

SUMMARY
Mice lacking Junctional Adhesion Molecule A (JAM-A, encoded by F11r) exhibit enhanced intestinal epithelial permeability, bacterial translocation, and elevated colonic lymphocyte numbers, yet do not develop colitis. To investigate the contribution of adaptive immune compensation in response to increased intestinal epithelial permeability, we examined the susceptibility of F11r-/-Rag1-/- mice to acute colitis. Although negligible contributions of adaptive immunity in F11r-/-Rag1-/- mice were observed, F11r-/-Rag1-/- mice exhibited increased microflora-dependent colitis. Elimination of T cell subsets and cytokine analyses revealed a protective role for TGF-β-producing CD4+ T cells in F11r-/- mice. Additionally, loss of JAM-A resulted in elevated mucosal and serum IgA that was dependent upon CD4+ T cells and TGF-β. Absence of IgA in F11r+/+Igha-/- mice did not affect disease whereas F11r-/-Igha-/- mice displayed markedly increased susceptibility to acute injury induced colitis. These data establish a role for adaptive immune mediated protection from acute colitis under conditions of intestinal epithelial barrier compromise.
INTRODUCTION
The pathogenesis of many inflammatory conditions of mucosal surfaces involves the combined dysfunction of the mucosal barrier and local immune responses to gut luminal contents. For example, compromised intestinal barrier function reported in people with inflammatory bowel disease (IBD) (Mankertz and Schulzke, 2007; Welcker et al., 2004) has been linked to alterations in expression of barrier forming tight junction molecules (Dogan et al., 1995; Gassler et al., 2001; Jankowski et al., 1998; Karayiannakis et al., 1998; Kucharzik et al., 2001). One transmembrane protein component of tight junctions that has been linked to regulation of intestinal permeability and has been linked to IBD is Junctional Adhesion Molecule A (JAM-A) (Laukoetter et al., 2007; Vetrano et al., 2008). JAM-A (encoded by F11r) has been implicated in a number of cellular functions including regulation of paracellular permeability, cell migration, and proliferation (Corada et al., 2005; Liu et al., 2000; Mandell et al., 2005; Martin-Padura et al., 1998; Nava et al.; Severson et al., 2008; Severson et al., 2009). JAM-A is expressed mainly at cell-cell junctions between epithelia and endothelia and is also expressed on the surface of certain immune cells including neutrophils and dendritic cells (Cera et al., 2004; Liu et al., 2000). We have previously shown that JAM-A deficiency in mice leads to a 10-fold increase in intestinal epithelial permeability (Laukoetter et al., 2007). Another unique feature of JAM-A-deficient mice that we previously observed is increased numbers of mucosal and submucosal isolated lymphoid aggregates in the colon, which predominantly contain B220+ cells.
Interestingly, despite a 10-fold increase in colonic permeability and lymphoid follicular hyperplasia in the colon, JAM-A-deficient mice do not develop spontaneous colitis. Although many reports have emphasized the pathogenic role of T and B lymphocytes in the development of colitis (Davidson et al., 1996; Mombaerts et al., 1993), subsets of T cells such as CD4+CD25+Foxp3+ T regulatory (Treg) cells are clearly involved in limiting or suppressing intestinal inflammation (Barnes and Powrie, 2009). In addition to their direct role in maintaining intestinal homeostasis, T cells also play a crucial role in the induction of IgA-mediated mucosal immunity. The main function of luminal IgA antibodies is to neutralize and prevent bacteria from penetrating deeper into mucosal tissues and causing immune activation (Boullier et al., 2009; Fernandez et al., 2003; Robinson et al., 2001). Such adaptive immune protection may be critical in the face of increased intestinal permeability where there is enhanced exposure to luminal microbial products, yet immune compensatory mechanisms in response to compromised intestinal permeability remain poorly understood.
In this study, we used JAM-A-deficient (F11r-/-) mice to investigate how adaptive immune pathways may compensate for a major increase in intestinal permeability to prevent spontaneous intestinal inflammation. Our results demonstrate that enhanced mucosal T and B cell responses, TGF-β production and IgA secretion compensate for the leaky barrier and increased bacterial translocation in JAM-A-deficient mice. Elimination of individual adaptive immune components revealed a critical protective role for TGFβ-producing CD4+ T cells promoting IgA secretion in JAM-A-deficient mice. Altogether, our findings establish a role for adaptive immune responses in limiting severe, acute mucosal injury in the context of intestinal barrier compromise.
RESULTS
Loss of T and B cells in JAM-A-deficient mice increases susceptibility to acute colitis
Given our previous observations that JAM-A-deficient mice display a 10-fold increase in intestinal epithelial permeability and greatly increased numbers of colonic lymphoid aggregates, we investigated whether adaptive immune cells play a compensatory role in preventing JAM-A-deficient mice from developing spontaneous colitis. We first evaluated the lymphocyte composition in the large intestine of F11r+/+ and F11r-/- (JAM-A-deficient) mice. As shown in Fig. 1A, F11r-/- mice exhibited a significant increase in T and B cells. The lamina propria of F11r-/- mice displayed a 2.4±0.8 fold increase in TCRβ+ cells and a 5.2±1.0 fold increase in B220+ cells compared to F11r+/+ mice. Additionally, we observed a 4.5±1.3 fold increase in CD4+IL-17A+ T cells in F11r-/- mice compared to F11r+/+ controls (Fig. 1B). In contrast, no significant differences were observed in the numbers of CD4+IL-4+ or CD4+IFN-γ+ T cells between F11r-/- and F11r+/+ mice. Notably, CD3+CD4+, CD3+CD8+ T cells and B220+ cells did not express cell surface JAM-A (data not shown), excluding the possibility that absence of JAM-A expression on lymphocytes in F11r-/- mice could directly alter their accumulation and/or function. We then generated F11r-/- mice deficient in T and B cells by crossing F11r-/- mice with Rag1-/- mice. F11r-/-Rag1-/- mice did not develop spontaneous colitis, however, approximately 15% of the animals developed spontaneous severe mucocutaneous infections from commensal Pasteurella microorganisms requiring sacrifice. All experiments presented here were performed using healthy mice that had no sign of infection.
Figure 1. Lack of adaptive immunity in JAM-A-deficient mice results in enhanced susceptibility to DSS-induced acute colitis.
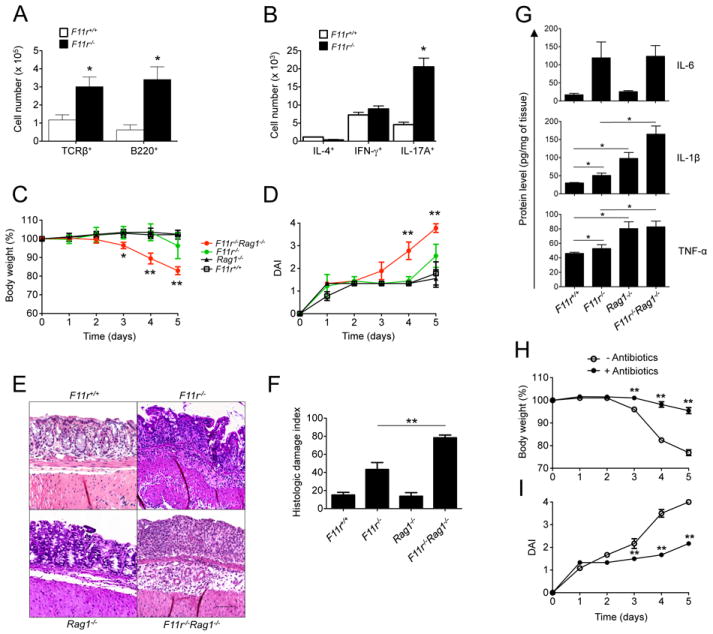
(A) Absolute cell numbers of T and B lymphocytes, determined from F11r+/+ or F11r-/- (JAM-A-deficient) colonic lamina propria using flow cytometry based on TCRβ and B220 surface expression, respectively. Data are representative of three independent experiments (n=4) and are presented as means ± SD. (B) Absolute numbers of IL-4+, IFN-γ+ and IL-17A+ cells in F11r+/+ or F11r-/- colonic lamina propria as determined by flow cytometry. T cell subsets were first gated based on TCRβ and CD4 surface expression, then based on IL-4, IFN-γ or IL-17A intracellular expression following PMA and ionomycin restimulation ex-vivo. Data are shown as means ± SD and are representative of two independent experiments (n=4). (C) Body weight changes and (D) disease activity index scores (DAI) from F11r+/+, F11r-/-, Rag1-/- and F11r-/-Rag1-/- mice following 2% DSS treatment (n=5). Body weights are calculated as percent of the initial body weight at day 0 and DAI scores are calculated as described in the Experimental Procedures. Data are shown as means ± SD and are representative of at least three independent experiments. (E) Representative hematoxylin and eosin staining photomicrographs from F11r+/+, F11r-/-, Rag1-/- and F11r-/-Rag1-/- mice at day 5 following 2% DSS treatment. Images show sections of colon highlighting predominant histological findings in each of the experimental groups that ranged from mild (F11r+/+Rag1-/-), to moderate (F11r-/-), to severe epithelial injury with extensive mucosal ulceration (F11r-/-Rag1-/-). Scale represents 100 μm. (F) Histologic damage index scores from swiss roll mounts of whole mouse colons collected after 5 days of DSS administration. Data are from 3 mice per group and represented as mean ± SD. *, P < 0.05; **, P < 0.01. (G) IL-6, IL-1β and TNF-α protein levels in F11r+/+, F11r-/- and F11r-/-Rag1-/- mice (n = 4), measured in large intestine homogenates by ELISA as described in the Experimental Procedures. *, P < 0.05. (H) Body weight change represented as percent of the initial body weight at day 0 in F11r-/-Rag1-/- mice treated with a broad spectrum antibiotic cocktail as described in the Experimental Procedures, prior to DSS treatment, or left untreated (n = 5). (I) Disease activity index scores for antibiotic-treated and untreated mice. **, P < 0.01.
In order to evaluate whether adaptive immunity played a role in the response to acute mucosal injury, we treated F11r-/-Rag1-/- mice with DSS and monitored disease activity. F11r-/-Rag1-/- mice were far more susceptible to DSS induced colitis when compared to F11r-/- mice, characterized by significant body weight loss (4.8±0.07% of initial body weight) and presence of blood in their stools as early as day 3 of DSS treatment (Fig. 1C/D). By day 5, disease activity in the F11r-/-Rag1-/- mice was so severe that sacrifice was necessary, with animals consistently losing more than 20% of the initial body weight (23±0.2%) and displaying severe diarrhea and macroscopic signs of bleeding. Histological analyses at day 5 revealed extensive colonic injury in F11r-/-Rag1-/- mice compared to F11r-/-, Rag1-/- or F11r+/+ littermate control mice (Fig. 1E/F). The extent of mucosal injury characterized by crypt loss, epithelial damage and ulceration, was significantly less in other groups at the same time point. Colonic inflammation was mainly restricted to the mucosa and submucosa, however, focal areas of transmural inflammation were also observed in F11r-/-Rag1-/- mice. Analysis of several proinflammatory cytokines in colonic homogenates from F11r-/-Rag1-/- mice revealed increased levels of IL-1β, IL-6, and TNF-α (Fig. 1G).
Next, we investigated whether the enhanced susceptibility to DSS-mediated acute colitis in F11r-/-Rag1-/- mice was driven by the local increased exposure to the intestinal microflora. F11r-/-Rag1-/- mice were administered a mixture of broad-spectrum antibiotics for 7 days prior to DSS treatment to limit the gut flora. Following DSS treatment, we found that antibiotic-treated F11r-/-Rag1-/- mice were significantly less susceptible to DSS compared with mice not treated with antibiotics. The reduced susceptibility to DSS was evidenced by a general decrease in body weight loss (Fig. 1H) and disease activity index (Fig. 1I). The same trend was observed in F11r-/- mice where there was a decrease in disease activity index in antibiotic-treated mice compared to untreated controls (data not shown). Altogether, these results demonstrate that adaptive immunity is a critical compensatory component that limits bacterial-driven acute colitis in JAM-A-deficient mice.
CD4+ T cells are a key component in protecting JAM-A-deficient mice from acute mucosal injury
Since we observed that lack of adaptive immune cells in JAM-A-deficient mice promote intestinal inflammation during acute DSS-mediated colitis, we examined the specific role of T cells in controlling intestinal inflammation in JAM-A-deficient mice using an antibody-based depletion approach. To this end, we used CD4 and CD8α antibodies to deplete the respective T cell subsets in vivo and tested whether they were critical in preventing excessive intestinal inflammation in F11r-/- mice following injury induced acute colitis. F11r-/- mice treated with anti-CD4 were significantly more susceptible to DSS than untreated F11r+/+ mice, losing 6.1% of their initial body weight starting at day 3 and 21.0% by day 6 (Fig. 2A). Anti-CD4 treated F11r-/- mice exhibited disease symptoms as early as day 4, similar to what was observed for F11r-/-Rag-/- mice, including loose stool and presence of macroscopic, fecal blood. The disease was more severe at day 5 and 6 with signs of diarrhea and further body weight loss (Fig. 2B). Histological analyses of the large intestine at day 6 revealed severe mucosal ulceration reminiscent of the histopathology associated with F11r-/-Rag-/- mice (Fig. 2C/D). In contrast, treatment of F11r-/- mice with anti-CD8 did not have any effect on the susceptibility to DSS, with anti-CD8 treated F11r-/- mice displaying disease activity indices and histopathology scores similar to untreated F11r-/- mice. Importantly, T cell subset depletion in the spleens and colons was confirmed by flow cytometry (data not shown). Furthermore, depleting antibodies did not affect DSS-induced colitis in F11r+/+ mice whether administered in combination or individually (data not shown).
Figure 2. CD4+ T cell depletion in JAM-A-deficient mice leads to enhanced susceptibility to DSS-induced acute colitis.
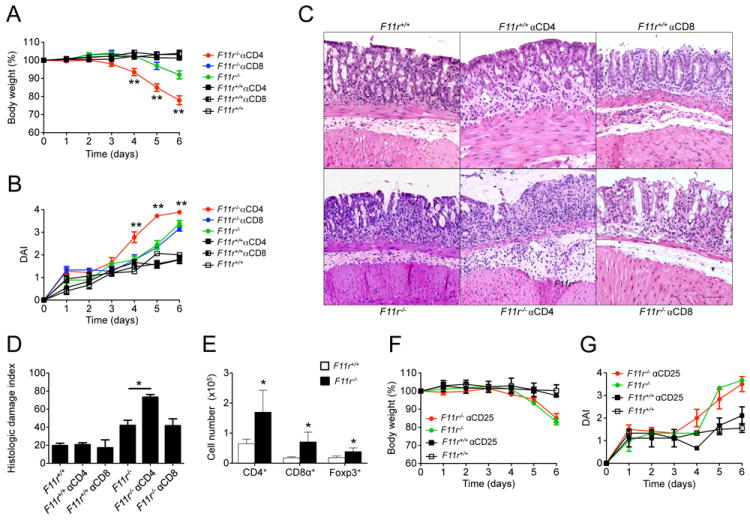
(A) Body weight changes represented as percent of the initial body weight at day 0 from F11r+/+ and F11r-/- (JAM-A-deficient) mice treated with 500 μg of anti-CD4 (GK1.5) or anti-CD8 (YTS169.4) antibodies at day -2 and day -1 prior to DSS treatment, or left untreated. (B) Disease activity index scores for anti-CD4 or anti-CD8-treated mice and untreated controls. Data are shown as means ± SEM and are pooled from three independent experiments (n=6). (C) Photomicrographs of representative hematoxylin and eosin stained colon sections from F11r+/+ and F11r-/- mice treated with anti-CD4 or anti-CD8 antibodies, or untreated control groups at day 5 following 2% DSS treatment. Images show sections of colon highlighting predominant histological findings in each of the experimental groups. Scale represents 100 μm. (D) Histologic damage index scores from swiss roll mounts of whole mouse colons collected after 5 days of DSS treatment. Data are from 3 mice per group and represented as mean ± SD. *, P < 0.05; **, P < 0.01. (E) Absolute cell numbers of CD4+, CD8+ and Foxp3+ T cell subsets, measured by flow cytometry on lamina propria cell preparations from F11r+/+ and F11r-/- mice (n=4). (F) Body weight changes in F11r+/+ and F11r-/- mice treated with 100 μg of anti-CD25 antibody at day -5 and day -2 prior to DSS treatment, or left untreated. (G) Disease activity index scores for anti-CD25 antibody-treated and untreated animals. Data are pooled from three independent experiments (n = 9) and are represented as means ± SEM.
To further investigate if particular CD4+ T cell subsets were responsible for limiting excessive intestinal inflammation in JAM-A-deficient mice during acute mucosal injury, we investigated the role of CD4+CD25+Foxp3+ Treg cells in compensating for defective barrier function. We first determined the numbers of CD4+CD25+Foxp3+ Treg cells in F11r-/- lamina propria cell preparations using flow cytometry. F11r-/- mice had a 2.5±0.6 fold increase in the numbers of CD4+CD25+Foxp3+ Treg cells in the lamina propria compared to F11r+/+ mice (Fig. 2E). We then treated F11r-/- mice with anti-CD25 following a previously published protocol to neutralize intestinal CD4+CD25+ Treg cells (Cong et al., 2009), and disease activity was monitored after DSS administration. There were no significant differences between anti-CD25 - treated F11r-/- mice and untreated F11r-/- controls (Fig. 2F/G). Weight loss during DSS-induced acute colitis in anti-CD25 treated F11r-/- mice followed a similar pattern as that of untreated F11r-/- control mice. Also, no differences were observed in terms of stool consistency and presence of occult blood. Depletion of CD4+Foxp3+ Treg cells in the intestinal lamina propria was incomplete (approximately 30% depletion), thus limiting definitive assessment of the role of Foxp3+ Treg cells in this protective response. Altogether, these results highlight an important role of CD4+ T cells in adaptive immune compensation to acute injury under conditions of chronic barrier compromise.
CD4-dependent TGF-β production suppresses acute colitis in JAM-A-deficient mice
In addition to CD4+ Treg cells, TGFβ-producing CD4+ T cells are also well appreciated to regulate intestinal immune responses (Thorstenson and Khoruts, 2001; Zhang et al., 2001). Interestingly, we observed that baseline TGF-β mRNA and protein levels in colonic homogenates were more than two-fold higher in F11r-/- mice when compared to F11r+/+ mice (Fig. 3A, B). To investigate the role for enhanced TGFβ expression in F11r-/- mice, animals were treated with neutralizing TGFβ antibodies at day -5 and day -2 prior to DSS treatment. Antibody-treated F11r-/- mice were far more susceptible to DSS-induced acute colitis than untreated F11r-/- mice, beginning to lose weight from day 3 post DSS treatment (6.6±1.1% weight loss) and losing close to 20% of their initial body weight by day 6 (20.6±4.4% weight loss) (Fig. 3C). In addition, anti-TGFβ treated F11r-/- mice also developed diarrhea at a much earlier time point (day 4 post DSS treatment) than their untreated counterparts, which contributed to a significantly higher disease activity index (3.2±0.1 vs. 2.1±0.1) (Fig. 3D). Histologic analyses revealed extensive ulceration, mucosal injury and inflammation in the anti-TGFβ treated- F11r-/- mice that were similar to that observed with F11r-/-Rag-/- mice treated with DSS (Fig. 3E/F). In contrast, the increased susceptibility to DSS following anti-TGFβ administration was not observed in F11r+/+ control mice. In addition, treatment of F11r-/- mice with an IgG1 isotype control antibody did not have any effect on the course of the disease (data not shown).
Figure 3. TGFβ signaling protects from acute colonic inflammation JAM-A-deficient mice.
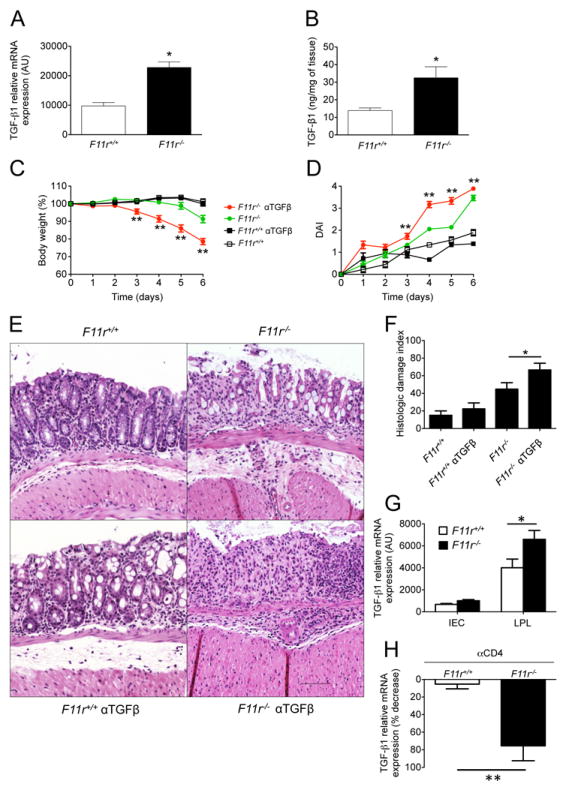
(A) TGFβ mRNA levels in F11r+/+ and F11r-/- (JAM-A-deficient) mice were evaluated by real-time PCR using total RNA from colon as described in the Experimental Procedures. Data are presented as relative expression following normalization with respect to gapdh. (B) TGFβ protein levels in F11r+/+ and F11r-/- mice were determined by ELISA using colonic tissue homogenates. (C) Body weight changes and (D) disease activity index scores (DAI) from F11r+/+ and F11r-/- mice treated with 100 μg of anti-TGFβ neutralizing antibodies at day -5 and day -2 prior to DSS treatment, or left untreated. Data are represented as means ± SEM and are pooled from three independent experiments (n=10). (E) Photomicrographs of representative hematoxylin and eosin stained colon sections from F11r+/+ and F11r-/- mice treated with anti-TGFβ neutralizing antibodies, or untreated control groups at day 5 following 2% DSS treatment. Images show sections of colon highlighting predominant histological findings in each of the experimental groups. Scale represents 100 μm. (F) Histologic damage index scores from swiss roll mounts of whole mouse colons collected after 5 days of DSS treatment. Data are from 3 mice per group and represented as mean ± SD. *, P < 0.05; **, P < 0.01. (G) TGFβ mRNA levels were measured in colonic intestinal epithelial cells (IEC) and lamina propria lymphocyte (LPL) samples by real-time PCR. Data are normalized to the endogenous control gapdh. (H) TGFβ mRNA levels in colons of F11r+/+ and F11r-/- mice treated with anti-CD4 depleting antibodies (GK1.5) at day -2 and day -1 with 500 μg of antibody per mouse per injection, or left untreated, followed by 6 days of DSS treatment. Data are shown as the mean ± SEM (n=3) percent reduction in TGFβ mRNA levels after CD4 T cell depletion. Expression levels were normalized to the endogenous control gapdh. *, P < 0.05; **, P < 0.01.
Because JAM-A-deficient mice were observed to have increased mucosal TGFβ production, and anti-CD4 as well as anti-TGFβ administration led to enhanced susceptibility to DSS, we investigated whether CD4+ T cells were the source of TGFβ. Real-time PCR data from lamina propria lymphocyte fractions, which are highly enriched in CD4+ T cells, indeed revealed that lymphocytes are a major source of TGFβ (Fig. 3G). Furthermore, to evaluate whether CD4+ T cells contribute to the elevated TGFβ amounts observed in F11r-/- mice, we quantified cytokine mRNA in the colons of DSS treated F11r+/+ and F11r-/- mice after CD4+ T cell depletion. Depletion of CD4+ T cells in DSS treated F11r-/- mice resulted in nearly an 80% reduction of TGFβ compared to that of F11r+/+ controls (Fig. 3H), consistent with CD4+ T cells being the major source for TGFβ in F11r-/- mice.
Increased IgA and bacterial translocation in JAM-A-deficient mice
We previously demonstrated that JAM-A-deficient mice have increased numbers of isolated lymphoid follicles in the large intestine when compared to wild-type mice (Laukoetter et al., 2007). Since these lymphoid aggregates are mainly composed of B220 staining B cells, we examined whether this could be associated with increased IgA production. By immunofluorescence staining, we found elevated amounts of IgA in colonic mucosa from F11r-/- mice compared to F11r+/+ control mice (Fig. 4A/B/C). Consistent with the elevated levels of IgA in the colonic mucosa, there were abundant collections of lamina propria CD138+IgA+ plasma cells in F11r-/- mice (Fig. 4D). Furthermore, quantification of CD138+IgA+ plasma cells revealed a significant increase in F11r-/- mice compared to F11r+/+ control mice (Fig. 4D). These elevated local amounts of IgA were also associated with higher amounts of systemic IgA as demonstrated by ELISA of serum samples from F11r-/- mice (Fig. 4E). The increased amounts of IgA were isotype specific since there was no significant difference in serum IgM or IgG. Furthermore, no difference in IgM staining was observed between F11r+/+ and F11r-/- colonic mucosa (data not shown).
Figure 4. Increased IgA levels and bacterial translocation in JAM-A-deficient mice.
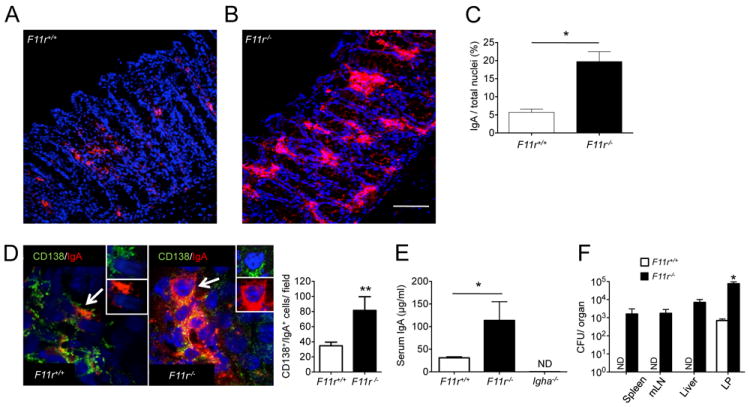
(A, B) Immunofluorescence labeling of IgA in F11r+/+ and F11r-/- (JAM-A-deficient) colonic mucosa. Frozen sections (7 μm thickness) were stained with a monoclonal anti-IgA antibody directly conjugated with PE (red). Tissues were counterstained with the nuclei stain TO-PRO-3 (blue). Scale represents 50 μm. In (C), quantification of IgA staining was performed in F11r+/+ and F11r-/- colonic mucosa on ten different fields per section (n = 3) as described in the Experimental Procedures. (D) Representative images of colonic mucosa from F11r+/+ and F11r-/- mice showing CD138/IgA double positive cells. Insets show CD138 staining (green, inset) and IgA staining (red, inset) on representative double positive cells indicated by arrows. Tissues were counterstained with TO-PRO-3 (blue). Scale bar is 10mm. Lower bar graph represents the mean± SD of the number of CD138/IgA double positive cells per high power field (40x) in the colonic lamina propria from 40 determinations (two mice per group). ** P <0.005. (E) Serum IgA levels as determined by ELISA assays on F11r+/+ and F11r-/- mice samples. Negative controls include serum samples from Igha-/- mice (ND, none detected). * P < 0.05. (F) Bacterial CFU counts from spleen, mLN, liver and colonic lamina propria (LP) samples from F11r+/+ and F11r-/- mice (n=3). Organs were collected and tissue homogenates were prepared as described in the Experimental Procedures. Serial dilutions were plated onto blood agar medium and the numbers of bacterial CFU were enumerated. No bacterial growth was observed for F11r+/+ spleen, mLN and liver samples (ND). * P < 0.05.
Because JAM-A-deficient mice have increased colonic permeability, we examined whether the increased IgA levels correlated with enhanced translocation of luminal bacteria into the colonic LP. Bacterial counts were determined by plating serial dilutions and enumerating CFU in colonic LP tissue homogenates derived from washed mucosa that was processed to remove the epithelium. We found a significantly higher number of bacteria in the colonic LP from F11r-/- mice compared to F11r-/- control mice with a two log-difference in the number of CFU (Fig. 4F). In addition, bacterial growth was also detected in the spleen, mesenteric lymph nodes and in the liver from F11r-/- mice but not from F11r-/- control mice, suggesting that in F11r-/- mice bacteria that translocated into the lamina propria further disseminated into secondary lymphoid organs and peripheral tissues.
IgA production limits acute colitis in JAM-A-deficient mice
Given the observation that CD4+ T cell-mediated responses are critical to limit the susceptibility of JAM-A-deficient mice to DSS-induced colitis and that IgA levels are significantly increased, we crossed F11r-/- mice with IgA-deficient (Igha-/-) mice and measured their susceptibility to acute injury induced colitis. F11r-/-Igha-/- mice were far more susceptible to DSS-induced acute colitis than F11r-/- mice or Igha-/- mice. F11r-/-Igha-/- mice weighed only 83.5±1.0% of their initial body weights compared to 92.1±1.7% for F11r-/- mice at day 6 (Fig. 5A), and had significantly higher disease activity indices than F11r-/- mice beginning at day 4 of DSS treatment (Fig. 5B). Histologic analyses of colonic mucosa confirmed significantly increased disease in the F11r-/-Igha-/- mice as evident by extensive mucosal injury and ulceration (Fig. 5C/D). Given that TGFβ is important for IgA class switch recombination and plasma cell differentiation, we explored whether TGFβ impacted IgA production in F11r-/- mice. First, we examined IgA in DSS-treated F11r-/- mice that were depleted of CD4+ T cells, since these cells are the predominant producers of TGFβ. We observed a significant decrease in IgA protein levels in CD4+ T cell-depleted JAM-A-deficient mice compared to untreated F11r-/- mice (Fig. 5E). Moreover, TGFβ neutralization also resulted in a significant reduction of IgA protein levels in anti-TGFβ treated F11r-/- mice compared with untreated F11r-/- mice (Fig. 5F). Altogether, these data demonstrate that increased production of IgA is a unifying protective mechanism promoted by TGFβ-producing CD4+ T cells that compensates for increased colonic permeability under conditions of acute mucosal injury.
Figure 5. TGFβ-producing CD4+ T cells induce IgA production and limit acute colitis in JAM-A-deficient mice.
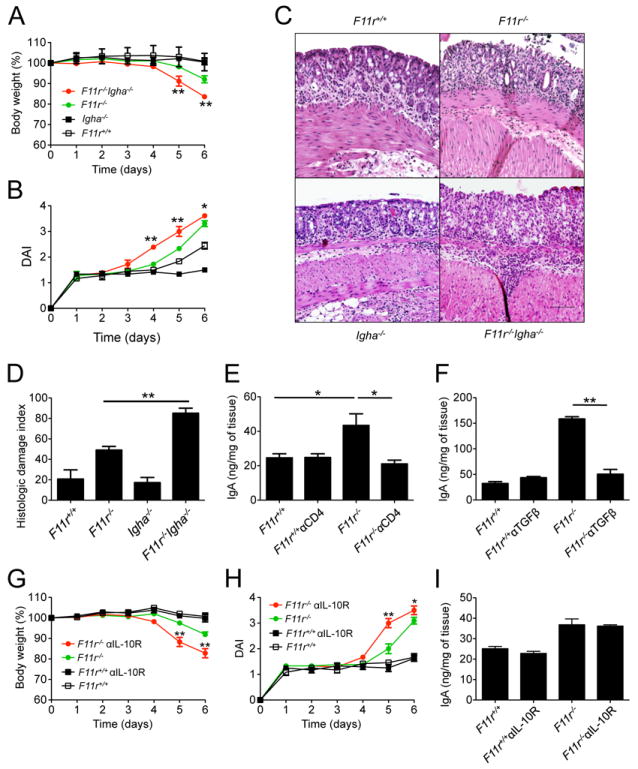
(A) Body weight changes and (B) disease activity index scores (DAI) in F11r+/+, F11r-/- (JAM-A-deficient), Igha-/- and F11r-/-Igha-/- mice following 2% DSS treatment. Data are pooled from three independent experiments and are represented as means ± SEM (n=9). (C) Representative hematoxylin and eosin staining photomicrographs of colonic tissue sections from in F11r+/+, F11r-/-, Igha-/- and F11r-/-Igha-/- mice at day 5 of DSS treatment. Images show sections of colon highlighting predominant histological findings in each of the experimental groups. Scale represents 100 μm. (D) Histologic damage index scores from swiss roll mounts of whole mouse colons collected after 5 days of DSS treatment. Data are from 3 mice per group and represented as mean ± SD. (E) IgA protein levels in F11r+/+ and F11r-/- colonic tissues, with or without anti-CD4 antibody treatment (E), or with or without anti-TGFβ treatment (F), as measured by ELISA. Representative data are shown as mean ± SEM (n=4). *, P < 0.05; **, P < 0.01. Body weight loss (G) and disease activity index (H) in F11r+/+ and F11r-/- mice following anti-IL-10 receptor antibody administration. Control groups included mice not treated with the antibodies. Data are pooled from two independent experiments (n=9) and are represented as means ± SEM. (I) IgA protein levels measured by ELISA in F11r+/+ and F11r-/- mice treated with anti-IL-10 receptor antibodies. Representative data are shown as means ± SEM (n=4). *, P < 0.05; **, P < 0.01.
Since TGFβ was shown to induce IgA and limit colonic disease induced by DSS, we also investigated the role of IL-10, another cytokine involved in maintaining intestinal homeostasis. Blockade of IL-10 signaling in F11r-/- mice using IL-10 receptor antibodies resulted in a significant increase in disease susceptibility compared to untreated F11r-/- mice, albeit to a lesser extent compared to anti-TGFβ treatment (Fig. 5G/H). However, treatment of F11r-/- mice with anti-IL-10R antibodies did not have any significant effects on colonic IgA levels as determined by ELISA (Fig. 5I), suggesting that the protective effects of IL-10 in F11r-/- mice are independent of IgA.
DISCUSSION
Intestinal immune cells are constantly challenged to maintain homeostasis in the face of stimulation by gut microbiota. In a situation of defective epithelial barrier function as encountered in JAM-A-deficient mice, one would predict that spontaneous disease might develop due to increased bacterial translocation across the epithelium. Interestingly, despite defects in barrier function, JAM-A-deficient mice do not develop clinical signs of spontaneous colitis, which suggests that protective mechanisms are in place to suppress inflammatory cues and prevent spontaneous disease. In the present study, we provide evidence that adaptive immunity, which plays a negligible role in protecting from acute intestinal inflammation in the context of a normal epithelial barrier, is indeed essential in protecting from acute, injury-induced colitis in the context of increased intestinal permeability. In particular, this adaptive immunity-based “protective” effect is dependent on TGFβ-producing CD4+ T cells that serve to promote IgA secretion in JAM-A-deficient mice. Altogether, these observations establish a role for adaptive immune responses in limiting severe, acute mucosal injury in the context of intestinal barrier compromise.
Our findings that acute intestinal inflammation is markedly enhanced in JAM-A-deficient mice lacking CD4+ T cells are in accordance with the well established role of immunoregulatory T cells in suppressing colitis (Groux et al., 1997; Kim et al., 2007; Powrie et al., 1996; Rubtsov et al., 2008; Sakaguchi et al., 1985; Zhang et al., 2001). Thus, it would be tempting to speculate that the enhanced susceptibility of JAM-A-deficient mice to DSS-induced acute colitis is due to the absence of functional CD4+CD25+ Treg cells. A critical role for CD4+CD25+ Treg cells in mediating IgA production in response to commensal flora has been reported (Cong et al., 2009). Here we used anti-CD25 to attempt to eliminate CD4+Foxp3+ Treg cells but only achieved partial depletion and observed no increase in susceptibility to colitis. While our findings preclude definitive assessment of the role of Foxp3+ Treg cells in regulating the adaptive immune compensation observed here, they are consistent with those reported by Cong et al (Cong et al., 2009) and highlight a critical role for intestinal permeability in regulating TGFβ-producing Treg cell-mediated IgA production.
We also evaluated the relative contribution of TGF-β, which is well appreciated for its role in mediating Treg cell immunosuppressive functions (Li et al., 2007; Marie et al., 2005). Neutralization of TGF-β in JAM-A-deficient mice led to the striking finding that compensation for intestinal barrier defects also depends on TGFβ signaling. It is possible that the increased TGFβ production in JAM-A-deficient mice compared with wild-type controls represents a secondary response to increased bacterial translocation and subsequent immune activation. Given the fact that TGFβ amounts in JAM-A/Rag1-deficient mice are greatly reduced compared to JAM-A-deficient mice (data not shown), we demonstrated that TGFβ producing T cells are a major source of TGFβ in JAM-A-deficient mice. We further showed that increased colonic TGFβ production in JAM-A-deficient mice during acute mucosal injury-induced colitis is CD4+ T cell-dependent. These findings suggest that TGF-β secreting cells (Weiner, 2001) may be a critical component of immune-mediated protection during increased intestinal permeability.
The role of T cells in promoting TGFβ-mediated immune regulation of intestinal inflammation has been previously described in a model of CD4+CD45RBhi-induced colitis. T cells that express dnTGFβRII escape immunoregulation by Treg cells, indicating that TGFβ-mediated control of inflammation is T-cell dependent (Fahlen et al., 2005). Thus, in JAM-A-deficient mice, TGFβ-producing CD4+ T cells may regulate pathogenic CD4+ T cells to limit intestinal inflammation. However, it is also conceivable that TGFβ exerts its effect via T cell-independent mechanisms. Our data are also consistent with TGFβ-producing CD4+ T cells playing a critical role in IgA isotype class-switch recombination in B cells, thereby promoting IgA-mediated immune responses and control of intestinal inflammation in the gut induced by antigen exposure (Borsutzky et al., 2004; Cazac and Roes, 2000; Cerutti, 2008).
Importantly, loss of IgA appears to be a unifying mechanism by which depletion of CD4+ T cells and neutralization of TGFβ increases susceptibility to acute mucosal damage in the context of a leaky epithelial barrier. Based on our data, it is clear that IgA-associated responses are critical for immune mediated-compensation during enhanced intestinal permeability. Murine studies have demonstrated an important role for IgA antibodies in immune exclusion. For example, mice deficient in the polymeric immunoglobulin receptor, which are unable to secrete IgA into the gut lumen, display increased mucosal leakiness and antigen uptake (Johansen et al., 1999; Sait et al., 2007). The current study highlights the observations that: a) IgA production is augmented in mice with a leaky gut barrier, most likely representing a humoral compensatory mechanism in response to the increased antigen uptake across the epithelium, and b) that regulation of intestinal inflammation in the specific context of increased epithelial permeability and acute mucosal injury relies on IgA-mediated immunity. The exact mechanisms by which IgA antibodies participate in dampening tissue inflammation in JAM-A-deficient mice remains unclear but may involve increased immune exclusion at the interface between lumen and epithelial cells, and/or increased transport of antigens across the epithelium and subsequent recognition and phagocytosis by antigen-presenting cells.
It is important to note that the development of intestinal inflammation during DSS-induced acute mucosal injury is a lymphocyte-independent process in wild-type mice (Dieleman et al., 1994). However, in JAM-A-deficient mice, which have enhanced colonic permeability, severity of colitis depends on adaptive immune cells, as demonstrated by the increased susceptibility to DSS in JAM-A/Rag1-deficient mice compared to JAM-A-deficient mice. This finding implicates a crucial role of adaptive immunity under conditions of existing defects in barrier function.
In summary, our findings provide new insights into the regulation of mucosal homeostasis in the context of impaired intestinal epithelial barrier function. JAM-A-deficient mice represent an important tool to study of immune mechanisms implicated in the containment of acute inflammatory responses triggered by enhanced exposure to luminal antigens and immune activation. We have identified a essential role for TGFβ-producing CD4+ T cells promoting IgA secretion in controlling intestinal inflammation in the presence of a compromised intestinal epithelial barrier.
EXPERIMENTAL PROCEDURES
Mice
JAM-A-deficient mice (F11r-/-) and littermate controls were bred onsite. Rag1tm1Mom (Rag1-/-) mice on a C57BL/6 background were purchased from The Jackson Laboratories (Bar Harbor). IgA-deficient mice (Igha-/-) (Harriman et al., 1999) were a kind gift from Dr. L. Eckmann. Rag1-/- and Igha-/- mice were crossed with JAM-A-deficient mice mice to generate F11r-/-Rag1-/- and F11r-/-Igha-/- mice. Mice were maintained under specific pathogen-free conditions at Emory University. All animal procedures were reviewed and approved by the Emory University Institutional Animal Care and Use Committee.
Genotyping
PCRs were performed on tail genomic DNA using the following primers: F11r forward 5’-TCTTTTCACCAATCGGAACG-3’ and reverse 5’-CGGCATTAATCCCAGAAGGT-3’; Rag1 forward common 5’-CCGGACAAGTTTTTCATCGT-3’, reverse wild-type allele 5’-GAGGTTCCGCTACGACTCTG-3’ and reverse mutant allele 5’-TGGATGTGGAATGTGTGCGAG-3’.
DSS-induced colitis
Mice were provided 2% (wt/vol) DSS (molecular mass = 36 – 50 kD) (MP Biomedicals) for 5 to 7 days and then colons were assessed for weight, length, and histology. Daily clinical assessment of DSS-treated animals included evaluation of stool consistency, detection of blood in stool, and body weight loss measurement. An individual score (ranging from 0 to 4) was attributed for each one of these parameters, and a disease activity index (DAI) ranging from 0 to 4 was calculated by combining all three scores (Laukoetter et al., 2007).
Histology
Histological examination was performed on whole colons of experimental and control mice. In addition, colonic tissue samples were embedded in OCT for immunofluorescence staining and stored at -80°C. Histological parameters were quantified in a blinded fashion by two experienced gastrointestinal pathologists using parameters as previously described (Nava et al.) with some modification. Briefly, hematoxylin and eosin sections of Swiss roll mounts of entire mouse colons were assessed for the percentage of the mucosa containing ulceration or crypt epithelial injury/damage/active inflammation comprising at least 1/3 of the thickness of the mucosa. The ulceration and injury/damage values were added and reported as a histologic damage index.
Flow cytometry
Lamina propria lymphocyte isolation was performed as previously described (Denning et al., 2011; Medina-Contreras et al., 2011). Intestinal cell preparations included lymphoid follicles. Cells were stained with PerCP-conjugated anti-CD45 (30-F11), FITC-conjugated anti-CD19 (1D3), allophycocyanin-conjugated TCRβ (H57-597), eFluor® 450-conjugated anti-CD4 (L3T4) (eBioscience) and PE/Texas Red-conjugated anti-CD8α (5H10) (Invitrogen) monoclonal antibodies. Data was acquired using a LSR II flow cytometer (Becton Dickinson) and analyzed using Flowjo software (Treestar). For intracellular cytokine detection, cells were restimulated with PMA and ionomycin ex-vivo in the presence of GolgiPlug (BD Biosciences).
Cytokine and IgA assays
For TGFβ1 protein level analyses, proximal colonic tissues were homogenized in sterile 1X PBS supplemented with 1% Triton X-100 and protease and phosphatase inhibitor cocktails (Sigma), and centrifuged for 20 min at maximum speed at 4°C. Supernatants were used to measure TGFβ1, IL-6, IL-1β and TNF-α protein levels using ELISA kits (eBioscience). IgA protein levels were evaluated on colonic homogenate supernatants or serum samples using an ELISA kit (Immunology Consultants Laboratory, Inc.).
Broad-spectrum antibiotic treatment
Mice were treated using a modified protocol described elsewhere (Rakoff-Nahoum et al., 2004).
Bacterial translocation
Organs were weighed and colon length was measured. For the spleen and mLN, the whole organ was homogenized in 500 μl of sterile PBS. For the liver, a piece (200 mg) of the right lobe was collected and homogenized. For colonic bacterial counts, fecal contents were removed by rinsing longitudinally opened colons with sterile PBS. Colons were then cut into 1.5 cm segments and epithelial cells were removed by vigorous horizontal shaking at 37°C for 20 min in HBSS containing 5% FBS, 10 mM HEPES and 2 mM EDTA pH 8.0. This procedure was repeated two times. Samples were subsequently homogenized in 500 μl of sterile PBS using an Omni-Prep homogenizer. Serial dilutions were then plated onto blood agar plates, incubated at 37°C for 36 hr and colony-forming units (CFU) were enumerated.
Antibody-mediated depletion
Anti-CD4 (clone GK1.5) and anti-CD8α (YTS169.4) neutralizing antibodies (Bio X Cell) were used. For each antibody, two 500 μg injections were given i.p. at day -2 and day -1 prior to DSS treatment. CD25 depletion was performed as previously described (Cong et al., 2009). To assess depletion efficiency, leukocytes were isolated from large intestine, small intestine, spleen and mLN and subjected to flow cytometric analyses. TGFβ depletion prior to DSS treatment was carried out by performing two 100 μg injections i.p. at day -5 and day -2 prior to DSS treatment using anti-TGFβ (1D11.16.8) monoclonal antibody (Bio X Cell). For IgA protein level measurement, chronic TGFβ depletion was conducted by injecting one week-old mice with 100 μg of anti-TGFβ neutralizing antibodies twice weekly for the first 2 weeks followed by 100 μg once every week for a total of 7 weeks.
Real-time PCR analysis
Total RNA was extracted from proximal colons of F11r-/- and F11r+/+ mice using RNeasy® Mini kit (Invitrogen) and genomic DNA contamination was removed following DNaseI treatment (Qiagen). Real-time PCR reactions were set up using the iScript™ One step RT-PCR kit with SYBR® Green (Bio-Rad Laboratories) and the following primers: forward tgfβ1 5’-ACCATGCCAACTTCTGTCTG-3’ and reverse tgfβ1 5’-CGGGTTGTGTTGGTTGTAGA-3’; forward gapdh 5’-TCATTGACCTCAACTACATGGTCTA-3’ and reverse gapdh 5’-ACACCAGTAGACTCCACGACATACT-3’. For the preparations of IECs and lamina propria-derived lymphocytes, cell suspensions were processed through a Percoll gradient and individual fractions were collected and used for RNA extraction. Reactions were run on a MyiQ Single color Real-time PCR Detection system (Bio-Rad Laboratories) and Ct values were used to calculate relative expression.
Confocal microscopy
Frozen tissue sections (7 μm thickness) from large intestines were fixed in cold ethanol for 20 min at -20°C. Sections were then blocked with 3% BSA prior to incubation with PE-conjugated rat anti-mouse IgA (eBioscience) for 20 min. Plasma cells were stained using anti-mouse CD138 (eBioscience). Tissues were counterstained with TO-PRO-3 for 3 min and mounted on coverslips. Images were acquired with a LSM 510 confocal microscope (Zeiss) and analyzed using Image J software (National Institutes of Health).
Data analysis
Results are expressed as means ± SEM unless otherwise indicated. P values were calculated using one-way ANOVA and unpaired two-tailed Student’s t-test. P < 0.05 was considered statistically significant.
Acknowledgments
The authors thank Dr. L. Eckmann for providing Igha-/- mice, Dr. R. Ahmed and Dr. R.S. Mittler for providing GK1.5 and YTS169.4 antibodies, and Winston Lee and Christopher Capaldo for discussions. This work was supported by grants from the NIH (DK061379 and DK72564 to C.A.P.; DK055679 and DK59888 to A.N.; and AA017870 and AI083554 to T.L.D.), core support from a Digestive Diseases Minicenter grant (DK 064399), a Emory Egleston Children’s Research Center seed grant to T.L.D., a Crohn’s and Colitis Foundation of America Career Development Award and an AGA Foundation for Digestive Health and Nutrition Research Scholar Award to P.N., and Crohn’s and Colitis Foundation of America Research Fellowship and senior research awards to M.K and B.F, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Borsutzky S, Cazac BB, Roes J, Guzman CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol. 2004;173:3305–3309. doi: 10.4049/jimmunol.173.5.3305. [DOI] [PubMed] [Google Scholar]
- Boullier S, Tanguy M, Kadaoui KA, Caubet C, Sansonetti P, Corthesy B, Phalipon A. Secretory IgA-mediated neutralization of Shigella flexneri prevents intestinal tissue destruction by down-regulating inflammatory circuits. J Immunol. 2009;183:5879–5885. doi: 10.4049/jimmunol.0901838. [DOI] [PubMed] [Google Scholar]
- Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- Cera MR, Del Prete A, Vecchi A, Corada M, Martin-Padura I, Motoike T, Tonetti P, Bazzoni G, Vermi W, Gentili F, et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J Clin Invest. 2004;114:729–738. doi: 10.1172/JCI21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A. 2009;106:19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102:10634–10639. doi: 10.1073/pnas.0500147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson NJ, Leach MW, Fort MM, Thompson-Snipes L, Kuhn R, Muller W, Berg DJ, Rennick DM. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med. 1996;184:241–251. doi: 10.1084/jem.184.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, Karp CL, Pulendran B. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. 2011;187:733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- Dogan A, Wang ZD, Spencer J. E-cadherin expression in intestinal epithelium. J Clin Pathol. 1995;48:143–146. doi: 10.1136/jcp.48.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MI, Pedron T, Tournebize R, Olivo-Marin JC, Sansonetti PJ, Phalipon A. Anti-inflammatory role for intracellular dimeric immunoglobulin a by neutralization of lipopolysaccharide in epithelial cells. Immunity. 2003;18:739–749. doi: 10.1016/s1074-7613(03)00122-5. [DOI] [PubMed] [Google Scholar]
- Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermuller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–228. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, Zhang Y, Mbawuike IN. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J Immunol. 1999;162:2521–2529. [PubMed] [Google Scholar]
- Jankowski JA, Bedford FK, Boulton RA, Cruickshank N, Hall C, Elder J, Allan R, Forbes A, Kim YS, Wright NA, Sanders DS. Alterations in classical cadherins associated with progression in ulcerative and Crohn’s colitis. Lab Invest. 1998;78:1155–1167. [PubMed] [Google Scholar]
- Johansen FE, Pekna M, Norderhaug IN, Haneberg B, Hietala MA, Krajci P, Betsholtz C, Brandtzaeg P. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J Exp Med. 1999;190:915–922. doi: 10.1084/jem.190.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiannakis AJ, Syrigos KN, Efstathiou J, Valizadeh A, Noda M, Playford RJ, Kmiot W, Pignatelli M. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J Pathol. 1998;185:413–418. doi: 10.1002/(SICI)1096-9896(199808)185:4<413::AID-PATH125>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113(Pt 13):2363–2374. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J Biol Chem. 2005;280:11665–11674. doi: 10.1074/jbc.M412650200. [DOI] [PubMed] [Google Scholar]
- Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol. 2007;23:379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, Parkos CA, Denning TL. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–282. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA, Nusrat A. JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep. 12:314–320. doi: 10.1038/embor.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 32:392–402. doi: 10.1016/j.immuni.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Robinson JK, Blanchard TG, Levine AD, Emancipator SN, Lamm ME. A mucosal IgA-mediated excretory immune system in vivo. J Immunol. 2001;166:3688–3692. doi: 10.4049/jimmunol.166.6.3688. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Sait LC, Galic M, Price JD, Simpfendorfer KR, Diavatopoulos DA, Uren TK, Janssen PH, Wijburg OL, Strugnell RA. Secretory antibodies reduce systemic antibody responses against the gastrointestinal commensal flora. Int Immunol. 2007;19:257–265. doi: 10.1093/intimm/dxl142. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–1925. doi: 10.1091/mbc.E08-10-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorstenson KM, Khoruts A. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J Immunol. 2001;167:188–195. doi: 10.4049/jimmunol.167.1.188. [DOI] [PubMed] [Google Scholar]
- Vetrano S, Rescigno M, Cera MR, Correale C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–214. doi: 10.1034/j.1600-065x.2001.1820117.x. [DOI] [PubMed] [Google Scholar]
- Welcker K, Martin A, Kolle P, Siebeck M, Gross M. Increased intestinal permeability in patients with inflammatory bowel disease. Eur J Med Res. 2004;9:456–460. [PubMed] [Google Scholar]
- Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001;167:4245–4253. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]