

Abstract
Leukocyte adhesion deficiencies are rare clinical syndromes of impaired host defense that provide novel insights into regulation of immune and inflammatory responses (1,2). Leukocyte adhesion deficiency (LAD)-I variant (LAD-Iv), also called LAD-III, is a unique disorder in which inside-out signaling of β1, β2, and β3 integrins on leukocytes and platelets is disrupted, leading to impaired cellular adhesion, recurrent infections, and bleeding [1-3]. We originally reported the second patient with this disorder to be identified and characterized the adhesive deficiencies and functional phenotype of this subject's leukocytes [4]. Here we show that the molecular defect in this index subject is a new mutation in FERMT3 (KINDLIN-3) which encodes KINDLIN-3, a cytoskeletal protein that interacts with the cytoplasmic tails of β1, β2, and β3 integrins and is required for inside-out and outside-in signaling of these heterodimers [5, 6]. We also report clinical features and previously-unrecognized defects in cells from a new patient, a sibling of the original subject that we described who carries the same FERMT3 mutation. Mutations in FERMT3 have now been shown to be the basis for LAD-Iv/LAD-III in each of the four original patients or families that established this syndrome[4, 7-9], including the family that we describe.
LAD syndromes provide critical insights into molecular regulation of integrin- and selectin-mediated leukocyte adhesion, and also reveal other key features of integrin and selectin biology [1, 5, 6, 10]. LAD-I is caused by mutations in the β2 subunit of the leukocyte integrin heterodimer family (“β2 integrins”), leading to absent or dramatically reduced levels of β2 integrins on the surfaces of myeloid leukocytes and lymphocytes; in contrast, LAD-II is a rare disorder of selectin ligand fucosylation [1, 11]. LAD-I variant (LAD-Iv), also termed LAD-III (abbreviated LAD-Iv/LAD-III here), is also extremely rare with fewer than two dozen patients reported in the world's literature. LAD-Iv/LAD-III results from molecular defects in inside-out signaling of integrins with preserved surface expression of integrin heterodimers [1-3]. Patients with LAD-I, LAD-II, and LAD-Iv/LAD-III have recurrent, often life-threatening, local and systemic infections secondary to impaired leukocyte adhesion, absent or dramatically reduced accumulation of polymorphonuclear leukocytes (PMNs; neutrophils) at extravascular sites of microbial invasion (deficient pus formation), and other defects in anti-microbial defensive functions [1-3, 11]. A striking feature unique to LAD-Iv is bleeding due to impaired platelet adhesive functions. Defective platelet adhesion in LAD-Iv is similar to that in Glanzmann Thrombasthenia, and can be severe and life-threatening [1-3].
Original description of LAD-Iv/LAD-III and early characterization of defects in adhesive functions of leukocytes and platelets in this syndrome came from reports of four index patients [4, 7-9]. Manifestations in these subjects defined a unique disease phenotype [1, 2], with subsequent reports of additional rare patients establishing the natural history of the disorder and its common and variable features [3]. The original index patient that we identified yielded the first evidence for defective inside-out signaling of β1, as well as β2 and β3, integrins[4]. This pattern, together with intact expression and structure of these integrins on leukocytes and platelets, became a defining characteristic of the defective adhesive phenotype in the new syndrome of LAD-Iv/LAD-III[1, 3].
The index subject (Patient 1), a boy, is the 3rd child of Hispanic parents who emigrated to the U.S. from Mexico. He presented with recurrent infections, leukocytosis, absent pus formation, mucosal bleeding, impaired platelet aggregation, and absent clot retraction at three weeks of age and underwent bone marrow transplantation at 8 months. Analysis of defective inside-out signaling of β1 and β2 integrins on his primary PMNs and EBV-transformed lymphoblasts were reported in detail [4]. He has been free of complications of immunodeficiency and bleeding since bone marrow transplantation. Two older sisters were healthy at birth and had no manifestations of immunodeficiency or bleeding problems. Additional information collected after the original report indicated no family history of immunodeficiency but revealed that the grandfathers were cousins.
Subsequently, other investigators reported impaired activation of Rap-1 due to decreased expression of the RAP guanine nucleotide exchange factor, CALDAG-GEF1, as a cause of LAD-Iv/LAD-III [12, 13]. Therefore, we examined EBV-transformed lymphoblasts from Patient 1 for this defect. We found normal levels of CALDAG-GEF-1 and Rap-1 in these cells, and no abnormalities when the sequence of CALDAG-GEF1 was analyzed. Furthermore, overexpression of CALDAG-GEF1 in transformed lymphoblasts from Patient 1 did not rescue defective integrin function (not shown). Expression of other candidate factors that might alter integrin signaling if absent or deficient, including IAP (CD47), UPAR (CD87), CD98, ICP-1, ILK, and RACK-1, was comparable to that in control cells (not shown).
Patient 2, a male sibling born 5 years after Patient 1, was noted to have cutaneous bleeding at sites of minor injury, ecchymoses, and petechiae shortly after uneventful term delivery. He was treated for sepsis at 1 wk of age. LAD-Iv/LAD-III was immediately considered because of the clinical presentation and family history; further evaluation revealed leukocytosis and hepatosplenomegaly, consistent with LAD-Iv/LAD-III [3] [4]. LAD-I was excluded by flow cytometric analysis, which demonstrated normal expression of β2 integrins. Defects in inside-out signaling of leukocyte integrins (see below) confirmed the diagnosis of LAD-Iv/LAD-III. Patient 2 was treated for additional infections including oral candidiasis, omphalitis, and two episodes of staphylococcal bacteremia before bone marrow transplantation at 15 weeks of age. Like Patient 1, he remains well after transplantation, with no further evidence of leukocyte dysfunction or thrombasthenia.
In vitro analysis of leukocytes from Patient 2, using approaches similar to those for characterization of cellular defects in Patient 1 [4], demonstrated impaired stimulated adhesion of isolated PMNs to purified fibrinogen and other ligands for β2 integrins (Fig 1A and data not shown). In addition, there was a defect in stimulated adhesion of primary PMNs to fibronectin, a ligand for β1 integrins, whereas an activating anti-β1 mAb, TS 2/16 [4], induced sub-maximal adhesion (Figure 1B). Transformed lymphoblasts from Patient 2 also failed to adhere to immobilized fibronectin when activated with phorbol myristate acetate (PMA) (Figure 1C). In contrast, lymphoblast adhesion to fibronectin increased 4 to 8-fold in response to exogenous Mn2+, which induces integrins to shift to the active allosteric conformation and allows them to recognize and bind ligands without a requirement for inside-out signaling [4, 8], or when they were incubated with the integrin activating antibody (Figure 1C). These findings indicate that the basally-expressed surface integrins are functional and that the phenotype of impaired adhesion is due to defective inside-out signaling [3, 4]. Isolated PMNs from the parents of Patients 1 and 2 adhered as expected when stimulated with fMLP or PMA (not shown).
Figure 1.

Leukocytes from Patient 2 demonstrated the LAD-Iv/LAD-III phenotype of impaired inside-out signaling of β1 and β2 integrins.
A. PMNs from Patient 2 did not adhere to β2 integrin ligands when activated. PMNs from Patient 2 or a healthy age-matched infant were incubated for 10 min at 37°C in wells coated with bovine serum albumin (BSA), gelatin, or fibrinogen (FB) in the absence of an agonist or with n-formyl-methionyl-phenylalanine (fMLP) (10−7M). Adhesion was quantitated as described in Methods. This figure illustrates results from a single experiment (performed in triplicate). The same pattern was demonstrated in two other experiments in which PMNs from Patient 2 and control subjects were activated with fMLP or PMA (10−7M) (data not shown).
B. Activated PMNs from Patient 2 did not adhere to fibronectin. PMNs from Patient 2 or a control subject were incubated on wells coated with fibronectin in the presence or absence of PMA or fMLP as in panel A. In parallel, PMNs in some wells were treated with the β1 integrin activating antibody TS 2/16 (“act mAb”; 10μg/ml). This figure indicates the pattern of adhesion in a single experiment (performed in triplicate) and is representative of two additional experiments examining primary PMNs from Patient 2.
C. Adhesion of EBV-transformed lymphoblasts from Patient 2 to immobilized ligands was induced by extracellular Mn++ or mAb TS 2/16 (“activating mAb”; see legend to panel B). Lymphoblasts from Patient 2 or a control subject were incubated for 15 min at 37°C in wells coated with fibronectin with buffer alone, or with PMA (10−7M) (performed in triplicate). In parallel wells lymphoblasts were treated with activating mAb, Mn++ (1μM), or Mn++ together with a blocking anti- β1 mAb (mAb p4C10, 10 μg/ml). A similar pattern was seen in a second experiment. In additional experiments, extracellular Mn++ induced adhesion of lymphoblasts from Patient 2 to immobilized ICAM-1, a ligand for β2 integrins; this was inhibited by a blocking anti- β2 mAb (mAb 60.3; 10μg/ml) (N=2, data not shown).
While PMNs, lymphocytes, and transformed lymphoblasts from patients with LAD-Iv/LAD-III have been examined intensively, there is a paucity of studies of monocytes. We found that there was a dramatic decrease in adhesion of monocytes from Patient 2 (Figure 2). In addition, the few residual cells failed to spread and differentiate in culture (Figure 2) under conditions in which monocytes develop into macrophages [14], suggesting a defect in outside-in signaling as well as inside-out activation of integrins [5], and that deficiencies in monocyte adhesion and differentiation to macrophages may have accounted for some of the features of the clinical phenotype of Patient 2. Nevertheless, neither Patient 2 nor Patient 1 had evidence for osteopetrosis, which has been reported in some patients with LAD-Iv/LAD-III [15-18] and has recently been ascribed to defective integrin signaling in monocyte-derived osteoclasts [19]. We were not able to perform additional studies of monocytes from Patient 2 because of semiemergent bone marrow transplantation.
Figure 2.
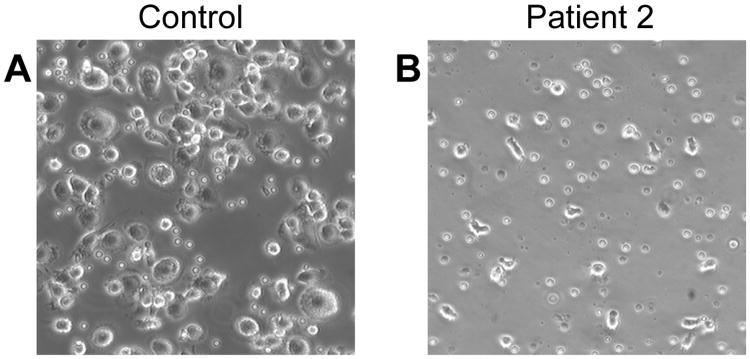
Monocytes from Patient 2 did not adhere or differentiate in culture. Mononuclear cells from a healthy volunteer (A) or a Patient 2 (B) were incubated in culture wells for 45 min, nonadherent cells were removed, and the adherent monocytes were washed, covered with fresh medium, and incubated at 37°C in 5% CO2 for an additional 48 hr. Representative fields were then photographed. These figures illustrate the cellular morphology in one experiment examining monocytes from this subject.
Subsequently reports by other investigators indicated that kindlin-3 is required for activation of β2 integrins and αIIbβ3 on murine leukocytes and platelets [20, 21]. Kindlin-3-deficient murine platelets and PMNs have adhesive defects that phenocopy those of cells from subjects with LAD-Iv/LAD-III [20, 21]. In addition, mutations in FERMT3 (KINDLIN 3), which encodes human KINDLIN-3, were identified in patients with LAD-Iv/LAD-III [15, 22-24]. KINDLIN-3 is largely restricted to hematopoietic cells and activates integrins by directly interacting with the cytoplasmic tails of β subunits, as do other members of the evolutionarily- considered Kindlin family [5, 6]. Therefore, we examined FERMT3 by sequence analysis of RT-PCR products from transformed lymphoblasts from Patient 1 and found a point mutation in codon 476 of exon 12, CAG→TAG (Supplemental Figure 1). Sequence analysis of RT-PCR products from genomic DNA in samples from Patients 1 and 2 again revealed this mutation, whereas DNA from each parent demonstrated heterozygosity for the CAG→TAG variation (Supplemental Figure 1). The mutation in Patients 1 and 2, which has not been previously reported in LAD-Iv/LAD-III patients, is predicted to create a premature stop codon in the sequence of FERMT3 encoding the FERM F2 subdomain of KINDLIN-3 [5]. Consistent with this, KINDLIN-3 was undetectable in lysates of transformed lymphoblasts from Patient 1 by western analysis (Figure 3).
Figure 3.

KINDLIN-3 is absent in EBV-transformed lymphoblasts from Patient 1. Lymphoblasts from Patient 1 and two control subjects were lysed, subjected to electrophoresis, and immunoblotted for KINDLIN-3 or actin as described in Methods.
Mutations in FERMT3 have now been identified in the four index LAD-Iv/LAD-III patients [2-4, 7-9], each with similar clinical features, including subjects of Hispanic [4] (this report), Turkish [7], Maltese [8], and Israeli Palestinian [9] descent. These mutations are in exon 12 [22], the splice acceptor site of exon 14 [24], and exon 2 [25] (A. Etzioni, personal communication) of the FERMT3 gene, in addition to the new mutation in exon 12 that we report here. Each leads to reduced levels or absence of KINDLIN3 mRNA and KINDLIN-3 protein in leukocytes and/or platelets [22, 24, 25] (Figure 3). Transfection of one subject's EBV-transformed lymphoblasts with murine kindlin-3 complementary DNA rescued the adhesion defect in these cells [24]. In addition to these four original subjects and, in two instances, siblings of these index patients [25] (this report), mutations in FERMT3 have been found in all LAD-Iv/LAD-III patients identified to date, including subjects of Turkish [15, 22, 24], Arabic [23], African-American [17, 18], and Gypsy [26] heritage. Thus, the unifying molecular basis for LAD-Iv/LAD-III from currently available genotyping information is mutation of FERMT3 in one or more [17] regions that disrupts expression of KINDLIN-3, alone or together with other sequence variations [22, 26]. Nevertheless, there is also variability in severity, bony involvement, and natural history among LAD-Iv/LAD-III patients [3, 22, 23], suggesting that there may be genetic or molecular alterations that are not yet apparent [27, 28].
Methods
Cells and cellular assays
Isolation of primary PMNs, preparation of EBV-transformed lymphoblasts, adhesion assays, and antibodies and reagents were previously described in detail [4]. These studies were approved by the University of Utah Institutional Review Board. Adhesion of monocytes and their culture to monocyte-derived-macrophages were done as previously reported [14].
Sequence analysis of CALDAG-GEF1 and FERMT3
Messenger RNA (mRNA) was isolated as previously described [29]. We analyzed mRNA-derived cDNA from transformed lymphoblasts from Patient 1 (Supplementary Figure 1, Panel A) and from control EBV-transformed lymphoblasts [4]. Genomic DNA isolated from Patients 1 and 2 (buccal mucosal cells) and both parents (peripheral blood leukocytes) (Supplementary Figure 1, Panel B) was also examined for sequence abnormalities in all exons and intron-exon boundaries for FERMT3 using primer sequences provided by Kuijpers and colleagues [22]. Sequencing was accomplished in the University of Utah School of Medicine sequencing core facility using standard methods. Genomic DNA from transformed lymphoblasts from Patient 1 and control EBV-transformed lymphoblasts was also analyzed for sequence alterations in all exons and intron-exon boundaries of CALDAG-GEF1 using sequences from GENBANK (not shown).
Western Analysis
EBV-transformed lymphoblasts (2×107/mL) were lysed in RIPA (50mM Tris-HCl [pH 7.5], 150mM NaCl, 0.1% SDS, 2mM EDTA, 1% Triton X-100) and mammalian protease inhibitor cocktail (Sigma-Aldrich). Thirty five μg of cleared protein lysate were reduced in Laemmli buffer and separated by 12% Tris-glycine SDS-polyacrylamide gel electrophoresis, and transferred to Hybond ECL Nitrocellulose membranes (GE Healthcare). Blots were probed with anti-KINDLIN3 polyclonal antibody (Sigma-Aldrich) or anti-ACTIN monoclonal antibody (MP biomedicals) and goat anti-mouse IgG conjugated to HRP as a secondary antibody (Invitrogen) and detected using ECL western blot detection reagents (GE Healthcare).
Supplementary On-Line Material
FERMT3 (KINDLIN-3) is mutated in Patients 1 and 2. A. Sequence analysis of mRNA isolated from transformed lymphoblasts from Patient 1 and a control subject revealed a point mutation in exon 12 (arrows) of Patient 1 that was not present in the healthy control. A CAG→TAG substitution created a stop codon in place of the normal glutamine (GLN) in this position. B. A homozygous 1426 C→T substitution results in a Q476X stop codon in genomic DNA from Patients 1 and 2. Sequencing of genomic PCR products amplified from exon 12 in samples from Patients 1 and 2 verified the C→T mutation. Analysis of genomic DNA samples from the mother and father of Patients 1 and 2 demonstrated that the parents are heterozygous for this variation.
Acknowledgments
The authors thank Jenny Pierce and Alex Greer for preparation of the manuscript, Diana Lim for drafting of the figures and consultation on data display, and Donelle Benson and Jessica Phibbs for technical assistance. We appreciate receiving FERMT3 primer sequence information from Dr. Taco Kuijpers and his group, sequence information regarding CALDAG-GEF1 from Dr. Hiraoki Kawasaki (Department of Neuropsychiatry, Graduate School of Medical Sciences, Krushu University, Kukuoka, Japan), and information from Dr. Amos Etzioni regarding details of LAD-Iv/LAD-III patients that his group has evaluated. We also greatly appreciate informative discussions with our colleagues at the University of Utah, and their suggestions regarding this study. This work was supported by the National Institutes of Health: 5R37HL044525-22, 2R01HL066277-10, and 2T32-DK007115-36.
References
- 1.Bunting M, Harris ES, McIntyre TM, et al. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving beta 2 integrins and selectin ligands. Curr Opin Hematol. 2002;9:30–35. doi: 10.1097/00062752-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Alon R, Etzioni A. LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol. 2003;24:561–566. doi: 10.1016/j.it.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Kuijpers TW, van Bruggen R, Kamerbeek N, et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood. 2007;109:3529–3537. doi: 10.1182/blood-2006-05-021402. [DOI] [PubMed] [Google Scholar]
- 4.Harris ES, Shigeoka AO, Li W, et al. A novel syndrome of variant leukocyte adhesion deficiency involving defects in adhesion mediated by beta1 and beta2 integrins. Blood. 2001;97:767–776. doi: 10.1182/blood.v97.3.767. [DOI] [PubMed] [Google Scholar]
- 5.Moser M, Legate KR, Zent R, et al. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 6.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuijpers TW, Van Lier RA, Hamann D, et al. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. The Journal of clinical investigation. 1997;100:1725–1733. doi: 10.1172/JCI119697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDowall A, Inwald D, Leitinger B, et al. A novel form of integrin dysfunction involving beta1, beta2, and beta3 integrins. J Clin Invest. 2003;111:51–60. doi: 10.1172/JCI14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alon R, Aker M, Feigelson S, et al. A novel genetic leukocyte adhesion deficiency in subsecond triggering of integrin avidity by endothelial chemokines results in impaired leukocyte arrest on vascular endothelium under shear flow. Blood. 2003;101:4437–4445. doi: 10.1182/blood-2002-11-3427. [DOI] [PubMed] [Google Scholar]
- 10.Evans R, Patzak I, Svensson L, et al. Integrins in immunity. J Cell Sci. 2009;122:215–225. doi: 10.1242/jcs.019117. [DOI] [PubMed] [Google Scholar]
- 11.Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. The New England journal of medicine. 2000;343:1703–1714. doi: 10.1056/NEJM200012073432307. [DOI] [PubMed] [Google Scholar]
- 12.Kinashi T, Aker M, Sokolovsky-Eisenberg M, et al. LAD-III, a leukocyte adhesion deficiency syndrome associated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood. 2004;103:1033–1036. doi: 10.1182/blood-2003-07-2499. [DOI] [PubMed] [Google Scholar]
- 13.Pasvolsky R, Feigelson SW, Kilic SS, et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J Exp Med. 2007;204:1571–1582. doi: 10.1084/jem.20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elstad MR, Stafforini DM, McIntyre TM, et al. Platelet-activating factor acetylhydrolase increases during macrophage differentiation. A novel mechanism that regulates accumulation of platelet-activating factor. The Journal of biological chemistry. 1989;264:8467–8470. [PubMed] [Google Scholar]
- 15.Mory A, Feigelson SW, Yarali N, et al. Kindlin-3: a new gene involved in the pathogenesis of LAD-III. Blood. 2008;112:2591. doi: 10.1182/blood-2008-06-163162. [DOI] [PubMed] [Google Scholar]
- 16.Jurk K, Schulz AS, Kehrel BE, et al. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb Haemost. 2010;103:1053–1064. doi: 10.1160/TH09-10-0689. [DOI] [PubMed] [Google Scholar]
- 17.McDowall A, Svensson L, Stanley P, et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood. 2010;115:4834–4842. doi: 10.1182/blood-2009-08-238709. [DOI] [PubMed] [Google Scholar]
- 18.Sabnis H, Kirpalani A, Horan J, et al. Leukocyte adhesion deficiency-III in an African-American patient. Pediatr Blood Cancer. 2010;55:180–182. doi: 10.1002/pbc.22386. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt S, Nakchbandi I, Ruppert R, et al. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J Cell Biol. 2011;192:883–897. doi: 10.1083/jcb.201007141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moser M, Nieswandt B, Ussar S, et al. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 21.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15:300–305. doi: 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 22.Kuijpers TW, van de Vijver E, Weterman MA, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood. 2009;113:4740–4746. doi: 10.1182/blood-2008-10-182154. [DOI] [PubMed] [Google Scholar]
- 23.Malinin NL, Zhang L, Choi J, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. 2009;15:313–318. doi: 10.1038/nm.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306–312. doi: 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manevich-Mendelson E, Feigelson SW, Pasvolsky R, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. 2009;114:2344–2353. doi: 10.1182/blood-2009-04-218636. [DOI] [PubMed] [Google Scholar]
- 26.Robert P, Canault M, Farnarier C, et al. A Novel Leukocyte Adhesion Deficiency III Variant: Kindlin-3 Deficiency Results in Integrin- and Nonintegrin-Related Defects in Different Steps of Leukocyte Adhesion. J Immunol. 2011;186:5273–5283. doi: 10.4049/jimmunol.1003141. [DOI] [PubMed] [Google Scholar]
- 27.Kilic SS, Etzioni A. The clinical spectrum of leukocyte adhesion deficiency (LAD) III due to defective CalDAG-GEF1. Journal of clinical immunology. 2009;29:117–122. doi: 10.1007/s10875-008-9226-z. [DOI] [PubMed] [Google Scholar]
- 28.Zimmerman GA. LAD syndromes: FERMT3 kindles the signal. Blood. 2009;113:4485–4486. doi: 10.1182/blood-2009-01-198853. [DOI] [PubMed] [Google Scholar]
- 29.Yost CC, Denis MM, Lindemann S, et al. Activated polymorphonuclear leukocytes rapidly synthesize retinoic acid receptor-alpha: a mechanism for translational control of transcriptional events. The Journal of experimental medicine. 2004;200:671–680. doi: 10.1084/jem.20040224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FERMT3 (KINDLIN-3) is mutated in Patients 1 and 2. A. Sequence analysis of mRNA isolated from transformed lymphoblasts from Patient 1 and a control subject revealed a point mutation in exon 12 (arrows) of Patient 1 that was not present in the healthy control. A CAG→TAG substitution created a stop codon in place of the normal glutamine (GLN) in this position. B. A homozygous 1426 C→T substitution results in a Q476X stop codon in genomic DNA from Patients 1 and 2. Sequencing of genomic PCR products amplified from exon 12 in samples from Patients 1 and 2 verified the C→T mutation. Analysis of genomic DNA samples from the mother and father of Patients 1 and 2 demonstrated that the parents are heterozygous for this variation.