

The structure of the FnI-EGF-like tandem domain was solved to 1.6 Å using data sets of native and holmium chloride bound protein. Putative biological roles of this tandem domain are discussed based on the structure.
Keywords: FXII, Hageman factor, experimental phasing, fibronectin type I domain, EGF-like domain
Abstract
Coagulation factor XII (FXII) is a key protein in the intrinsic coagulation and kallikrein–kinin pathways. It has been found that negative surfaces and amyloids, such as Aβ fibrils, can activate FXII. Additionally, it has been suggested that FXII simulates cells and that it plays an important role in thrombosis. To date, no structural data on FXII have been deposited, which makes it difficult to support any hypothesis on the mechanism of FXII function. The crystal structure of the FnI-EGF-like tandem domain of FXII presented here was solved using experimental phases. To determine the phases, a SIRAS approach was used with a native and a holmium chloride-soaked data set. The holmium cluster was coordinated by the C-terminal tails of two symmetry-related molecules. Another observation was that the FnI domain was much more ordered than the EGF-like domain owing to crystal packing. Furthermore, the structure shows the same domain orientation as the homologous FnI-EGF-like tandem domain of tPA. The plausibility of several proposed interactions of these domains of FXII is discussed. Based on this FXII FnI-EGF-like structure, it could be possible that FXII binding to amyloid and negatively charged surfaces is mediated via this part of FXII.
1. Introduction
Decades after its discovery, the blood coagulation factor factor XII (FXII), also known as Hageman factor, remains without a clear physiological function. Its essential role in in vitro blood clotting is much better understood. Extensive research has been performed on the type of surfaces and molecules that activate FXII in vitro, leading to initiation of the intrinsic coagulation pathway and the kallikrein–kinin system (Griep et al., 1986 ▶; Tazi et al., 1992 ▶; Matata et al., 1996 ▶). The search for a physiological function of FXII has resulted in a number of possible roles, such as the induction of mitosis (Schmeidler-Sapiro et al., 1991 ▶; Gordon et al., 1996 ▶), angiogenesis (LaRusch et al., 2010 ▶), inflammation reactions (Jansen et al., 1996 ▶) and complement activation (Ghebrehiwet et al., 1981 ▶). Additionally, FXII has been found to have an important role in thrombus formation (Renné et al., 2005 ▶; Meijden et al., 2009 ▶).
Besides negatively charged artificial surfaces, several ‘natural’ surfaces have been proposed to activate FXII in vivo, such as amyloid β aggregates (Yasuhara et al., 1994 ▶; Bergamaschini et al., 1998 ▶, 2001 ▶), polyphosphates (Smith et al., 2006 ▶; Müller et al., 2009 ▶), collagen (Meijden et al., 2009 ▶) and misfolded proteins (Maas, Govers-Riemslag et al., 2008 ▶). Remarkably, not all of these surfaces result in activation of both the intrinsic coagulation pathway and the kallikrein–kinin system. Recently, it has been shown that FXII can also be activated by hydrophobic surfaces. However, the addition of other proteins to the buffer inhibited activation (Zhuo et al., 2006 ▶). This implies that FXII is not predisposed to interact with and be activated by hydrophobic surfaces.
Not only has clarifying its physiological function been a difficult task, but determination of the importance of the various FXII domains in the binding to activating surfaces has also been difficult. FXII consists of six domains, a fibronectin type II (FnII) domain, two EGF-like domains, a fibronectin type I (FnI) domain, a kringle domain and a serine protease domain, and it also contains a proline-rich region with little homology to other proteins (Cool et al., 1985 ▶; Fig. 1 ▶ a). Most evidence points to the FnII domain as the domain which interacts with negatively charged surfaces, but the N-terminal peptide, the FnI domain and the kringle or proline-rich domain have also been implicated in binding (Pixley et al., 1987 ▶; Clarke et al., 1989 ▶; Citarella et al., 1996 ▶, 2000 ▶). Another study showed that the FnI domain of FXII can bind cross-β structure, as found in amyloid and misfolded proteins (Maas, Schiks et al., 2008 ▶).
Figure 1.

(a) The domain composition of FXII shows the seven domains, with FnII as the N-terminal domain and SP as the C-terminal domain. The amino-acid sequence of the crystallized FXII-FnIE is depicted under the domain composition. The underlined sequences are the FnI domain (green; Glu114–Ala154) and the EGF domain (magenta; Ser155–Ala194); the residues that are not underlined are cloning artifacts. (b) Coomassie-stained SDS–PAGE gel showing FXII-FnIE after purification. (c, d) Crystals of FXII-FnIE obtained using 0.1 M Tris–HCl pH 8.5 (c) or 0.1 M bis-tris–HCl pH 6.5 (d) with 17% PEG 4K and 0.2 M potassium acetate
FXII has also been implicated in the stimulation of a number of cell types. The binding of FXII to the surface of these cells results in the activation of several kinase signalling pathways, leading to mitosis. Some studies have attributed this to interaction between one of the EGF-like domains of FXII and the EGF receptor (EGFR) on these cells (Schmeidler-Sapiro et al., 1991 ▶; Gordon et al., 1996 ▶). An indirect activation model has also been proposed in which FXII binds to the uPA receptor via its FnII domain, after which a multiple receptor complex is formed (LaRusch et al., 2010 ▶). The activation of kinase signalling pathways was found to be independent of the protease activity of FXII (LaRusch et al., 2010 ▶), ruling out any form of enzymatic activation of this pathway, as is the case in the coagulation and kallikrein–kinin pathways.
Currently, no structures of FXII or of FXII domains have been deposited in the PDB. Here, we describe the structure of the putative amyloid-binding domain (the FnI domain) in combination with the second EGF domain (FXII-FnIE) and compare it with the structure of the FnI-EGF domain of tissue-type plasminogen activator (tPA), which is also involved in amyloid binding (Maas, Schiks et al., 2008 ▶). Based on this structure, we discuss the possible binding mode to negatively charged surfaces and amyloid surfaces and the possibility that FXII binds EGFR via the EGF domain.
2. Methods
2.1. Cloning of FXII-FnIE
The FnI-EGF tandem-domain DNA was amplified from FXII cDNA (SNP variant rs17876030) by a standard PCR reaction using a forward primer containing a BamHI restriction site (GGATCCGAGAAGTGCTTTGAGCCTCAGCTTCTCC), a reverse primer containing a NotI restriction site (GCGGCCGCGGTGCCACGTCGCAGAAGGGTC) and PfuTurbo DNA polymerase. The resulting product encoded amino acids 114–194 of mature FXII (or 133–213 of FXII including the signal peptide; UniProt entry P00748). The construct was designed based on the UniProt domain boundaries in combination with the domain boundaries of tPA FnI-EGF (Smith et al., 1995 ▶). Owing to the restriction sites for BamHI and NotI, two additional residues at the N-terminus (Gly-Ser) and two additional Ala residues at the C-terminus were present in the construct. The BamHI- and NotI-digested product was ligated into pPICZα plasmid (Invitrogen); after amplification, this plasmid was linearized with SacI. Picha pastoris X-33 cells were transformed with the plasmid by electroporation. Colonies were screened for expression of FXII-FnIE using 1 ml buffered glycerol medium (BMG; see the Pichia Expression Manual, Invitrogen; http://www.invitrogen.com/content/sfs/manuals/pich_man.pdf) for biomass production and 1 ml buffered methanol medium (BMM) for protein expression. The cells were grown in BMM for 72 h at 295–298 K with shaking and 20 µl 50% methanol was added to the cultures every day. The highest expressing clone was selected and used for large-scale production of FXII-FnIE.
2.2. Expression and purification of FXII-FnIE
The selected P. pastoris clone was grown in BMG for 36 h at 301 K to create biomass. This culture was used to inoculate BMM at a 1:50 ratio. This culture was incubated for 72 h at 301 K and 7.5 ml methanol was added to the culture every 12 h to sustain expression. The incubation was performed in a flask at a shaking rate of 250 rev min−1.
The expression medium was harvested, filtered over a 0.45 µm filter and concentrated to 150 ml using a hollow-fibre column with a 5 kDa molecular-weight cutoff (MWCO). Using the same hollow-fibre column, the medium was exchanged to 25 mM MES–NaOH pH 6, 50 mM NaCl. The FXII-FnIE solution was loaded onto a Capto S column (GE Healthcare) and eluted with a linear gradient from 50 to 500 mM NaCl in 100 column volumes. Fractions containing FXII-FnIE were collected, concentrated to 0.4 ml and loaded onto a Superdex 75 column equilibrated in 10 mM HEPES–NaOH pH 7.5, 75 mM NaCl. FXII-FnIE eluted as a single peak, which was collected and concentrated to ∼10 mg ml−1.
2.3. Crystallization of FXII-FnIE
Several commercially available crystallization screens were used for the initial screening. 150 nl FXII-FnIE solution and 150 nl reservoir solution were mixed in Corning 3550 sitting-well plates using a HoneyBee liquid-handling robot (Genomic Solutions Ltd). Conditions containing crystals were further optimized using the hanging-drop vapour-diffusion technique by combining 1 µl FXII-FnIE solution and 1 µl well solution. Crystals for data collection were grown in 0.1 M bis-tris–HCl pH 6.5 or 0.1 M Tris–HCl pH 8.5 with 16–18% PEG 4000 and 0.2 M potassium acetate at 291 K. Before cryocooling, some crystals were soaked for several hours in the same solution with 5 mM potassium gold cyanide at pH 8.5 or for shorter time periods of 10–60 min with 3 mM potassium tetranitroplatinate(II) at pH 6.5, 3 mM potassium hexachloroplatinate (IV) at pH 6.5, 10 mM holmium chloride at pH 6.5 or pH 8.5 or 2 mM lead nitrate at pH 8.5. Subsequently, the crystals were back-soaked in several drops of the crystallization condition supplemented with 25% glycerol.
Crystals grown at pH 8.5 were also soaked with cross-β-forming peptides, with the aim of the determination of the structure of FXII-FnIE in complex with these peptides. The peptides Aβ14–24 (HQKLVFFAEDV), FP6 (IDIKIR), FP13 (KRLEVDIDIKIRS) and the Sup35-fragment GNNQQNY were dissolved in DMSO at a concentration of 20 mM and diluted in 0.1 M Tris–HCl pH 8.5, 17% PEG 4000, 0.2 M potassium acetate to a final concentration of 1 mM peptide and 5% DMSO. The molar ratio of peptide to FXII-FnIE was approximately 1:1. The crystals were quickly back-soaked in two drops of mother liquor supplemented with 25% glycerol.
2.4. X-ray diffraction and structure determination
The diffraction experiments were performed on the X06SA beamline at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland). Data sets were collected from well diffracting crystals; for the crystals soaked with heavy atoms a fluorescence scan was performed, and if the heavy atom was present a data set was recorded at the wavelength of the absorption peak. The data were processed with XDS (Kabsch, 2010 ▶) and were scaled and merged in AIMLESS (Winn et al., 2011 ▶). An automated sequential approach of heavy-atom search, SIRAS and initial model building was performed using phenix.autosol from the PHENIX suite (Adams et al., 2010 ▶). Both an anomalous holmium data set and a native data set were used as input files together with a sequence file. The resulting initial model was built to near-completion using ARP/wARP (Langer et al., 2008 ▶) with the density-modified electron-density map from RESOLVE. The native structure was refined with phenix.refine (Afonine et al., 2012 ▶) using TLS and isotropic B factors. The holmium structure was refined using simulated annealing in the initial rounds and the refinement run additionally included TLS and isotropic B-factor refinement. In the later rounds of refinement the anomalous signal was used to refine the dispersive f′ and anomalous f′′ scattering contributions. The TLS groups were generated based on the TLSMD-like program in the PHENIX suite; this resulted in three groups for the FnI domain and two groups for the EGF domain. Manual model building and real-space refinement was performed in Coot (Emsley et al., 2010 ▶). Images were produced using PyMOL (Schrödinger) and density maps for the images were calculated using FFT from the CCP4 suite (Winn et al., 2011 ▶).
2.5. Calculation of the holmium–oxygen distance
An advanced search for ‘HO’ as a ligand was performed using the PDBe web portal and resulted in nine hits [PDB entries 2y9x (2.78 Å resolution; Ismaya et al., 2011 ▶), 2y9w (2.3 Å; Ismaya et al., 2011 ▶), 3eeu (2.0 Å; Messing et al., 2009 ▶), 2olc (2.0 Å; Ku et al., 2007 ▶), 2bpu (1.35 Å; Jakoncic et al., 2006 ▶), 1rer (3.2 Å; Gibbons et al., 2004 ▶), 1psr (1.05 Å; Brodersen et al., 1998 ▶), 1buu (1.9 Å; Ng et al., 1998 ▶) and 1msb (2.3 Å; Weis et al., 1991 ▶)]. The distances between Ho atoms and protein atoms were measured in Coot and the mean distance and the standard deviation were calculated. A mean bond length of 2.5 ± 0.3 Å was calculated. A search for holmium–oxygen bonds in the Cambridge Structural Database (Allen, 2002 ▶) was also performed. This resulted in 267 bonds with a mean bond length of 2.376 ± 0.002 Å.
3. Results and discussion
3.1. Expression and purification
FXII-FnIE was well expressed by the selected P. pastoris X-33 clone and was secreted into the expression medium. It could be purified to high homogeneity from concentrated medium using a two-step protocol. The first step was cation-exchange chromatography at pH 6, which resulted in a highly pure FXII-FnIE sample (Fig. 1 ▶ b). An additional size-exclusion chromatography step was used to change the buffer and to separate the monomeric FXII-FnIE from possible aggregates. The protein consisted of residues Glu114–Thr194 of mature FXII with the naturally occurring mutation A188P. Owing to cloning artifacts FXII-FnIE had additional amino acids at the N- and C-termini: a glycine and a serine at the N-terminus and two alanine residues at the C-terminus (Fig. 1 ▶ a).
3.2. Structure determination
Crystals of FXII-FnIE grew within a day in conditions containing 16–18% PEG 4K at pH 8.5 or 6.5 (Figs. 1 ▶ c and 1 ▶ d). The crystals diffracted to 2.3 Å resolution and belonged to the same space group regardless of the pH. Initial attempts to obtain phases by molecular replacement using either the FnI-EGF structure of tPA or trimmed models of the FnI domains or EGF-like domains were unsuccessful. The low sequence identity of related structures, <43% for the EGF-like domain and <26% for the FnI domain, was the most probable cause. In order to obtain phases, crystals were soaked in different heavy-atom solutions at either pH 6.5 or 8.5. This resulted in a 2.5 Å resolution data set containing anomalous data to 2.9 Å resolution for crystals soaked with holmium chloride at pH 8.5. Crystals soaked with other heavy metals did not result in data sets with anomalous signal; however, a gold soak at pH 8.5 resulted in a crystal that diffracted to higher resolution than the native crystals: 1.6 Å compared with 2.3 Å. The gold data set was used as the native data. All crystals belonged to space group I4122, contained one protein molecule per asymmetric unit and had a solvent content of ∼58% (see Table 1 ▶ for diffraction data and refinement statistics). Space group I41 was also considered as a possible space group, but analysis with phenix.xtriage (Zwart et al., 2005 ▶) did not reveal twinning in the data and I4122 was suggested as the space group.
Table 1. Crystallographic data-collection and refinement statistics for FXII-FnIE.
Values in parentheses are for the highest resolution shell.
| Native | Holmium-bound | |
|---|---|---|
| PDB code | 4bdx | 4bdw |
| Data-collection statistics | ||
| Wavelength () | 1.03961 | 1.53520 |
| Space group | I4122 | I4122 |
| Unit-cell parameters | ||
| a = b () | 96.8 | 96.3 |
| c () | 47.5 | 47.3 |
| = = () | 90 | 90 |
| Resolution | 48.391.62 (1.651.62) | 48.122.50 (2.622.50) |
| No. of reflections | 343282 | 44075 |
| No. of unique reflections | 14626 | 4061 |
| R merge † (%) | 7.5 (68.4) | 5.2 (30.8) |
| R merge † (lowest resolution shell‡) (%) | 6.2 | 3.9 |
| Completeness (%) | 99.8 (96.0) | 99.6 (97.1) |
| Multiplicity | 23.5 (10.7) | 10.9 (5.7) |
| I/(I) | 27.6 (3.5) | 28.4 (4.3) |
| CC1/2 | 0.999 (0.862) | 0.998 (0.911) |
| Anomalous completeness | 98.6 (91.0) | |
| Anomalous multiplicity | 5.8 (2.8) | |
| Wilson B factor (2) | 21.3 | 47.4 |
| Refinement statistics | ||
| R work/R free (%) | 21.6/24.9 | 21.9/23.5 |
| No. of atoms | ||
| Protein | 681 | 653 |
| Water | 67 | 15 |
| Acetate | 4 | |
| Holmium | 2 | |
| Chloride | 1 | |
| B factor (2) | ||
| Protein | 40.6 | 63.3 |
| FnI domain | 28.2 | 55.8 |
| EGF domain | 52.3 | 70.8 |
| Water | 41.3 | 55.8 |
| Acetate | 31.3 | |
| Holmium | 78.9 | |
| R.m.s. deviations | ||
| Bond lengths () | 0.008 | 0.003 |
| Bond angles () | 1.11 | 0.70 |
| Ramachandran plot§ | ||
| Favoured (%) | 98 | 95 |
| Outliers (%) | 0 | 0 |
R
merge = 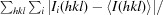
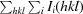 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the weighted average of all i observations of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the weighted average of all i observations of reflection hkl.
The lowest resolution shell was 48.398.72 for the native crystal and 48.398.29 for the holmium-bound crystal.
The Ramachandran plot statistics were obtained from the MolProbity server (Chen et al., 2010 ▶).
A SIRAS approach with the holmium and native data sets using phenix.autosol was used to solve the structure. Several plausible solutions for the heavy-atom sites were found using a combination of the HySS and SOLVE modules. The solution that led to the final structure has a figure of merit of 0.35 after SOLVE, which is low but is still well within the acceptable range of 0.25–0.45. This solution contained two heavy-atom sites. The BAYES-CC (Terwilliger et al., 2009 ▶) for this solution was 69.7 ± 11.8 and was the highest of all of the solutions. Additional rounds of density modification and model building by RESOLVE resulted in a well defined electron-density map that was much clearer than the initial SOLVE map (Fig. 2 ▶). The final RESOLVE map was used to build a nearly complete model with ARP/wARP.
Figure 2.

(a) The electron-density map calculated using the observed amplitudes and the experimental phases obtained from SOLVE shows some peptide-backbone density. (b) After density modification and initial model building by RESOLVE the electron-density map shows clear backbone-density and side-chain features. Both maps were produced using Coot and contoured at an r.m.s.d. of 1.4.
During the refinement of this model against the native data set, it became clear that the electron density for the EGF-like domain was less well defined than that of the FnI domain (Figs. 3 ▶ a and 3 ▶ b) and that the C-terminal part of the EGF-like domain in particular was hard to model. This uncertainty is reflected in the higher B factors of the EGF-like domain compared with those of the FnI domain (Figs. 3 ▶ c and 3 ▶ d) and is also the reason for the relatively high refinement R values of the native structure compared with other 1.6 Å resolution structures in the PDB.
Figure 3.

The characteristics of the FXII-FnIE crystal structure. Electron density for the EGF-like domain (a) is less well defined compared with the density for the FnI domain (b) in the |2F o − F c| map contoured at an r.m.s.d. of 1.8. The B factors for the EGF-like domain are higher than those for the FnI domain, as can be seen in (c), where the Cα B factors are shown using a colour range from blue (low B factor) to red (high B factor). The FnI domain is mainly coloured blue, whereas the EGF-like domain is mostly coloured green. The difference in the B factors of the domains can also be seen in the B-factor plot (d). The average B factors of the backbone atoms of the native FXII-FnIE structure are plotted in black and the all-atom average B factors are plotted in grey. The β-sheet residues are depicted as arrows above the B-factor plot.
The crystal contains large solvent channels running along the c axis. One channel is lined by the EGF-like domain and the other channel is lined by the fifth β-strand (strand E) of the FnI domain (Fig. 4 ▶ a). This particular crystal packing of FXII-FnIE was the most probable cause for the differences in flexibility between the two domains. Some remarkable contacts were found for the FnI domain, the N-terminal β-sheet of the FnI domain was extended by the N-terminal β-sheet of a symmetry-related FnI domain (Fig. 4 ▶ b) and the stacking of the phenylalanine side chains stabilizes a crystallographic tetramer (Fig. 4 ▶ c). Together with the other crystal contacts made by the FnI domain with symmetry-related FnI domains, these extensive contacts gave a buried surface area of 900 Å2 per molecule (Figs. 4 ▶ b, 4 ▶ c and 4 ▶ d) as calculated with PISA (Krissinel & Henrick, 2007 ▶). In contrast, the EGF-like domain had only one region which made crystal contacts in the crystal, involving two hydrogen bonds (Fig. 4 ▶ e), leaving the EGF-like domain more mobile.
Figure 4.

The packing of FXII-FnIE viewed along the c axis shows large solvent channels (a): one lined by the EGF domain (magenta) and the other lined by β-strand E of the FnI domain (green). The FnI domain makes extensive crystal contacts on two sides of the domain (b, c, d), whereas the EGF domain has only one crystal-packing interface on a twofold axis, stabilized by a hydrogen bond between Arg160 O and His166 N and depicted in cyan (e). Some of the contacts made by the FnI domain are remarkable; the N-terminal β-sheet extends itself with a symmetry-related β-sheet (b) and a tetramer is formed via stacking of phenylalanine side chains (c). In the holmium-bound structure two symmetry-related FXII-FnIE molecules are connected via a holmium cluster of three Ho atoms (f). The Ho atoms are shown with the coordinating protein atoms and the anomalous map is contoured at an r.m.s.d. of 4; other coordinating atoms, such as water, are not displayed because their positions are uncertain.
3.3. The holmium coordination
In the holmium-bound structure the density for the C-terminus of the EGF domain was better defined owing to the coordination of a holmium cluster connecting two EGF domains. One holmium is coordinated by the side-chain carboxyl group of Asp82, the backbone O atom of Thr83 and the carboxyl-terminus. The water and/or Cl atoms that coordinate the second holmium are not well defined owing to the limited resolution. The distances between the Ho and O atoms are comparable with the distances found in the PDB. In the FXII-FnIE structure the mean distance is 2.6 ± 0.2 Å and in the structures found in the PDB the mean distance is 2.5 ± 0.3 Å. However, both of these mean distances are larger than the mean distance found in the CSD, which is 2.376 ± 0.002 Å. The second holmium was placed on a twofold rotation axis at the heart of an anomalous peak which was found on this axis (Fig. 4 ▶ f).
3.4. The FXII-FnIE structure is similar to that of tPA-FnIE
The FnI domain of FXII showed the canonical domain fold, as did the EGF-like domain. The finger domain has two-stranded minor and three-stranded major β-sheets, which are stacked on each other by a disulfide bond and hydrophobic interactions. The EGF-like domain has an antiparallel β-strand followed by a double-hairpin region. The overall structure of FXII-FnIE was similar to that of the tPA-FnIE structure determined by NMR (Smith et al., 1995 ▶; PDB entry 1tpg); the r.m.s.d. between the Cα atoms of the two structures was 2.95 Å (Fig. 5 ▶ a). The superposition was performed using SUPERPOSE (CCP4 suite; Winn et al., 2011 ▶), which aligns secondary-structure elements followed by fitting of Cα positions. The alignment indicated that the orientation of the domains with respect to each other found in both structures is preferred. The interface between the domains is formed by hydrophobic residues in both structures; however, these residues are not identical (Figs. 5 ▶ b and 5 ▶ c).
Figure 5.

(a) The superposition of FXII-FnIE (green) with tPA-FnIE (blue) shows no large changes in the domain–domain orientation. The r.m.s.d. of the Cα atoms of this alignment is 2.95 Å. The residues that form the hydrophobic core are shown as sticks (b) and are coloured green in the sequence alignment (c). The asterisks under the sequence alignment indicate conserved residues and the residues shown in green for tPA-FnIE are based on the hydrophobic core determined by Smith et al. (1995 ▶). The residues that form the hydrophobic core are not conserved, but the hydrophobic core itself is.
3.5. Interaction of FnI domains with cross-β structure
FnI domains have been found to bind to amyloid and protein aggregates that are rich in cross-β structure (Maas, Schiks et al., 2008 ▶). However, the molecular mechanism of this interaction is unclear. A similar interaction mode was found in structures of N-terminal FnI domains of fibronectin in complex with bacterial fibronectin-binding peptides (Schwarz-Linek et al., 2003 ▶; Bingham et al., 2008 ▶), as well as for a collagen peptide binding to the eighth FnI domain of fibronectin (Erat et al., 2009 ▶). The peptides interacted with the FnI domain by extending the three-stranded β-sheet at β-strand E (Fig. 6 ▶), resulting in a four-stranded antiparallel β-sheet. We hypothesized that this could also be the mode of interaction of the FnI domains with the β-sheet ends of cross-β structures. The large solvent channel next to FnI β-strand E harbours sufficient space for small peptides to bind. Despite the fact that it has been reported that the FnI domain only binds to aggregated amyloid peptides in an ELISA (Maas, Schiks et al., 2008 ▶), it might be possible that the FnI domain also binds to smaller oligomeric species. We tried to soak FXII-FnIE crystals with multiple cross-β-forming peptides in a 1:1 molar ratio, but this did not lead to crystals of a complex.
Figure 6.

The binding of a collagen peptide (in blue) to the eighth FnI domain of fibronectin shows a common interaction mode for the FnI domain with peptides (PDB entry 3ejh; Erat et al., 2009 ▶). The extension of the three-stranded β-sheet by an extra strand is found in all the FnI structures that have been cocrystallized. Such an extension could also be the interaction mode of the FnI domains with amyloid/cross-β structures.
3.6. Interaction with the EGF receptor
The putative interaction of FXII with the EGF receptor has been implicated in stimulating the division of cells (Schmeidler-Sapiro et al., 1991 ▶; Gordon et al., 1996 ▶). The structures of EGFR in complex with EGF and TGFa (Garrett et al., 2002 ▶; Ogiso et al., 2002 ▶) served as templates to dock FXII-FnIE to EGFR. A superposition of the FXII-FnIE structure onto that of EGF bound to EGFR showed that the FnI domain overlaps with domain I of EGFR (Fig. 7 ▶). Furthermore, a conserved arginine that is a crucial residue for the interaction of EGF with domain III of EGFR (Engler et al., 1992 ▶) is replaced by a phenylalanine in the second EGF-like domain of FXII. These two observations make it unlikely that the second EGF-like domain directly interacts with EGFR even if large domain rearrangements take place upon binding. Whether the first EGF-like domain of FXII is able to interact with EGFR remains to be seen; however, this EGF-like domain also does not contain the arginine important for binding. These data imply that indirect EGFR stimulation via the uPAR receptor, as suggested by LaRusch et al. (2010 ▶), might be more plausible.
Figure 7.

A superposition of FXII-FnIE (green) with EGF (pale yellow) bound to EGFR (orange) shows that in the absence of any large conformational changes FXII-FnIE is not able to interact with EGFR because the FnI domain clashes with domain I of EFGR. The conserved arginine in EGF makes a salt bridge with Asp355 of EGFR; the phenylalanine in the EGF-like domain of FXII does not facilitate such an interaction (EGF-EGFR structure; PDB entry 1ivo; Ogiso et al., 2002 ▶).
3.7. Binding of FXII-FnIE to negative surfaces
One of most well known properties of FXII is its binding to negatively charged surfaces via complementary charges. The FnII domain is thought to be an important contributor to this interaction, but it is not essential for binding (Citarella et al., 1996 ▶). To determine whether FXII-FnIE could make a contribution to the binding of negatively charged surfaces based on complementary charges, the charge distribution of FXII-FnI was calculated using the APBS plug-in in PyMOL (Baker et al., 2001 ▶). A continuous patch of positive potential was found on the surface of FXII-FnIE. This patch was made up of the FnI residues Lys127, Arg141, Lys145, His150 and Arg153 and was just over 20 Å in length (Fig. 8 ▶). This patch resembled the 20–25 Å long patches found in llama antibodies that bind to Aβ fibrils via complementary charges (Haupt, Morgado et al., 2011 ▶; Beringer & Dorresteijn, manuscript in preparation). Furthermore, Haupt and coworkers showed that the antibody B10 was also able to bind to a wide variety of negatively charged biopolymers such as DNA and heparin (Haupt, Bereza et al., 2011 ▶). Additional experiments are needed to determine whether this patch in FXII-FnIE contributes to the binding to negatively charged surfaces and whether it is required for binding to misfolded proteins or amyloid deposits.
Figure 8.
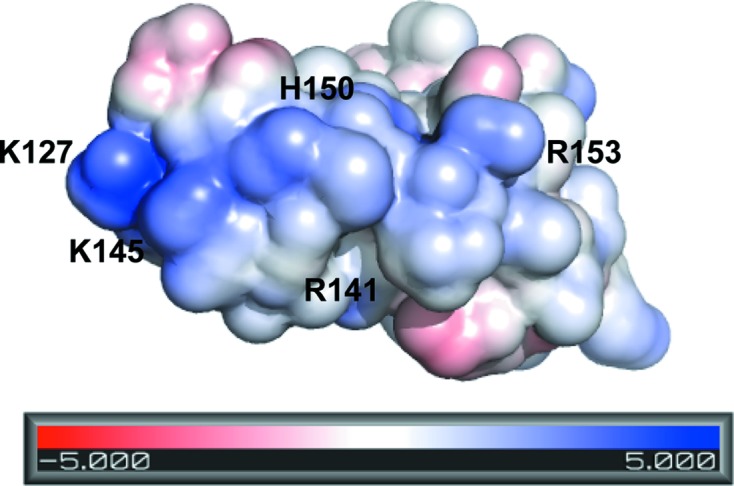
The surface potential of FXII-FnIE shows an elongated positive patch on the surface along β-strand E of the FnI domain, which might be involved in binding to negatively charged surfaces.
4. Conclusion
In this paper, we describe the structure of FXII-FnIE and its possible involvement in the proposed functions of FXII. It has been found that FXII-FnIE interacts with amyloid fibrils and cross-β structures and we suggest two putative interactions based on this structure. The first one is via β-sheet extension at β-strand E of the FnI domain, which is found in the structures of other FnI domain–peptide complexes. The current data unfortunately do not allow any conclusion in this respect. The second possible interaction mechanism is via electrostatic interaction, as found for some llama antibodies. The FnI domain has a similar patch of five positively charged residues stretching over 20 Å to these antibodies, which might interact with negatively charged residues on amyloid fibrils. This latter mechanism could also be responsible for the binding of FXII to negatively charged surfaces in general. The well described interaction of the FnII domain with negatively charged surfaces could thus be supplemented by that of FnI. In addition to its interaction with surfaces, it has been suggested that FXII can bind to EGFR via one of its EGF-like domains. Here, we show that without any conformational change the binding of the second EGF-like domain of FXII to EGFR is very unlikely owing to steric hindrance of the FnI domain and the lack of an important arginine residue in the EGF-like domain. For a better understanding of the interactions of FXII with a wide variety of putative binding partners, further structural studies are needed of FXII itself or in complex with binding partners.
Supplementary Material
PDB reference: FXII-FnIE, native, 4bdx
PDB reference: holmium-bound, 4bdw
Acknowledgments
We thank T. Stegmann for her assistance in cloning the construct and Dr A. J. Jakobi for his assistance in purification. We thank the Netherlands Organization for Scientific Research (NWO-CW) for financial support through grant 700.56.015.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Bergamaschini, L., Donarini, C., Foddi, C., Gobbo, G., Parnetti, L. & Agostoni, A. (2001). Neurobiol. Aging, 22, 63–69. [DOI] [PubMed]
- Bergamaschini, L., Parnetti, L., Pareyson, D., Canziani, S., Cugno, M. & Agostoni, A. (1998). Alzheimer Dis. Assoc. Disord. 12, 102–108. [DOI] [PubMed]
- Bingham, R. J., Rudiño-Piñera, E., Meenan, N. A., Schwarz-Linek, U., Turkenburg, J. P., Höök, M., Garman, E. F. & Potts, J. R. (2008). Proc. Natl Acad. Sci. USA, 105, 12254–12258. [DOI] [PMC free article] [PubMed]
- Brodersen, D. E., Etzerodt, M., Madsen, P., Celis, J. E., Thogersen, H. C., Nyborg, J. & Kjeldgaard, M. (1998). Structure, 6, 477–489. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Citarella, F., Ravon, D. M., Pascucci, B., Felici, A., Fantoni, A. & Hack, C. E. (1996). Eur. J. Biochem. 238, 240–249. [DOI] [PubMed]
- Citarella, F., te Velthuis, H., Helmer-Citterich, M. & Hack, C. E. (2000). Thromb. Haemost. 84, 1057–1065. [PubMed]
- Clarke, B. J., Côté, H. C., Cool, D. E., Clark-Lewis, I., Saito, H., Pixley, R. A., Colman, R. W. & MacGillivray, R. T. (1989). J. Biol. Chem. 264, 11497–11502. [PubMed]
- Cool, D. E., Edgell, C. J., Louie, G. V., Zoller, M. J., Brayer, G. D. & MacGillivray, R. T. (1985). J. Biol. Chem. 260, 13666–13676. [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engler, D. A., Campion, S. R., Hauser, M. R., Cook, J. S. & Niyogi, S. K. (1992). J. Biol. Chem. 267, 2274–2281. [PubMed]
- Erat, M. C., Slatter, D. A., Lowe, E. D., Millard, C. J., Farndale, R. W., Campbell, I. D. & Vakonakis, I. (2009). Proc. Natl Acad. Sci. USA, 106, 4195–4200. [DOI] [PMC free article] [PubMed]
- Garrett, T. P., McKern, N. M., Lou, M., Elleman, T. C., Adams, T. E., Lovrecz, G. O., Zhu, H. J., Walker, F., Frenkel, M. J., Hoyne, P. A., Jorissen, R. N., Nice, E. C., Burgess, A. W. & Ward, C. W. (2002). Cell, 110, 763–773. [DOI] [PubMed]
- Ghebrehiwet, B., Silverberg, M. & Kaplan, A. P. (1981). J. Exp. Med. 153, 665–676. [DOI] [PMC free article] [PubMed]
- Gibbons, D. L., Vaney, M.-C., Roussel, A., Vigouroux, A., Reilly, B., Lepault, J., Kielian, M. & Rey, F. A. (2004). Nature (London), 427, 320–325. [DOI] [PubMed]
- Gordon, E. M., Venkatesan, N., Salazar, R., Tang, H., Schmeidler-Sapiro, K., Buckley, S., Warburton, D. & Hall, F. L. (1996). Proc. Natl Acad. Sci. USA, 93, 2174–2179. [DOI] [PMC free article] [PubMed]
- Griep, M. A., Fujikawa, K. & Nelsestuen, G. L. (1986). Biochemistry, 25, 6688–6694. [DOI] [PubMed]
- Haupt, C., Bereza, M., Kumar, S. T., Kieninger, B., Morgado, I., Hortschansky, P., Fritz, G., Röcken, C., Horn, U. & Fändrich, M. (2011). J. Mol. Biol. 408, 529–540. [DOI] [PubMed]
- Haupt, C., Morgado, I., Kumar, S. T., Parthier, C., Bereza, M., Hortschansky, P., Stubbs, M. T., Horn, U. & Fändrich, M. (2011). J. Mol. Biol. 405, 341–348. [DOI] [PubMed]
- Ismaya, W. T., Rozeboom, H. J., Weijn, A., Mes, J. J., Fusetti, F., Wichers, H. J. & Dijkstra, B. W. (2011). Biochemistry, 50, 5477–5486. [DOI] [PubMed]
- Jakoncic, J., Di Michiel, M., Zhong, Z., Honkimaki, V., Jouanneau, Y. & Stojanoff, V. (2006). J. Appl. Cryst. 39, 831–841.
- Jansen, P. M., Pixley, R. A., Brouwer, M., de Jong, I. W., Chang, A. C. K., Hack, C. E., Taylor, F. B. Jr & Colman, R. W. (1996). Blood, 87, 2337–2344. [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Ku, S.-Y., Smith, G. D. & Howell, P. L. (2007). Acta Cryst. D63, 493–499. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- LaRusch, G. A., Mahdi, F., Shariat-Madar, Z., Adams, G., Sitrin, R. G., Zhang, W. M., McCrae, K. R. & Schmaier, A. H. (2010). Blood, 115, 5111–5120. [DOI] [PMC free article] [PubMed]
- Maas, C., Govers-Riemslag, J. W. P., Bouma, B., Schiks, B., Hazenberg, B. P. C., Lokhorst, H. M., Hammarström, P., Ten Cate, H., De Groot, P. G., Bouma, B. N. & Gebbink, M. F. B. G. (2008). J. Clin. Invest. 118, 3208–3218. [DOI] [PMC free article] [PubMed]
- Maas, C., Schiks, B., Strangi, R. D., Hackeng, T. M., Bouma, B. N., Gebbink, M. F. B. G. & Bouma, B. (2008). Amyloid, 15, 166–180. [DOI] [PubMed]
- Matata, B. M., Courtney, J. M., Sundaram, S., Wark, S., Bowry, S. K., Vienken, J. & Lowe, G. D. (1996). J. Biomed. Mater. Res. 31, 63–70. [DOI] [PubMed]
- Meijden, P. E. van der, Munnix, I. C., Auger, J. M., Govers-Riemslag, J. W., Cosemans, J. M., Kuijpers, M. J., Spronk, H. M., Watson, S. P., Renné, T. & Heemskerk, J. W. (2009). Blood, 114, 881–890. [DOI] [PubMed]
- Messing, S. A., Gabelli, S. B., Liu, Q., Celesnik, H., Belasco, J. G., Pineiro, S. A. & Amzel, L. M. (2009). Structure, 17, 472–481. [DOI] [PMC free article] [PubMed]
- Müller, F., Mutch, N. J., Schenk, W. A., Smith, S. A., Esterl, L., Spronk, H. M., Schmidbauer, S., Gahl, W. A., Morrissey, J. H. & Renné, T. (2009). Cell, 139, 1143–1156. [DOI] [PMC free article] [PubMed]
- Ng, K. K., Park-Snyder, S. & Weis, W. I. (1998). Biochemistry, 37, 17965–17976. [DOI] [PubMed]
- Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., Kim, J.-H., Saito, K., Sakamoto, A., Inoue, M., Shirouzu, M. & Yokoyama, S. (2002). Cell, 110, 775–787. [DOI] [PubMed]
- Pixley, R. A., Stumpo, L. G., Birkmeyer, K., Silver, L. & Colman, R. W. (1987). J. Biol. Chem. 262, 10140–10145. [PubMed]
- Renné, T., Pozgajová, M., Grüner, S., Schuh, K., Pauer, H. U., Burfeind, P., Gailani, D. & Nieswandt, B. (2005). J. Exp. Med. 202, 271–281. [DOI] [PMC free article] [PubMed]
- Schmeidler-Sapiro, K. T., Ratnoff, O. D. & Gordon, E. M. (1991). Proc. Natl Acad. Sci. USA, 88, 4382–4385. [DOI] [PMC free article] [PubMed]
- Schwarz-Linek, U., Werner, J. M., Pickford, A. R., Gurusiddappa, S., Kim, J.-H., Pilka, E. S., Briggs, J. A., Gough, T. S., Höök, M., Campbell, I. D. & Potts, J. R. (2003). Nature (London), 423, 177–181. [DOI] [PubMed]
- Smith, B. O., Downing, A. K., Driscoll, P. C., Dudgeon, T. J. & Campbell, I. D. (1995). Structure, 3, 823–833. [DOI] [PubMed]
- Smith, S. A., Mutch, N. J., Baskar, D., Rohloff, P., Docampo, R. & Morrissey, J. H. (2006). Proc. Natl Acad. Sci. USA, 103, 903–908. [DOI] [PMC free article] [PubMed]
- Tazi, S., Tans, G., Hemker, H. C. & Nigretto, J. M. (1992). Thromb. Res. 67, 665–676. [DOI] [PubMed]
- Terwilliger, T. C., Adams, P. D., Read, R. J., McCoy, A. J., Moriarty, N. W., Grosse-Kunstleve, R. W., Afonine, P. V., Zwart, P. H. & Hung, L.-W. (2009). Acta Cryst. D65, 582–601. [DOI] [PMC free article] [PubMed]
- Weis, W. I., Kahn, R., Fourme, R., Drickamer, K. & Hendrickson, W. A. (1991). Science, 254, 1608–1615. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yasuhara, O., Walker, D. G. & McGeer, P. L. (1994). Brain Res. 654, 234–240. [DOI] [PubMed]
- Zhuo, R., Siedlecki, C. A. & Vogler, E. A. (2006). Biomaterials, 27, 4325–4332. [DOI] [PubMed]
- Zwart, P. H., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl. Protein Crystallogr. 43, contribution 7.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: FXII-FnIE, native, 4bdx
PDB reference: holmium-bound, 4bdw