.
Figure 4. Schematic and structure of BoNT/A.
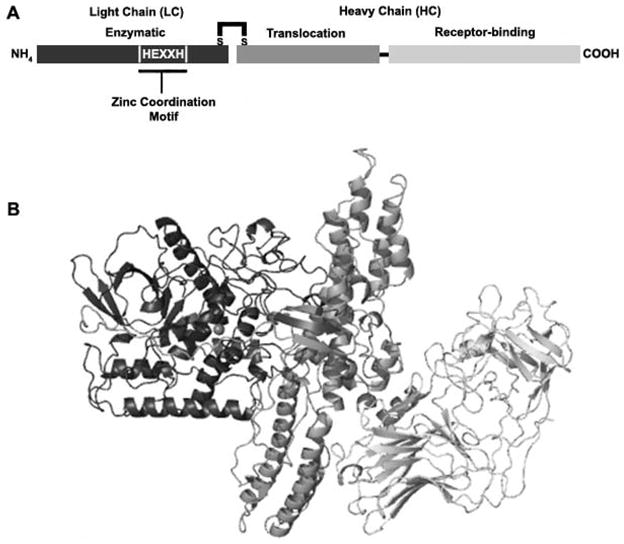
A. Schematic of BoNT/A as an A/B toxin, linked by a disulfide bond between the light (LC) and heavy (HC) chains. B. Crystal structure of BoNT/A. LC (black) is the catalytically active domain and contains a HEXXH motif for coordination of the Zn2+ atom. The HC contains the N-terminal translocation (grey) domain and the C-terminal receptor-binding domain (light grey). Grey sphere represents zinc atom within LC active site. Colors between schematic and structure are coordinated with each domain. Structure: PDB 3bta.
BoNTs Accessing the Bloodstream
There is debate on whether or not the haemagglutinin components of the BoNT proteins complexes disrupt the intestinal barrier to contribute to BoNT transport [150, 151]. Trans-epithelial cell transport progresses through temperature-dependent transport system that can be utilized by BoNT holotoxin alone, suggesting that haemagglutinin-and non-haemagglutinin components may increase the permeability of the epithelial barrier, but are not necessary for active BoNT transport to the bloodstream [152, 153]. Clathrin and dynamin-dependent internalization and transport mechanism have been implicated for BoNT entry into epithelial cells to an EEA-1 positive, non-acidified vesicle [154, 155]. The saturable nature of HC/A binding suggests specific receptor necessary for BoNT binding and trans-epithelial transport. Gangliosides and host synaptic vesicle proteins have been implicated in binding and internalization in the peripheral α-motor neurons, but it is unclear how these components contribute to trans-epithelial transport.
BoNT Neuronal Specificity
BoNT/HC is composed of two domains, the N-terminal translocation domain and the C-terminal receptor-binding domain (HCR). While there is considerable variability in the primary amino acid sequence of the BoNT/HCR among serotypes A–G, the BoNT/HCR have two conserved jelly roll and β-trefoil folding motifs. The HCR domain binds neurons via several mechanisms that are serotype specific, but includes a general dual receptor model for binding and entry into peripheral α-motor neurons. In this model, BoNT/HCR binds to gangliosides on the surface of a neuron and this complex binds to protein receptors as a high-affinity complex. Gangliosides consist of a polysaccharide group linked to a ceramide lipid, and BoNT/A, B, C, E, G, and tetanus neurotoxin require the binding to ganglioside to associate with neurons [156]. Active synaptic vesicle recycling is required for internalization of BoNTs, except for BoNT/C. This activity-dependent internalization supports a model where BoNT receptor(s) for BoNT are located within the luminal domain of the synaptic vesicle and exposed to the extracellular milieu after synaptic vesicle fusion with the plasma membrane. BoNT/A binds synaptic vesicle protein SV2 on loop 4 of the transmembrane protein and the ganglioside GT1b [157]. A role for glycosylation has been implicated in recent studies on BoNT/E binding to SV2 [158]. BoNT/B and BoNT/G utilize syntaptotagmin (I or II) as the protein component for neuron receptors [159, 160]. BoNT/C HC binds GT1b and GD1b, but the protein receptor has not been resolved [161]. Tetanus neurotoxin binds two molecules of gangliosides, but a role for a protein receptor component remains to be determined [162]. HCR/A binding to ganglioside demonstrates an increase in affinity over time, suggesting a conformational change in the ganglioside binding site to a higher-affinity interaction [163], but how this conformational change modulates affinity to the protein receptor is not clear. BoNT/D HCR associates with phosphatidylethanolamine (PE) and shows a ganglioside-independent toxicity of cerebellar granule cells, suggesting a difference in binding specificity compared to the other BoNT serotypes [161]. Recent research suggests that a third binding site, with specificity to lipids, may exist within the N-terminal domain of the HCR[164]. BoNT/A HCR was shown to bind phosphatidylinositol phosphates (PIP), however the binding site was not defined through mutational binding experiments and the structural model needed to be adjusted to accommodate the suggested receptor binding model, therefore how this domain is utilized during BoNT binding and internalization is unclear [164].
BoNT-neuronal Cell Interactions
BoNT entry into neurons is activity dependent, except for BoNT/C, demonstrating the necessity of vesicle recycling for BoNT uptake into neurons [157, 160]. SV2 and Synaptotagmin (I and II) on the synaptic vesicle are integral components of synaptic vesicle fusion with the plasma membrane [165, 166]. Recent studies implicate the binding of the BoNT/HCRs to a protein complex in synaptic vesicles that included the proton ATPase, synaptophysin, SV2 and synaptotagmin [167]. After fusion with the plasma membrane, the BoNT-receptor complex is endocytosed in a clathrin-dependent, dynamin-dependent pathway [165]. The N terminus of the HC is involved in translocation of the LC from the endosome into the cytosol. The translocation event is pH-dependent, which suggests that pore formation or LC translocation requires a conformation change to BoNT structure [168]. Indirect evidence through chimeras of BoNT/A and BoNT/E suggests that translocation speed can vary depending on the HC involved [169]. Recent research suggests that the isolated translocation domain of BoNT inserts into the membrane independent of pH, but acidification may be required to unfold and transport the LC through the HC pore [170, 171].
Mechanism of BoNT Action in Neurons
BoNTs share ~65% primary amino acid similarity and ~35% amino acid identity with Tetanus toxin [172]. BoNT/ A, /E, and /C cleave SNAP25; BoNT/C also cleaves Syntaxin 1a; and BoNT/B, /D, /F, /G, and tetanus toxin cleave VAMP-2 [173–175] (Table 1). Cleavage sites within each SNARE substrate differ among the BoNTs, suggesting specific residues on the LC are necessary for recognition of substrate. Recent studies have identified amino acids necessary for substrate recognition by LC/A, LC/B, LC/D, LC/E, LC/F, and tetanus toxin [176–185], mechanistically supporting a crystal structure of a non-catalytic LC/A bound to SNAP25 through exosite and active site interactions [179, 180, 185]. Initial interactions of LC/A with SNAP25 were predicted with the LC/A-SNAP25 co-crystal, and functionally showed that LC/A interacts with a long stretch of amino acids N-terminal to the cleavage site (Q197/R198) of SNAP25. Structural and biochemical studies suggest that SNAP25 wraps around the LC via a mechanism that mimics the binding of the HC loop to LC[186]. The LC-HC interaction is proposed to inactivate cleavage activity until the LC is released into the cytoplasm of the host cell. Kinetic data utilizing mutagenized SNAP25 identified specific amino acids N-terminal to the active site that contribute to SNAP25 binding, as well as amino acids immediately surrounding the active site that orients SNAP25 within the active site and contributes to cleavage activity. Corresponding studies involving mutations in LC/A demonstrated specific amino acid interactions with SNAP25 along with the minimum length of substrate, defining the specificity of cleavage [180]. LC/E showed a similar mechanism of recognition for SNAP25, in which amino acids N-terminal to the LC/E active site on SNAP25 were identified as well as amino acids immediately surrounding the cleavage site that when mutated, affected catalysis [179]. LC/B, which cleaves VAMP, demonstrated a slightly different mechanism of recognition and catalysis where amino acids N-terminal to the cleavage site (Q76/F77) affected binding and catalysis when mutated. Tetanus toxin, which cleaves VAMP-2 at the same residue at BoNT/B and has ~ 40% primary amino acid homology with BoNT/B, showed similar substrate recognition, but included an extension of the binding domain upstream of the cleavage site [177]. These combinations of amino acid interactions involved in binding and catalysis as a mechanism for substrate recognition seems to explain the specificity of neurotoxin substrate recognition and cleavage site specificity among BoNT serotypes and tetanus toxin despite a highly identical tertiary structure.
Table 1.
BoNT and TeNT SNARE substrates and cleavage sites.
Botulinum Neurotoxin Subtypes
Each BoNT serotype comprises subtypes that can vary between 3 and 32% at the primary amino acid level [187] (Figure 5). Currently, BoNT/A includes five subtypes, termed A1 through A5. BoNT/A1 and BoNT/A2 showed similar cleavage of SNAP25 and possessed~ 95% primary amino acid homology [188]. DNA sequencing analyses revealed two additional subtypes BoNT/A: BoNT/A3 and BoNT/A4 [188, 189]. Clostridia producing BoNT/A3 was isolated from a botulism outbreak in Scotland in 1922, while clostridia producing BoNT/A4 was isolated from a case of infant botulism in 1988 [190, 191]. LC/A3 shows 81% identity to LC/A1, while LC/A4 shows 88% identity to LC/A1 [188] (Table 2). Phylogenetic analysis of the four subtypes suggested that residue changes in LC/A3 and LC/A4 could potentially affect binding (Km) and catalysis (kcat) of these two subtypes relative to LC/A1[189]. Previous modeling studies suggested that LC/A3 and LC/A4 may possess different catalytic activities for SNAP25 relative to LC/A1 [188]. Biochemcial studies showed that LC/A3 had similar kinetic properties as LC/A1 and amino acids that differed with LC/A1 within the binding site did not change the binding or catalytic activity of LC/A3 for SNAP25, while LC/A4 showed a ~80-fold decrease in catalytic activity compared to LC/A1, suggesting a defect in SNAP25 cleavage. This change in catalytic activity is believed to be due to a single residue substitution indirectly causing a loss of coordination with the catalytic zinc in the active site, as a point mutation to LC/A4 restores catalytic activity to the level of LC/A1[192]. Another phenotype of LC/A1, autocatalysis, is observed with LC/A2, but not LC/A3 and LC/A4, demonstrating a difference in self-substrate recognition. Finally, a synthetic diagnostic peptide replicating the substrate cleavage site, SNAPtide™, demonstrates differences in cleavage activity between the subtypes, potentially suggesting differences in substrate recognition [192]. Thus, while LC/A subtypes retained the capacity to cleave SNAP25, efficiency and mechanisms for the cleavage reactions appear different which will influence the ability to generate serotype-specific inhibitors of catalysis, challenging that ability to establish common steps in catalysis that can be targeted for inactivation. The HCs of BoNT/A subtypes are ~87% identical and polyclonal antisera to HCR/A1 neutralizes BoNT/A1 and BoNT/A2 [187, 193], but there are sufficient amino acid differences where neutralizing monoclonal antibodies against BoNT/A1 are not effective in neutralizing BoNT/A2 [187]. Current vaccine strategies involve recombinant hepta-HC serotype vaccination, producing a polyclonal antibody response against the seven BoNT serotypes [193].
Figure 5. Phylogenetic dendogram of BoNT/A1–A5 amino acid identity.
Dendogram constructed using DNAStar® MegAlign.
Table 2.
Protein similarity of BoNT/A1–A4. Similarity of BoNT/A2–A4 compared to BoNT/A1.
| BoNT A1 versus | Holotoxin | LC 1-425 | HC 438–1295 | HN subdomain 438–872 | HCN subdomain 873–1093 | HCC subdomain 1094–1295 |
|---|---|---|---|---|---|---|
| A2 | 89 | 95 | 87 | 87 | 83 | 90 |
| A3 | 84 | 76 | 86 | 85 | 82 | 90 |
| A4 | 89 | 84 | 89 | 87 | 85 | 97 |
Adapted with permission from Arndt, J.W., et al., 2006, J. Mol. Biol., 362:733–742.
These data indicate that while many of the properties of the subtypes of BoNT/A are similar, there are unique properties. Continued characterization of the BoNTs serotypes may provide useful information on the development of strategies to generate vaccines and therapies against botulism and to develop novel BoNT derivatives that can extend the therapeutic utility.
Prospects
Since the first discovery of protein toxins as virulence factors of bacterial pathogens in the 19th century, scientific investigation has defined the biochemical and cellular aspects of toxin action that was facilitated with the resolution of the structural properties of protein toxins. The determination that protein toxins were often organized into discreet domains that comprises a catalytic, receptor binding, and translocation domain allowed rapid development of vaccines to control disease. The next chapter in the study of protein toxin action is to utilize our understanding of toxin structure function to utilize toxins for immunological and therapeutic intervention of human disease.
Acknowledgments
This work was supported by a grant from the Great Lakes Regional Center of Excellence (GLRCE) to JTB (NIH-NIAID U54 AI057153) and a grant to MRB (NIH-NINDS K99 NS061763).
References
- 1.Field M, GLH, Laird WJ, Smith PL. Heat-stable enterotoxin of Escherichia coli: In vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc Natl Acad Sci. 1978;75:2800–2804. doi: 10.1073/pnas.75.6.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giannella RA. Pathogenesis of acute bacterial diarrheal disorders. Ann Rev Med. 1981;32:341–357. doi: 10.1146/annurev.me.32.020181.002013. [DOI] [PubMed] [Google Scholar]
- 3.Crane JK, WMS, Bolen EJ, Sando JJ, Linden J, Guerrant RL, Sears CL. Regulation of intestinal guanylate cyclase by the heat-stable enterotoxin of Escherichia coli (STa) and protein kinase C. Infect Immun. 1992;60:5004–5012. doi: 10.1128/iai.60.12.5004-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takao T, HT, Aimoto S, Shimonishi Y, Hara S, Takeda T, Takeda Y, Miwatani T. Amino acid sequence of a heat-stable enterotoxin isolated from enterotoxigentic Escherichia coli strain 18D. FEBS Lett. 1983;152:1–5. doi: 10.1016/0014-5793(83)80469-4. [DOI] [PubMed] [Google Scholar]
- 5.Ozaki H, ST, Kubota H, Hata Y, Katsube Y, Shimonishi Y. Molecular structure of the toxic domain of heat-stable enterotoxin produced by enterotoxigenic Escherichia coli. J Biol Chem. 1991;266:5934–5941. [PubMed] [Google Scholar]
- 6.Hidaka Y, KH, Yoshimura S, Ito H, Takeda Y, Shimonishi Y. Disulfide linkages in heat-stable enterotoxin (STp) produced by a procine strain of enterotoxigenic Escherichia coli. Bull Chem Soc Jpn. 1988;61:1265–1271. [Google Scholar]
- 7.Waldman SA, OHP Influence of a glycine or proline substitution on the functional properties of a 14-amino acid analog of Escherichia coli heat-stable enterotoxin. Infect Immun. 1989;57:2420–2424. doi: 10.1128/iai.57.8.2420-2424.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamasaki S, ST, Hidaka Y, Ozaki H, Ito H, Hirayama T, Takeda Y, Sugimura T, Tai A, Shimonishi Y. Structure-activity relationship of Escherichia coli heat-stable enterotoxin: role of Ala residue at position 14 in toxin-receptor interaction. Bull Chem Soc Jpn. 1990;63:2063–2070. [Google Scholar]
- 9.Hasegawa M, SY Recognition and signal transduction mechanism of Escherichia coli heat-stable enterotoxin and its receptor, guanylate cyclase C. J Peptide Res. 2005;65:261–271. doi: 10.1111/j.1399-3011.2005.00218.x. [DOI] [PubMed] [Google Scholar]
- 10.Yoshimura S, IH, Watanabe H, Aimoto S, Shimonishi Y, Hara S, Takeda T, Miwatani T, Takeda Y. Essential structure for full enterotoxigenic activity of heat-stable enterotoxin produced by enterotoxigenic Escherichia coli. FEBS Lett. 1985;181:138–142. doi: 10.1016/0014-5793(85)81129-7. [DOI] [PubMed] [Google Scholar]
- 11.Ikemura H, Takagi H, Inouye M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J Biol Chem. 1987;262:7859–7864. [PubMed] [Google Scholar]
- 12.Gupta DD, SS S, Chakrabarti MK. Involvement of protein kinase C in the mechanism of action of Escherichia coli heat-stable enterotoxin (STa) in a human colonic carcinoma cell line, COLO-205. Toxicology and App Pharm. 2005;206:9–16. doi: 10.1016/j.taap.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein JL, SJ, Bhuva M, Layden TJ, Rao MC. Escherichia coli heat-stable enterotoxin-mediated colonic Cl-secretion is absent in cystic fibrosis. Gastroenterology. 1994;107:950–956. doi: 10.1016/0016-5085(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 14.Goncalves C, VV, Schwartz J-L, Dubreuil JD. The Escherichia coli enterotoxin STb permeabilizes piglet jejunal brush border membrane vesicles. Infect Immun. 2007;75:2208–2213. doi: 10.1128/IAI.01829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handl CE, FJI STb producing Escherichia coli are rarely associated with infantile darrhoea. J Diarrhoeal Dis Res. 1992;10:37–38. [PubMed] [Google Scholar]
- 16.Arriaga YL, HBA, Dreyfus LA. Contribution of individual disulfide bonds to biological action of Escherichia coli heat-stable enterotoxin B. Infect Immun. 1995;63:4715–4720. doi: 10.1128/iai.63.12.4715-4720.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrie V, HJ, Dubreuil JD. Oligomerization of Escherichia coli enterotoxin b through its C-terminal hydorphobic alpha-helix. Biochim Biophys Acta. 2001;1535:128–133. doi: 10.1016/s0925-4439(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto K, BT, Yamanaka H, Akashi N, Fujii Y. Disulfide bond formation and secretion of Escherichia coli heat-stable enterotoxin II. J Bacteriol. 1995;177:4579–4586. doi: 10.1128/jb.177.16.4579-4586.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rousset E, HJ, Dubreuil JD. Sulfatide from the pig jejunum brush border epithelial cell surface is involved in binding of Escherichia coli enterotoxin b. Infect Immun. 1998;66:5650–5658. doi: 10.1128/iai.66.12.5650-5658.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennedy DJ, GRN, Dunn JA, Abernathy R, Ryerse JS, Guerrant RL. Effects of Escherichia coli Heat-Stable Enterotoxin STb on Intestines of Mice, Rats, Rabbits, and Piglets. Infect Immun. 1984;46:639–643. doi: 10.1128/iai.46.3.639-643.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreyfus LA, HB, Howard DE, Shaban R, Beatty DM, Morris SJ. Calcium influx mediated by the Escherichia coli heat-stable enterotoxin B (STb) Proc Natl Acad Sci. 1993;90:3202–3206. doi: 10.1073/pnas.90.8.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erume J, Berberov EM, Kachman SD, Scott MA, Zhou Y, Francis DH, Moxley RA. Comparison of the contributions of heat-labile enterotoxin and heat-stable enterotoxin b to the virulence of enterotoxigenic Escherichia coli in F4ac receptor-positive young pigs. Infect Immun. 2008;76:3141–3149. doi: 10.1128/IAI.01743-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucas ML, Duncan NW, o’reilly NF, McIlvenny TJ, Nelson YB. Lack of evidence in vivo for a remote effect of Escherichia coli heat stable enterotoxin on jejunal fluid absorption. Neurogastroenterol Motil. 2008;20:532–538. doi: 10.1111/j.1365-2982.2007.01046.x. [DOI] [PubMed] [Google Scholar]
- 24.EG Channel-forming toxins: tales of transformation. Curr Opin. 1997;7:566–573. doi: 10.1016/s0959-440x(97)80123-6. [DOI] [PubMed] [Google Scholar]
- 25.Lesieur C, V-SB, Abrami L, Fivaz M, van der Goot FG. Membrane insertion: the strategy of toxins. Mol Membr Biol. 1997;14:45–64. doi: 10.3109/09687689709068435. [DOI] [PubMed] [Google Scholar]
- 26.Kurisu G, Zakharov SD, Zhalnina MV, Bano S, Eroukova VY, Rokitskaya TI, Antonenko YN, Wiener MC, Cramer WA. The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat Struct Biol. 2003;10:948–954. doi: 10.1038/nsb997. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita E, Zhalnina MV, Zakharov SD, Sharma O, Cramer WA. Crystal structures of the OmpF porin: function in a colicin translocon. Embo J. 2008;27:2171–2180. doi: 10.1038/emboj.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe S, Bennett MJ, Fujii G, Curmi PM, Kantardjieff KA, Collier RJ, Eisenberg D. The crystal structure of diphtheria toxin. Nature. 1992;357:216–222. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- 29.Ratts R, Zeng H, Berg EA, Blue C, McComb ME, Costello CE, vanderSpek JC, Murphy JR. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J Cell Biol. 2003;160:1139–1150. doi: 10.1083/jcb.200210028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tilley SJ, SHR The mechanism of pore formation by bacterial toxins. Curr Opin Struct Biol. 2006;16:230–236. doi: 10.1016/j.sbi.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Iacovache I, PP, Scheib H, Lesieur C, Sakai N, Matile S, Parker MW, van der Goot FG. A rivet model for channel formation by aerolysin-like pore-forming toxins. EMBO J. 2006;25:457–466. doi: 10.1038/sj.emboj.7600959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melton JA, CRJ, Finkelstein A. PA63 channel of anthrax toxin: An extended beta-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 33.Moniatte M, vdGFG, Buckley JT, Pattus F, van Dorsselaer A. Characterisation of the heptameric pore-forming complex of the Aeromonas toxin aerolysin using MALDI-TOF massspectrometry. FEBS Lett. 1996;384:269–272. doi: 10.1016/0014-5793(96)00328-6. [DOI] [PubMed] [Google Scholar]
- 34.Parker MW, FSC Pore-forming protein toxins: From structure to function. Prog Biophys Mol Bio. 2005;88:91–142. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Sekiya K, SR, Danbara H, Futaesaku Y. A ring-shaped structure with a crown formed by streptolysin O on the erythrocyte membrane. J Bacteriol. 1993;175:5953–5961. doi: 10.1128/jb.175.18.5953-5961.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park JM, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med. 2004;200:1647–1655. doi: 10.1084/jem.20041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, Voskoboinik I, Bird PI, Trapani JA, Whisstock JC, et al. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367:1227–1236. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.RKT Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramachanran R, TRK, Johnson AE. The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc Natl Acad Sci. 2005;102:7139–7144. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosado CJ, KS, Bull TE, Kulper MJ, Law RHP, Buckle AM, Voskobolnlk I, Bird PI, Trapani JA, Whisstock JC, Dunstone MA. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tilley SJ, SHR Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 44.Yoshino K, AJ, Murata H, Takao T, Kohsaka T, Shimonishi Y, Takeda T. Purification and characterization of a novel superantigen produced by a clinical isolate of Yersinia pseudotuberculosis. FEBS Lett. 1994;356:141–144. doi: 10.1016/0014-5793(94)01249-0. [DOI] [PubMed] [Google Scholar]
- 45.McCormick JK, YJM, Schlievert PM. Toxic Shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 46.MS B. Enterotoxins. In: Easom AC, CSF, editors. Staphyloccoi and Staphylococcal Infections. Academic Press; New York: 1983. pp. 559–598. [Google Scholar]
- 47.Fraser JD, PT The bacterial superantigen and superantigen-like proteins. Immun Rev. 2008;225:226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 48.Donadini R, LCW, Kwan AH, Mackay JP, Fields BA. Crystal and solution structures of a superantigen from Yersinia pseudotuberculosis reveal a jelly-roll fold. Structure. 2004;12:145–156. doi: 10.1016/j.str.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Al-Shangiti AM, NCE, Nair SP, Briggs DC, Henderson B, Chain BM. Structural relationships and cellular tropism of staphylococcal superantigen-like proteins. Infect Immun. 2004;72:4261–4270. doi: 10.1128/IAI.72.7.4261-4270.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arcus VL, LR, Proft T, Fraser JD, Baker EN. The Three-dimensional structure of a superantigen-like protein, SET3, from a pathogenicityisland of the Staphylococcus aureus genome. J Biol Chem. 2002;277:32274–32281. doi: 10.1074/jbc.M203914200. [DOI] [PubMed] [Google Scholar]
- 51.Chung MC, WBD, Baker H, Langley RJ, Baker EN, Fraser JD. The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol Microbiol. 2007;66:1342–1355. doi: 10.1111/j.1365-2958.2007.05989.x. [DOI] [PubMed] [Google Scholar]
- 52.Langley R, WB, Willoughby N, Basu I, Proft T, Fraser JD. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol. 2005;174:2926–2933. doi: 10.4049/jimmunol.174.5.2926. [DOI] [PubMed] [Google Scholar]
- 53.Kim J, URG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule. Science. 1994;266:1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 54.Gunther S, VAK, Moza B, Kasper KJ, Wyatt AW, Zhu P, Rahman AK, Li Y, Mariuzza RA, McCormick JK, Sundberg EJ. A novel loop domain in superantigens extends their T cell receptor recognition site. J Mol Biol. 2007;371:210–221. doi: 10.1016/j.jmb.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, LH, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14:93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 56.Zhao Y, Li Z, Drozd SJ, Guo Y, Mourad W, Li H. Crystal structure of Mycoplasma arthritidis mitogen complexed with HLA-DR1 reveals a novel superantigen fold and a dimerized superantigen-MHC complex. Structure. 2004;12:277–288. doi: 10.1016/j.str.2004.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sundberg EJ, DL, Mariuzza RA. TCR recognition of peptide/MHC class II complexes and superantigens. Semin Immunol. 2007;19:262–271. doi: 10.1016/j.smim.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pless DD, RG, Reinke EK, Ulrich RG, Bavari S. Persistence of zinc-binding bacterial superantigens at the surface of antigen-presenting cells contributes to the extreme potency of these superantigens as T-cell activators. Infect Immun. 2005;73:5358–5366. doi: 10.1128/IAI.73.9.5358-5366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundberg EJ, LH, Llera AS, McCormick JK, Tormo J, Schlievert PM, Karjalainen K, Mariuzza RA. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure. 2002;10:687–699. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 60.Sundberg EJ, Deng L, Mariuzza RA. TCR recognition of peptide/MHC class II complexes and superantigens. Semin Immunol. 2007;19:262–271. doi: 10.1016/j.smim.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang X, Buonpane RA, Moza B, Rahman AK, Wang N, Schlievert PM, McCormick JK, Sundberg EJ, Kranz DM. Neutralization of multiple staphylococcal superantigens by a single-chain protein consisting of affinity-matured, variable domain repeats. J Infect Dis. 2008;198:344–348. doi: 10.1086/589776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas D, Dauwalder O, Brun V, Badiou C, Ferry T, Etienne J, Vandenesch F, Lina G. Staphylococcus aureus superantigens elicit redundant and extensive human V{beta} patterns. Infect Immun. 2009 doi: 10.1128/IAI.01388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ostolaza H, SA, Goni FM. The binding of divalent cations to Escherichia coli alpha-haemolysin. Eur J Biochem. 1995;228:39–44. [PubMed] [Google Scholar]
- 64.Wolff N, GJM, Delepelaire P, Wandersman C, Delepierre M. C-terminal secretion signal of an Erwinia chrysanthemi protease secreted by a signal peptide-independent pathway: proton NMR and CD conformational studies in membrane-mimetic environments. Biochemistry. 1994;33:6792–6801. doi: 10.1021/bi00188a007. [DOI] [PubMed] [Google Scholar]
- 65.Allenby NE, O’Connor N, Pragai Z, Carter NM, Miethke M, Engelmann S, Hecker M, Wipat A, Ward AC, Harwood CR. Post-transcriptional regulation of the Bacillus subtilis pst operon encoding a phosphate-specific ABC transporter. Microbiology. 2004;150:2619–2628. doi: 10.1099/mic.0.27126-0. [DOI] [PubMed] [Google Scholar]
- 66.Delepelaire P. Type I secretion in gram-negative bacteria. Biochim Biophys Acta. 2004:149–161. doi: 10.1016/j.bbamcr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 67.Letoffe S, DP, Wandersman C. Protein secretion in gram-negative bacteria: assembly of the three components of ABC protein-mediated exporters is ordered and promoted by substrate binding. EMBO J. 1996;15:5804–5811. [PMC free article] [PubMed] [Google Scholar]
- 68.Glaser P, Danchin A, Ladant D, Barzu O, Ullmann A. Bordetella pertussis adenylate cyclase: the gene and the protein. Tokai J Exp Clin Med. 1988;13(Suppl):239–252. [PubMed] [Google Scholar]
- 69.Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, Ladant D, Leclerc C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18) J Exp Med. 2001;193:1035–1044. doi: 10.1084/jem.193.9.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weingart CL, Mobberley-Schuman PS, Hewlett EL, Gray MC, Weiss AA. Neutralizing antibodies to andenylate cyclase toxin promote phagocytosis of Bordetella pertussisby human neutrophils. Infect Immun. 2000;68:7152–7155. doi: 10.1128/iai.68.12.7152-7155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rogel A, Hanski E. Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J Biol Chem. 1992;267:22599–22605. [PubMed] [Google Scholar]
- 72.Cheung GYC, Kelly SM, Jess TJ, Prior S, Price NC, Parton R, Coote JG. Functional and structural studies on different forms of the adenylate cyclase toxin of Bordetella pertussis. Microbial Pathogenesis. 2009;46:36–42. doi: 10.1016/j.micpath.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Arnoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 74.Galgani M, De Rosa V, De Simone S, Leonardi A, D’Oro U, Napolitani G, Masci AM, Zappacosta S, Racioppi L. Cyclic AMP modulates the functional plasticity of immature dendritic cells by inhibiting Src-like kinases through protein kinase A-mediated signaling. J Biol Chem. 2004;279:32507–32514. doi: 10.1074/jbc.M403355200. [DOI] [PubMed] [Google Scholar]
- 75.Ehrmann IE, Gray MC, Gordon VM, Gray LS, Hewlett EL. Hemolytic activity of adenylate cyclase toxin from Bordetella pertussis. FEBS Lett. 1991;278:79–83. doi: 10.1016/0014-5793(91)80088-k. [DOI] [PubMed] [Google Scholar]
- 76.Chenal A, Guijarro JI, Raynal B, Delepierre M, Ladant D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: implication for protein secretion. J Biol Chem. 2009;284:1781–1789. doi: 10.1074/jbc.M807312200. [DOI] [PubMed] [Google Scholar]
- 77.Kamanova J, Kofronova O, Masin J, Genth H, Vojtova J, Linhartova I, Benada O, Just I, Sebo P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J Immunol. 2008;181:5587–5597. doi: 10.4049/jimmunol.181.8.5587. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe M, Blobel G. SecB functions as a cytosolic signal recognition factor for protein export in E. coli. Cell. 1989;58:695–705. doi: 10.1016/0092-8674(89)90104-9. [DOI] [PubMed] [Google Scholar]
- 79.Johnson TL, Abendroth J, Hol WG, Sandkvist M. Type II secretion: from structure to function. FEMS Microbiol Lett. 2006;255:175–186. doi: 10.1111/j.1574-6968.2006.00102.x. [DOI] [PubMed] [Google Scholar]
- 80.Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala’Aldeen D. Type V protein Secretion Pathway: the Autotransporter Story. Microbiolgy and Molecular Biology Reviews. 2004;68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 1998;8:410–415. doi: 10.1016/s0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- 82.Yanez ME, Korotkov KV, Abendroth J, Hol WG. Structure of the minor pseudopilin EpsH from the Type 2 secretion system of Vibrio cholerae. J Mol Biol. 2008;377:91–103. doi: 10.1016/j.jmb.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bachert C, Zhang N, Patou J, van Zele T, Gevaert P. Role of staphylococcal superantigens in upper airway disease. Curr Opin Allergy Clin Immunol. 2008;8:34–38. doi: 10.1097/ACI.0b013e3282f4178f. [DOI] [PubMed] [Google Scholar]
- 84.Heyningen SV. Cholera toxin: interaction of subunits with ganglioside GM1. Science. 1974;183:656–657. doi: 10.1126/science.183.4125.656. [DOI] [PubMed] [Google Scholar]
- 85.Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS MicrobiolLett. 2007;266:129–137. doi: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rodighiero C, Tsai B, Rapoport TA, Lencer WI. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moss J, Manganiello VC, Vaughan M. Hydrolysis of nicotinamide adenine dinucleotide by choleragen and its A protomer: possible role in the activation of adenylate cyclase. Proc Natl Acad Sci U S A. 1976;73:4424–4427. doi: 10.1073/pnas.73.12.4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gill DM. Involvement of nicotinamide adenine dinucleotide in the action of cholera toxin in vitro. Proc Natl Acad Sci U S A. 1975;72:2064–2068. doi: 10.1073/pnas.72.6.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- 90.Halm DR, Rechkemmer GR, Schoumacher RA, Frizzell RA. Apical membrane chloride channels in a colonic cell line activated by secretory agonists. Am J Physiol. 1988;254:C505–511. doi: 10.1152/ajpcell.1988.254.4.C505. [DOI] [PubMed] [Google Scholar]
- 91.Nystrom-Asklin J, Adamsson J, Harandi AM. The adjuvant effect of CpG oligodeoxynucleotide linked to the non-toxic B subunit of cholera toxin for induction of immunity against H. pylori in mice. Scand J Immunol. 2008;67:431–440. doi: 10.1111/j.1365-3083.2008.02085.x. [DOI] [PubMed] [Google Scholar]
- 92.Plano GV, Day JB, Ferracci F. Type III export: new uses for an old pathway. Mol Microbiol. 2001;40:284–293. doi: 10.1046/j.1365-2958.2001.02354.x. [DOI] [PubMed] [Google Scholar]
- 93.Marlovits TC, Kubori T, Sukhan A, Thomas DR, Galan JE, Unger VM. Structural insights into the assembly of the type III secretion needle complex. Science. 2004;306:1040–1042. doi: 10.1126/science.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kenjale R, Wilson J, Zenk SF, Saurya S, Picking WL, Picking WD, Blocker A. The needle component of the type III secreton of Shigella regulates the activity of the secretion apparatus. J Biol Chem. 2005;280:42929–42937. doi: 10.1074/jbc.M508377200. [DOI] [PubMed] [Google Scholar]
- 95.Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol Microbiol. 2007;63:1719–1730. doi: 10.1111/j.1365-2958.2007.05620.x. [DOI] [PubMed] [Google Scholar]
- 96.Picking WL, Nishioka H, Hearn PD, Baxter MA, Harrington AT, Blocker A, Picking WD. IpaD of Shigella flexneri is independently required for regulation of Ipa protein secretion and efficient insertion of IpaB and IpaC into host membranes. Infect Immun. 2005;73:1432–1440. doi: 10.1128/IAI.73.3.1432-1440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goehring UM, Schmidt G, Pederson KJ, Aktories K, Barbieri JT. The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J Biol Chem. 1999;274:36369–36372. doi: 10.1074/jbc.274.51.36369. [DOI] [PubMed] [Google Scholar]
- 98.Krall R, Sun J, Pederson KJ, Barbieri JT. In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect Immun. 2002;70:360–367. doi: 10.1128/IAI.70.1.360-367.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wurtele M, Wolf E, Pederson KJ, Buchwald G, Ahmadian MR, Barbieri JT, Wittinghofer A. How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat Struct Biol. 2001;8:23–26. doi: 10.1038/83007. [DOI] [PubMed] [Google Scholar]
- 100.Ganesan AK, Vincent TS, Olson JC, Barbieri JT. Pseudomonas aeruginosa Exoenzyme S disrupts Ras-mediated signal transduction by inhibiting guanine nucleotide exchange factor-catalyzed nucleotide exchange. J Biol Chem. 1999;274:21823–21829. doi: 10.1074/jbc.274.31.21823. [DOI] [PubMed] [Google Scholar]
- 101.Deng Q, Barbieri JT. Modulation of host cell endocytosis by the type III cytotoxin, Pseudomonas ExoS. Traffic. 2008;9:1948–1957. doi: 10.1111/j.1600-0854.2008.00808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maresso AW, Deng Q, Pereckas MS, Wakim BT, Barbieri JT. Pseudomonas aeruginosa ExoS ADP-ribosyltransferase inhibits ERM phosphorylation. Cell Microbiol. 2007;9:97–105. doi: 10.1111/j.1462-5822.2006.00770.x. [DOI] [PubMed] [Google Scholar]
- 103.Fu H, Coburn J, Collier RJ. The eukaryotic host factor that activates exoenzyme S of Pseudomonas aeruginosa is a member of the 14-3-3 protein family. Proc Natl Acad Sci U S A. 1993;90:2320–2324. doi: 10.1073/pnas.90.6.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y, Barbieri JT. A leucine-rich motif targets Pseudomonas aeruginosa ExoS within mammalian cells. Infect Immun. 2005;73:7938–7945. doi: 10.1128/IAI.73.12.7938-7945.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis. 2001;183:1767–1774. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- 106.Corech R, Rao A, Laxova A, Moss J, Rock MJ, Li Z, Kosorok MR, Splaingard ML, Farrell PM, Barbieri JT. Early immune response to the components of the type III system of Pseudomonas aeruginosa in children with cystic fibrosis. J Clin Microbiol. 2005;43:3956–3962. doi: 10.1128/JCM.43.8.3956-3962.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burns DL. Type IV transporters of pathogenic bacteria. Curr Opin Microbiol. 2003;6:29–34. doi: 10.1016/s1369-5274(02)00006-1. [DOI] [PubMed] [Google Scholar]
- 108.Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. Biogenesis, architecture, and function of bacterial type IV secretion systems. Ann Rev Microbiol. 2005;59:451–485. doi: 10.1146/annurev.micro.58.030603.123630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Saier MH. Protein secretion and membrane Insertion systems in Gram-Negative bacteria. J Membrane Biol. 2006;214:75–90. doi: 10.1007/s00232-006-0049-7. [DOI] [PubMed] [Google Scholar]
- 110.Planet PJ, Kachlany SC, DeSalle R, Figurski DH. Phylogney of genese for secretion NTPases: identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. PNAS. 2001;98:2503–2508. doi: 10.1073/pnas.051436598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J. 2003;22:1969–1980. doi: 10.1093/emboj/cdg223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV, Waksman G. Structure of a type IV secretion system core complex. Science. 2009;323:266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Backert S, Meyer TF. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr Opin Microbiol. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 114.de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 2008;4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. 2008;10:2416–2433. doi: 10.1111/j.1462-5822.2008.01219.x. [DOI] [PubMed] [Google Scholar]
- 116.Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala’Aldeen D. Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev. 2004;68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Henderson IR, Navarro-Garcia F, Nataro JP. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 1998;6:370–378. doi: 10.1016/s0966-842x(98)01318-3. [DOI] [PubMed] [Google Scholar]
- 118.Loveless BJ, Saier MH. A novel family of channel-forming, autotransporting, bacterial virulence factors. Mol Membr Biol. 1997;14:801–807. doi: 10.3109/09687689709048171. [DOI] [PubMed] [Google Scholar]
- 119.Yen MR, Peabody CR, Partovi SM, Zhai Y, Tseng YH, Saier MH. Protein-translocating outer membrane porins of Gram-negative bacteria. Biochim Biophys Acta. 2002;1562:6–31. doi: 10.1016/s0005-2736(02)00359-0. [DOI] [PubMed] [Google Scholar]
- 120.Oliver DC, Huang G, Nodel E, Pleasance S, Fernandez RC. A conserved region within the Bordetella pertussis autotransporter BrkA is necessary for folding of its passenger domain. Mol Microbiol. 2003;47:1367–1383. doi: 10.1046/j.1365-2958.2003.03377.x. [DOI] [PubMed] [Google Scholar]
- 121.Brunder W, Schmidt H, Karch H. EspP, a novel extracellular serine protease of enterohaemorrhagic Escherichia coli O157:H7 cleaves human coagulation factor V. Mol Microbiol. 1997;24:767–778. doi: 10.1046/j.1365-2958.1997.3871751.x. [DOI] [PubMed] [Google Scholar]
- 122.Leininger E, Roberts M, Kenimer JG, Charles IG, Fairweather N, Novotny P, Brennan MJ. Pertactin, an Arg-Gly-Asp-containing Bordetella pertussis surface protein that promotes adherence of mammalian cells. Proc Natl Acad Sci U S A. 1991;88:345–349. doi: 10.1073/pnas.88.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.St Geme JW, Cutter D. The Haemophilus influenzae Hia adhesin is an autotransporter protein that remains uncleaved at the C terminus and fully cell associated. J Bacteriol. 2000;182:6005–6013. doi: 10.1128/jb.182.21.6005-6013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Henderson IR, Cappello R, Nataro JP. Autotransporter proteins, evolution and redefining protein secretion. Trends Microbiol. 2000;8:529–532. doi: 10.1016/s0966-842x(00)01853-9. [DOI] [PubMed] [Google Scholar]
- 125.Jacob-Dubuisson F, Locht C, Antoine R. Two-partner secretion in Gram-negative bacteria: a thrifty, specific pathway for large virulence proteins. Mol Microbiol. 2001;40:306–313. doi: 10.1046/j.1365-2958.2001.02278.x. [DOI] [PubMed] [Google Scholar]
- 126.Grass S, St Geme JW. Maturation and secretion of the non-typable Haemophilus influenzae HMW1 adhesin: roles of the N-terminal and C-terminal domains. Mol Microbiol. 2000;36:55–67. doi: 10.1046/j.1365-2958.2000.01812.x. [DOI] [PubMed] [Google Scholar]
- 127.Guedin S, Willery E, Tommassen J, Fort E, Drobecq H, Locht C, Jacob-Dubuisson F. Novel topological features of FhaC, the outer membrane transporter involved in the secretion of the Bordetella pertussis filamentous hemagglutinin. J Biol Chem. 2000;275:30202–30210. doi: 10.1074/jbc.M005515200. [DOI] [PubMed] [Google Scholar]
- 128.Jacob-Dubuisson F, Buisine C, Willery E, Renauld-Mongenie G, Locht C. Lack of functional complementation between Bordetella pertussis filamentous hemagglutinin and Proteus mirabilis HpmA hemolysin secretion machineries. J Bacteriol. 1997;179:775–783. doi: 10.1128/jb.179.3.775-783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Klauser T, Pohlner J, Meyer TF. The secretion pathway of IgA protease-type proteins in gram-negative bacteria. Bioessays. 1993;15:799–805. doi: 10.1002/bies.950151205. [DOI] [PubMed] [Google Scholar]
- 130.Frangione B, Franklin EC. Chemical typing of the immunoglobulins IgM, IgA1 and IgA2. FEBS Lett. 1972;20:321–323. doi: 10.1016/0014-5793(72)80096-6. [DOI] [PubMed] [Google Scholar]
- 131.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. Type VII secretion--mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 134.Schiavo G, Rossetto O, Santucci A, DasGupta RR, Montecucco C. Botulinum Neurotoxins are Zinc Proteases. Journal of Biological Chemistry. 1992;267:23479–23483. [PubMed] [Google Scholar]
- 135.Tanzi MG, Gabay MP. Association between honey consumption and infant botulism. Pharmacotherapy. 2002;22:1479–1483. doi: 10.1592/phco.22.16.1479.33696. [DOI] [PubMed] [Google Scholar]
- 136.Dastoor SF, Misch CE, Wang HL. Botulinum toxin (Botox) to enhance facial macroesthetics: a literature review. J Oral Implantol. 2007;33:164–171. doi: 10.1563/0-835.1. [DOI] [PubMed] [Google Scholar]
- 137.Callaway JE. Botulinum toxin type B (Myobloc): pharmacology and biochemistry. Clin Dermatol. 2004;22:23–28. doi: 10.1016/j.clindermatol.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 138.Dressler D, Munchau A, Bhatia KP, Quinn NP, Bigalke H. Antibody-induced botulinum toxin therapy failure: can it be overcome by increased botulinum toxin doses? Eur Neurol. 2002;47:118–121. doi: 10.1159/000047963. [DOI] [PubMed] [Google Scholar]
- 139.Dressler D, Bigalke H. Botulinum toxin antibody type A titres after cessation of botulinum toxin therapy. Mov Disord. 2002;17:170–173. doi: 10.1002/mds.1238. [DOI] [PubMed] [Google Scholar]
- 140.Suen JC, Hatheway CL, Steigerwalt AG, Brenner DJ. Genetic confirmation of identities of neurotoxigenic Clostridium baratii and Clostridium butyricum implicated as agents of infant botulism. J Clin Microbiol. 1988;26:2191–2192. doi: 10.1128/jcm.26.10.2191-2192.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Popoff M, RaM JC. Structural and genomic features of clostridial neurotoxins. In: Freer JEAaJH., editor. The Comprehensive Sourcebook of Bacterial Portein Toxins. Academic Press; London: 1999. pp. 174–201. [Google Scholar]
- 142.Dineen SS, Bradshaw M, Karasek CE, Johnson EA. Nucleotide sequence and transcriptional analysis of the type A2 neurotoxin gene cluster in Clostridium botulinum. FEMS Microbiol Lett. 2004;235:9–16. doi: 10.1016/j.femsle.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 143.Oguma K, Inoue K, fuginaga Y, yokota K, Watanabe T, Ohyama T, Takeshi K, Inoue K. Structure and function of Clostridium botulinum protenitor toxin. J Toxicol. 1999;18:17–34. [Google Scholar]
- 144.Quinn CP, Minton NP. Clostridial neurotoxins. In: Durre HBaP., editor. Clostridia. Willey-VCH; Veinheim: 2001. pp. 211–250. [Google Scholar]
- 145.Sharma SK, Ramzan MA, Singh BR. Separation of the components of type A botulinum neurotoxin complex by electrophoresis. Toxicon. 2003;41:321–331. doi: 10.1016/s0041-0101(02)00309-4. [DOI] [PubMed] [Google Scholar]
- 146.Rodriguez Jovita M, Collins MD, East AK. Gene organization and sequence determination of the two botulinum neurotoxin gene clusters in Clostridium botulinum type A(B) strain NCTC 2916. Curr Microbiol. 1998;36:226–231. doi: 10.1007/s002849900299. [DOI] [PubMed] [Google Scholar]
- 147.East AK, Richardson PT, Allaway D, Collins MD, Roberts TA, Thompson DE. Sequence of the gene encoding type F neurotoxin of Clostridium botulinum. FEMS Microbiol Lett. 1992;75:225–230. doi: 10.1016/0378-1097(92)90408-g. [DOI] [PubMed] [Google Scholar]
- 148.Sugii S, Ohishi I, Sakaguchi G. Correlation between oral toxicity and in vitro stability of Clostridium botulinum type A and B toxins of different molecular sizes. Infect Immun. 1977;16:910–914. doi: 10.1128/iai.16.3.910-914.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bandyopadhyay S, Clark AW, DasGupta BR, Sathyamoorthy V. Role of the heavy and light chains of botulinum neurotoxin in neuromuscular paralysis. J Biol Chem. 1987;262:2660–2663. [PubMed] [Google Scholar]
- 150.Couesnon A, Pereira Y, Popoff MR. Receptor-mediated transcytosis of botulinum neurotoxin A through intestinal cell monolayers. Cell Microbiol. 2007;10:375–387. doi: 10.1111/j.1462-5822.2007.01051.x. [DOI] [PubMed] [Google Scholar]
- 151.Jin Y, Takegahara Y, Sugawara Y, Matsumura T, Fujinaga Y. Disruption of the epithelial barrier by botulinum haemagglutinin (HA) proteins--differences in cell tropism and the mechanism of action between HA proteins of types A or B, and HA proteins of type C. Microbiology. 2009;155:35–45. doi: 10.1099/mic.0.021246-0. [DOI] [PubMed] [Google Scholar]
- 152.Maksymowych AB, Simpson LL. Binding and transcytosis of botulinum neurotoxin by polarized human colon carcinoma cells. J Biol Chem. 1998;273:21950–21957. doi: 10.1074/jbc.273.34.21950. [DOI] [PubMed] [Google Scholar]
- 153.Park JB, Simpson LL. Inhalational poisoning by botulinum toxin and inhalation vaccination with its heavy-chain component. Infect Immun. 2003;71:1147–1154. doi: 10.1128/IAI.71.3.1147-1154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Couesnon A, Pereira Y, Popoff MR. Receptor-mediated transcytosis of botulinum neurotoxin A through intestinal cell monolayers. Cell Microbiol. 2008;10:375–387. doi: 10.1111/j.1462-5822.2007.01051.x. [DOI] [PubMed] [Google Scholar]
- 155.Couesnon A, Shimizu T, Popoff MR. Differential entry of botulinum neurotoxin A into neuronal and intestinalcells. Cell Microbiol. 2009;11:289–308. doi: 10.1111/j.1462-5822.2008.01253.x. [DOI] [PubMed] [Google Scholar]
- 156.Kitamura M, Takamiya K, Aizawa S, Furukawa K. Gangliosides are the binding substances in neural cells for tetanus and botulinum toxins in mice. Biochim Biophys Acta. 1999;1441:1–3. doi: 10.1016/s1388-1981(99)00140-7. [DOI] [PubMed] [Google Scholar]
- 157.Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, Chapman ER. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592–596. doi: 10.1126/science.1123654. [DOI] [PubMed] [Google Scholar]
- 158.Dong M, Liu H, Tepp WH, Johnson EA, Janz R, Chapman ER. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol Biol Cell. 2008;19:5226–5237. doi: 10.1091/mbc.E08-07-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rummel A, Karnath T, Henke T, Bigalke H, Binz T. Synaptotagmins I and II Act as Nerve Cell Receptors for Botulinum Neurotoxin G 10.1074/jbc.M403945200. J Biol Chem. 2004;279:30865–30870. doi: 10.1074/jbc.M403945200. [DOI] [PubMed] [Google Scholar]
- 160.Dong M, Richards DA, Goodnough MC, Tepp WH, Johnson EA, Chapman ER. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J Cell Biol. 2003;162:1293–1303. doi: 10.1083/jcb.200305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tsukamoto K, Kohda T, Mukamoto M, Takeuchi K, Ihara H, Saito M, Kozaki S. Binding of Clostridium botulinum type C and D neurotoxins to ganglioside and phospholipid. Novel insights into the receptor for clostridial neurotoxins. J Biol Chem. 2005;280:35164–35171. doi: 10.1074/jbc.M507596200. [DOI] [PubMed] [Google Scholar]
- 162.Rummel A, Bade S, Alves J, Bigalke H, Binz T. Two carbohydrate binding sites in the H(CC)-domain of tetanus neurotoxin are required for toxicity. J Mol Biol. 2003;326:835–847. doi: 10.1016/s0022-2836(02)01403-1. [DOI] [PubMed] [Google Scholar]
- 163.Yowler BC, Schengrund CL. Botulinum neurotoxin A changes conformation upon binding to ganglioside GT1b. Biochemistry. 2004;43:9725–9731. doi: 10.1021/bi0494673. [DOI] [PubMed] [Google Scholar]
- 164.Muraro L, Tosatto S, Motterlini L, Rossetto O, Montecucco C. The N-terminal half of the receptor domain of botulinum neurotoxin A binds to microdomains of the plasma membrane. Biochem Biophys Res Commun. 2009;380:76–80. doi: 10.1016/j.bbrc.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 165.Jung N, Haucke V. Clathrin-mediatedendocytosis at synapses. Traffic. 2007;8:1129–1136. doi: 10.1111/j.1600-0854.2007.00595.x. [DOI] [PubMed] [Google Scholar]
- 166.Morgans CW, Kensel-Hammes P, Hurley JB, Burton K, Idzerda R, McKnight GS, Bajjalieh SM. Loss of the Synaptic Vesicle Protein SV2B results in reduced neurotransmission and altered synaptic vesicle protein expression in the retina. PLoS ONE. 2009;4:e5230. doi: 10.1371/journal.pone.0005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baldwin MR, Barbieri JT. Association of Botulinum neurotoxins with synaptic vesicle protein complexes. Toxicon. 2009 doi: 10.1016/j.toxicon.2009.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fischer A, Mushrush DJ, Lacy DB, Montal M. Botulinum neurotoxin devoid of receptor binding domain translocates active protease. PLoS Pathog. 2008:4. doi: 10.1371/journal.ppat.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang J, Meng J, Lawrence GW, Zurawski TH, Sasse A, Bodeker MO, Gilmore MA, Fernandez-Salas E, Francis J, Steward LE, Aoki KR, Dolly JO. Novel chimeras of Botulinum Neurotoxins A and E unveil contributions from the binding, translocation, and protease domains to their functional characteristics. J Biol Chem. 2008;283:16993–17002. doi: 10.1074/jbc.M710442200. [DOI] [PubMed] [Google Scholar]
- 170.Fischer A, Montal M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. PNAS. 2007;104:10447–10452. doi: 10.1073/pnas.0700046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Montal M. Translocation of botulinum neurotoxin light chain protease by the heavy chain protein-conducting channel. Toxicon. 2008 doi: 10.1016/j.toxicon.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lacy DB, Stevens RC. Sequence homology and structural analysis of the clostridial neurotoxins. J Mol Biol. 1999;291:1091–1104. doi: 10.1006/jmbi.1999.2945. [DOI] [PubMed] [Google Scholar]
- 173.Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Sudhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem. 1994;269:1617–1620. [PubMed] [Google Scholar]
- 174.Foran P, Lawrence GW, Shone CC, Foster KA, Dolly JO. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry. 1996;35:2630–2636. doi: 10.1021/bi9519009. [DOI] [PubMed] [Google Scholar]
- 175.Schiavo G, Shone CC, Bennett MK, Scheller RH, Montecucco C. Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J Biol Chem. 1995;270:10566–10570. doi: 10.1074/jbc.270.18.10566. [DOI] [PubMed] [Google Scholar]
- 176.Arndt JW, Yu W, Bi F, Stevens RC. Crystal structure of botulinum neurotoxin type G light chain: serotype divergence in substrate recognition. Biochemistry. 2005;44:9574–9580. doi: 10.1021/bi0505924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen S, Hall C, Barbieri JT. Substrate recognition of VAMP-2 by botulinum neurotoxin B and tetanus neurotoxin. J Biol Chem. 2008;283:21153–21159. doi: 10.1074/jbc.M800611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ahmed SAOM, Ludivico ML, Gilsdorf J, Smith LA. Identification of Residues Surrounding the Active Site of Type A Botulinum Neurotoxin Important for Substrate Recognition and Catalytic Activity. Protein J. 2008;27:151–162. doi: 10.1007/s10930-007-9118-8. [DOI] [PubMed] [Google Scholar]
- 179.Chen S, Barbieri JT. Unique substrate recognition by botulinum neurotoxins serotypes A and E. Journal of Biological Chemistry. 2006;281:10906–10911. doi: 10.1074/jbc.M513032200. [DOI] [PubMed] [Google Scholar]
- 180.Chen S, Kim JP, Barbieri JT. Mechanism of Substrate Recognition by Botulinum Neurotoxin Serotype A. J Biol Chem. 2007;282:9621–9627. doi: 10.1074/jbc.M611211200. [DOI] [PubMed] [Google Scholar]
- 181.Rigoni M, Caccin P, Johnson EA, Montecucco C, Rossetto O. Site-Directed Mutagenesis Identifies Active-Site Residues of the Light Chain of Botulinum Neurotoxin Type A. Biochemical and Biophysical Research Communications. 2001;288:1231–1237. doi: 10.1006/bbrc.2001.5911. [DOI] [PubMed] [Google Scholar]
- 182.Schmidt JJ, Stafford RG. Botulinum neurotoxin serotype F: identification of substrate recognition requirements and development of inhibitors with low nanomolar affinity. Biochemistry. 2005;44:4067–4073. doi: 10.1021/bi0477642. [DOI] [PubMed] [Google Scholar]
- 183.Sikorra S, Henke T, Swaminathan S, Galli T, Binz T. Identification of the amino acid residues rendering TI-VAMP insensitive toward botulinum neurotoxin B. J Mol Biol. 2006;357:574–582. doi: 10.1016/j.jmb.2005.12.075. [DOI] [PubMed] [Google Scholar]
- 184.Arndt JW, Chai Q, Christian T, Stevens R. Structure of Botulinum Neurotoxin Type D Light Chain at 1.65 A Resolution: Repercussions for VAMP-2 Substrate Specificity. Biochemistry. 2006;45:3255–3262. doi: 10.1021/bi052518r. [DOI] [PubMed] [Google Scholar]
- 185.Breidenbach MA, Brunger AT. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature. 2004;432:925–929. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- 186.Chen S, Kim JJ, Barbieri JT. Mechanism of substrate recognition by botulinum neurotoxin serotype A. J Biol Chem. 2007;282:9621–9627. doi: 10.1074/jbc.M611211200. [DOI] [PubMed] [Google Scholar]
- 187.Smith TJ, Lou J, Geren IN, Forsyth CM, Tsai R, LaPorte SL, Tepp WH, Bradshaw M, Johnson EA, Smith LA, et al. Sequence Variation within Botulinum Neurotoxin Serotypes Impacts Antibody Binding and Neutralization. Infect Immun. 2005;73:5450–5457. doi: 10.1128/IAI.73.9.5450-5457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Arndt JW, Jacobson MJ, Abola EE, Forsyth CM, Tepp WH, Marks JD, Johnson EA, Stevens RC. A structural perspective of the sequence variability within botulinum neurotoxin subtypes A1–A4. J Mol Biol. 2006;362:733–742. doi: 10.1016/j.jmb.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 189.Hill KK, Smith TJ, Helma CH, Ticknor LO, Foley BT, Svensson RT, Brown JL, Johnson EA, Smith LA, Okinaka RT, et al. Genetic diversity among Botulinum Neurotoxin-producing clostridial strains. J Bacteriol. 2007;189:818–832. doi: 10.1128/JB.01180-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Edmond BJ, Guerra FA, Blake J, Hempler S. Case of infant botulism in Texas. Tex Med. 1977;73:85–88. [PubMed] [Google Scholar]
- 191.Leighton GR. Report to the Scottish Board of Health. H.M. Stationery Office; London: 1923. [Google Scholar]
- 192.Henkel JS, Jacobson M, Tepp W, Pier C, Johnson EA, Barbieri JT. Catalytic Properties of Botulinum Neurotoxin Subtypes A3 and A4 (dagger) Biochemistry. 2009;48:2522–2528. doi: 10.1021/bi801686b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Baldwin MR, Tepp WH, Przedpelski A, Pier CL, Bradshaw M, Johnson EA, Barbieri JT. Subunit vaccine against the seven serotypes of botulism. Infect Immun. 2008;76:1314–1318. doi: 10.1128/IAI.01025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 195.Schiavo G, Shone CC, Bennett MK, Scheller RH, Montecucco C. Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J Biol Chem. 1995;270:10566–10570. doi: 10.1074/jbc.270.18.10566. [DOI] [PubMed] [Google Scholar]
- 196.Williamson LC, Halpern JL, Montecucco C, Brown JE, Neale EA. Clostridial neurotoxins and substrate proteolysis in intact neurons: botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J Biol Chem. 1996;271:7694–7699. doi: 10.1074/jbc.271.13.7694. [DOI] [PubMed] [Google Scholar]
- 197.Schiavo G, Rossetto O, Catsicas S, Polverino de Laureto P, DasGupta BR, Benfenati F, Montecucco C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- 198.Schiavo G, Shone CC, Rossetto O, Alexander FC, Montecucco C. Botulinum neurotoxin serotype F is a zinc endopeptidase specific for VAMP/synaptobrevin. J Biol Chem. 1993;268:11516–11519. [PubMed] [Google Scholar]
- 199.Schiavo G, Malizio C, Trimble WS, Polverino de Laureto P, Milan G, Sugiyama H, Johnson EA, Montecucco C. Botulinum G neurotoxin cleaves VAMP/synaptobrevin at a single Ala-Ala peptide bond. J Biol Chem. 1994;269:20213–20216. [PubMed] [Google Scholar]