

Abstract
Protein maturation in the endoplasmic reticulum (ER) is subject to stringent quality control. Terminally misfolded polypeptides are usually ejected into the cytoplasm and targeted for destruction by the proteasome. Ubiquitin conjugation is essential for both extraction and proteolysis. Here, we discuss the role of the ubiquitin conjugation machinery in this pathway and focus on the role of ubiquitin ligase complexes as gatekeepers for membrane passage. We then examine the type of ubiquitin modification applied to the misfolded ER protein and the role of de-ubiquitylating enzymes in the extraction of proteins from the ER.
Where proteins fold
Proteins destined for secretion from the cell or for the endocytic system originate by translation in the cytoplasm, from which they enter the endoplasmic reticulum (ER), typically cotranslationally. The nascent polypeptide enters the ER via the Sec61 translocon and – when the requisite signals are present and can be recognized – engages the glycosylation machinery. Nascent chains encounter chaperones that govern the folding process and allow the introduction of disulfide bonds. For protein complexes composed of multiple subunits, their proper association is an essential criterion for quality control and must not be jeopardized by aggregation. This is all the more remarkable when different subunits of a multi-protein complex are produced from the correspondingly distinct and individually translated mRNAs. Newly synthesized polypeptides thus attain their final conformation – autonomously or in complex with binding partners – while protected from aggregation within the crowded ER environment through transient association with components of the folding machinery (Figure 1for detailed review see [1]).
Figure 1. Protein folding in the ER.

Schematic overview of a nascent polypeptide entering the ER lumen co-translationally, where it engages folding machinery to obtain its final conformation (folding cycle). Quality control check-point(s) establish the folding status of the poly-peptide which then either proceeds to its final destination or is selectively degraded, either via the dislocation pathway or via a bulk degradation mechanism (e.g. autophagy or lipid droplet formation).
Nonetheless, protein folding in the ER is inherently imperfect and errors made at any step en route to the final product reduce the fraction of proteins that reach their proper conformation. For some proteins, like the cystic fibrosis chloride conductance regulator (CFTR), more than half of the newly synthesized polypeptide may not reach maturity [2]. Any significant accumulation of misfolded proteins inside the ER entails the risk of aggregation, and is likely to compromise ER function. Polypeptides that fail to meet ER quality control and cannot be rescued must be degraded. Indeed, the build-up of misfolded proteins that can occur in either professional secretory cells or in cells treated with compounds such as tunicamycin or dithiothreitol (DTT) evokes the unfolded protein response (UPR), a stereotypic transcriptional response that ultimately adjusts the composition – both lipid and protein – of the ER [3]. These changes include upregulation of folding chaperones and quality control machinery, a decrease in protein synthesis and, if the damage is deemed beyond repair, induced cell death (apoptosis). We know of no ER-resident proteases that can deal with the onslaught of terminally misfolded proteins inside the ER lumen of cells exposed to tunicamycin or DTT. Instead, the consensus view is that misfolded proteins are ejected into the cytoplasm – a step we shall refer to as dislocation – where they are targeted for ubiquitin-dependent degradation by the proteasome [4]. The steps that contribute to this means of protein elimination are collectively referred to as ER-associated degradation (ERAD). Although this aspect of ER quality control has received the most attention by far, not all misfolded proteins follow this route; proteins with only slight imperfections may still enter the secretory pathway and eventually be targeted to endolysosomal compartments for degradation, just like proteins that sustain damage at other intracellular locations are delivered to lysosomes to be cannibalized for salvaging of their building blocks. We shall discuss the nature of the misfolded polypeptide and the role of the ubiquitylation machinery in its elimination.
Tracking misfolded proteins
What exactly constitutes a misfolded protein? Structural alterations caused by amino acid replacements, truncations of the polypeptide chain, or non-native disulfide bonds, to name a few examples – while evidently causing alterations in covalent structure – are difficult to characterize in conformational terms. Even more problematic are structural changes that result from a failure to engage the necessary folding assistants without alteration to the covalent structure of the newly synthesized protein itself. None of these products can be obtained in quantities that allow an assessment of their conformation by standard physico-chemical means (crystallography, NMR, CD). Instead, surrogate measures are used to diagnose the misfolded state, such as the failure to enter the secretory pathway and lack of terminal carbohydrate modifications [1], the loss or acquisition of epitopes recognized by antibodies, altered susceptibility to protease digestion and loss of enzymatic or binding activity. There likely exists a continuum of folded and misfolded states, with the tipping point for diagnosis as seriously damaged and terminally misfolded being different for each protein. It has been surprisingly difficult to design mutant versions of endogenous proteins that show drastically altered kinetics of turnover and so serve as substrates to study ER quality control. As a result, relatively few substrates have been analyzed in detail. In addition, such substrates are commonly expressed at high levels in the setting of a transfection experiment. While overexpression allows easy detection of the misfolded product, it has the drawback that such substrates can saturate or even overwhelm the quality control machinery that is the object of study, likely inducing UPR-mediated remodeling of the ER [3].
Dislocation across the ER membrane barrier is proposed to take place through a protein-conducting channel, akin to translocation into the ER. In contrast to the canonical translocon, the composition of which is largely agreed upon [5], no singular or definitive complex has been assigned to perform this task in dislocation (Box 1). Involvement of lipid droplet formation [6] as well as autophagy [7] have been proposed as a possible means to relieve the ER of proteinaceous waste, but so far with little hard evidence in support. At least in yeast, visible lipid droplet formation was dispensable for dislocation of tested substrates [8]. This observation does not formally exclude a role for a mechanism akin to lipid droplet formation in dislocation, and the detection of components of the dislocation machinery (AUP1, UBE2G2) [9,10] on lipid droplets continues to fuel this hypothesis. Autophagy of an ER folding compartment overwhelmed with misfolded proteins (perchance UPR-dependent) remains an attractive means to clear misfolded proteins in bulk and deserves further investigation (Figure 1). The fidelity of the ER membrane as a barrier impermeant to proteins unless facilitated by the appropriate channels is perhaps too easily assumed in light of the dynamic nature of the ER and the abundance of fission and fusion events that take place there [11]; perforations, even temporary, of the ER membrane could allow escape of unwanted products.
Box 1: Identity of a dislocon.
To achieve delivery to the proteasome, misfolded ER glycoproteins must be discharged across the ER membrane into the cytoplasm. This passage likely requires a proteinaceous channel, and several promising ‘dislocon’ candidates have been proposed.
Sec61: The first candidate is the translocon itself. This dual function was proposed on the basis of the rapid dislocation of Class I MHC products by the immuno-evasin US2, and the occurrence of a diagnostic deglycosylated dislocation intermediate in association with Sec61β [57]. Sec61 has since been found to engage in complex formation with different members of the dislocation machinery [58].
Derlins: In mammals, the family of Derlin proteins (whose name derives from its founding member, the yeast Der1p protein involved in degradation of misfolded CPY* [59]) consists of three members (Derlin1–3) that have all been implicated in ER quality control [40]. They bear no obvious functional domains, carry multiple transmembrane domains and form both homo-and heterodimers, giving rise to the hypothesis that they could form (part of) a protein-conducting channel.
Ubiquitin ligases: A sizeable group of E3 ubiquitin ligases is directly anchored to, or associates with, the ER membrane and a growing number are implicated in ER quality control (Table 2). Sizeable proteins, they often include multiple transmembrane domains that do not obviously contribute to their enzymatic function and are more likely important for their intracellular positioning. The strongest case for ubiquitin ligase-mediated transport has been made for Hrd1p in yeast, which forms oligomers with the aid of Usa1 [60], associates with Der1p [60], and has been site-specifically photo-crosslinked to a dislocation substrate at residues in the Hrd1p transmembrane domains [20,21].
Ubiquitylation is a general requirement for the dislocation of individual misfolded proteins. Attachment of a poly-ubiquitin chain triggers two important steps: first is the recruitment of the AAA (ATPase associated with a variety of cellular activities) ATPase p97 (VCP; Cdc48 in yeast), thought to provide the mechanical force to extract the misfolded polypeptide from the ER [12]; and, second, ubiquitylation flags the protein for targeting to the proteasome and thus its final demise [4]. The unity of function of poly-ubiquitylation has led to the proposal that dislocation and degradation are tightly coupled. This view requires modification, however; involvement of de-ubiquitylation enzymes (DUBs) in the dislocation reaction [13,14,15], demonstrates an uncoupling of dislocation from degradation [16]. The complexity of the mammalian ubiquitylation machinery, the build and type of ubiquitin linkages themselves, and the association of cytosolic chaperones with dislocation substrates [16], indicate that proteasomal degradation of misfolded ER proteins is more complex than previously considered.
Ubiquitylation drives dislocation
Where examined, the extraction of a misfolded ER glycoprotein requires its ubiquitylation. The poly-ubiquitin chain serves as a recognition handle for p97 through its cofactors UFD1 and NPL4, and recruits it to drive dislocation [12]. Accordingly, obstruction of ubiquitylation causes the misfolded protein to accumulate inside the lumen of the ER. Whether or not the requirement for poly-ubiquitylation applies universally, or whether in select cases even a single ubiquitin (or multiple single ubiquitins) would suffice to engage the dislocation machinery remains to be clarified. Recruitment of substrates to the proteasome is believed to require a minimum of 4 ubiquitin units for a single chain to result in productive engagement of the 19S cap [4], but whether this (Ub)4 rule applies generally is, again, not known. Turnover of at least Pax3 has been reported to rely on only a single ubiquitin moiety [17].
Ubiquitylation of proteins takes place via an E1-E2-E3 cascade [18]. In mammalian cells, two E1s have been identified, about 40 E2s (ubiquitin conjugating enzymes) and possibly as many as 1000 E3s (ubiquitin ligases), although for the vast majority of E3s their enzymatic activity remains to be verified experimentally. Notwithstanding striking homologies, their function as enzymatically active E3s may not simply be assumed, as proteins with near-identical folds may serve very different functions. Nonetheless, this hierarchy would obviously allow a great deal of specificity. Variables that control the operation of the ubiquitin system include the identity of the substrate and the E3 ligase that modifies it, the amino acid to which ubiquitin is conjugated, and the type of oligo- or poly-ubiquitin linkage made. The possibility of mono-, multi-, or poly-ubiquitylation adds yet further complexity. The fact that certain proteins can be ubiquitylated at multiple distinct acceptor residues implies that different E3-type activities target these substrates, or that a single E3 activity is capable of sequential and/or processive engagement of one and the same substrate. The relevance of this point will become clear when we discuss sequential rounds of ubiquitin addition and removal as a requirement for destruction of misfolded ER proteins.
In the case of poly-ubiquitylation, there is the variety in linkage to consider. Ubiquitin offers seven lysine residues to which a following ubiquitin molecule can be attached. Individual linkages are named for the position of the accepting lysine residue (e.g. K48 for attachment of ubiquitin to the lysine at position 48). Each type of linkage results in a characteristic and separate spatial structure. Ubiquitin is conjugated via its C-terminal di-glycine motif in thio-ester bond to the relevant E2 and is then transferred to an acceptor residue in the substrate polypeptide. Lysine is the preferred, but by no means the only, ubiquitin acceptor; ubiquitin conjugation to cysteine, threonine, serine or a protein’s N-terminus has been reported [19]. The genesis of the various types of poly-ubiquitin chains has been extensively reviewed [19].
A physical interaction between the dislocation substrate in the ER and the E3 of choice must occur to enable ubiquitin conjugation, as we discussed for the Hrd1p ligase in yeast (see Box 1) [20,21]. How a soluble ER protein crosses the membrane barrier to reach the enzymatic domain of the E3 ligase is unclear. The polypeptide would at least have to protrude partially from a putative dislocon to allow ubiquitylation. Therefore transfer of a part of the polypeptide to be destroyed must already have progressed to the point where a residue suitable for ubiquitylation is exposed and accessible to the E3. The force needed to extract a polypeptide from the ER is provided by cytosolic p97, which engages the substrate only after poly-ubiquitin attachment. In much the same way as yeast Kar2p (yeast BiP ortholog) ratchets proteins into the ER in the course of translation, a similar mechanism might be employed by p97 for extraction of proteins from the ER in dislocation. If we extend this parallel, there might even exist the need for a force that pushes the protein out of the ER, akin to the translating ribosome for protein import. If indeed there were such an ER-resident first mover, its identity is yet to be discovered.
Substrate ubiquitylation is more easily understood for proteins that span the ER membrane and so may provide a naturally exposed handle for ubiquitylation. For some substrates, cytosolic – not membrane-bound – E3 ligases participate in their removal; the Hsc70/CHIP E3 ligase complex is recruited to the ER to target CFTRΔF508 for degradation [22] and the HIV encoded protein Vpu recruits the cytosolic ligase complex SCFbeta-TrCP to target CD4 for dislocation and proteasomal degradation [23]. The number of mammalian E3 ligases implicated in ER quality control continues to expand, including both integral membrane as well as cytosolic E3 ligases (Tables 1 and 2). Propelled by studies of yeast Hrd1p, most of the current mechanistic information has been acquired for its mammalian ortholog HRD1, which may or may not represent all avenues open to dislocation.
Table 1. Mammalian and yeast ubiquitin conjugating enzymes and interacting ubiquitin ligases involved in ER dislocation.
UBC (ubiquitin conjugating domain); UBA (ubiquitin-associated domain). The ER membrane is represented by a shaded bar with the ER lumen in the upper right corner.
| Mammalian ubiquitin conjugating enzymes |
Topology/Functional Domains |
Interacting mammalian ubiquitin ligases |
|---|---|---|
| UBE2J1 (UBC6E, NCUBE1) [65] | 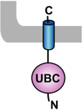 |
HRD1 [45] RMA1 [22] |
| UBE2J2 (UBC6, NCUBE2) [65] | 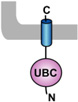 |
Parkin [66] |
| UBE2G2 (UBC7) [65] | Gp78 [65] HRD1 [67] Parkin [65] |
|
|
Yeast ubiquitin conjugating enzymes |
Interacting yeast ubiquitin ligases |
|
| Ubc1 [65] | Hrd1p [65] | |
| Ubc6 [65] | 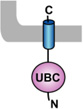 |
Doa10 [65] |
| Ubc7 [65] | Hrd1p [65] Doa10 [65] |
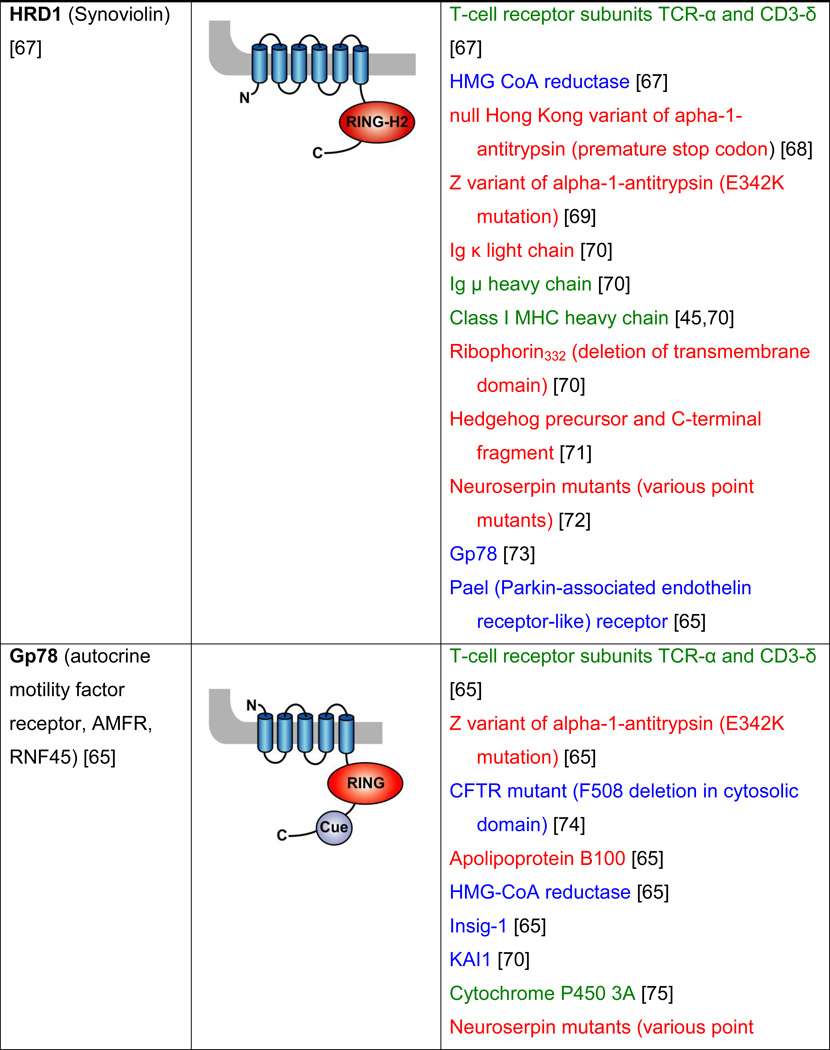
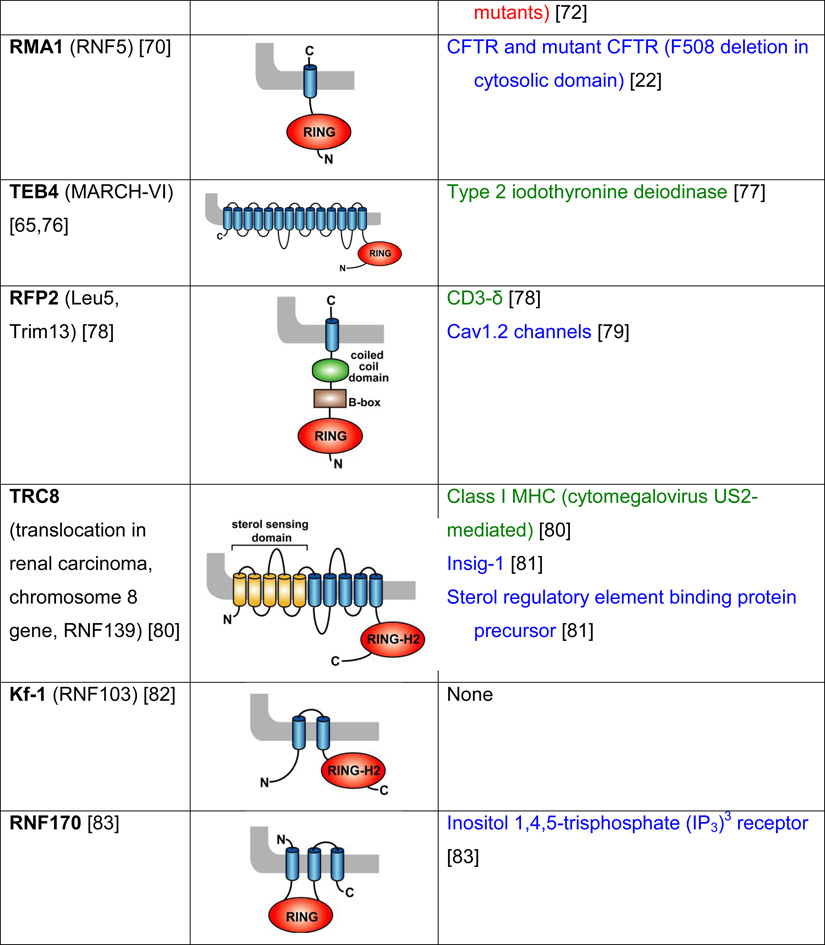
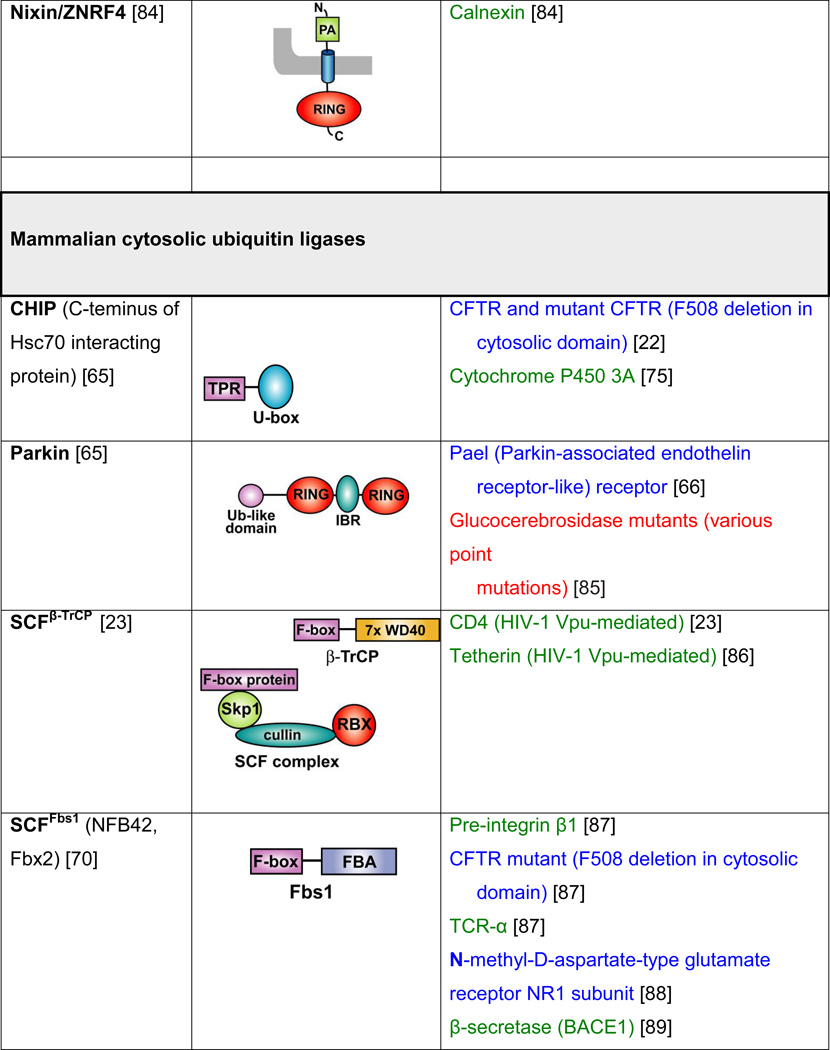
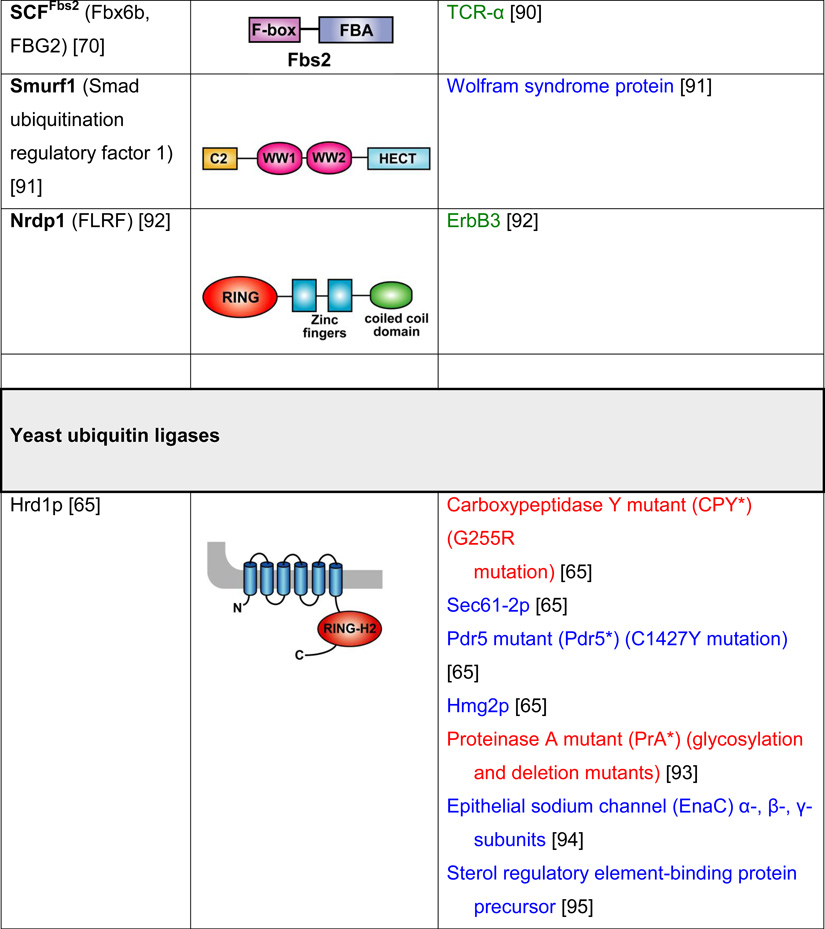
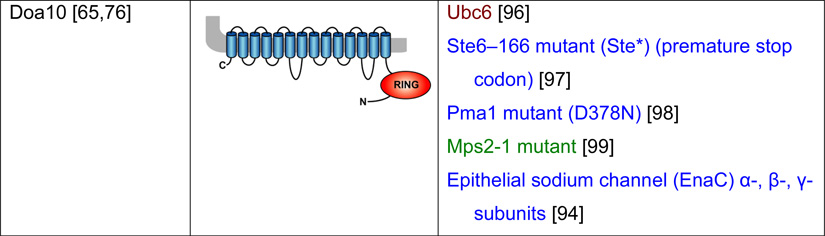
Table 2. Mammalian and yeast ubiquitin ligases involved in ER dislocation and corresponding in vivo substrates.
The ER membrane is represented by a shaded bar with the ER lumen in the upper right corner. Except for TEB4/Doa10, the indicated membrane topologies of polytopic ubiquitin ligases are predicted based on the sequence but not experimentally confirmed. Polytopic membrane substrates are in blue, single-pass membrane substrates are in green, ER luminal substrates are in red, and tail-anchored substrates are indicated in brown color. RING (really interesting new gene); PA (protease-associated domain); TPR (tetratricopeptide repeat domain); IBR (in-between RING domain); FBA (F-box-associated domain); HECT (homologous to the E6-AP carboxyl terminus).
 |
 |
 |
 |
 |
 |
It is common practice to test the involvement of any particular E3 ligase in turnover of known dislocation substrates and thus implicate the ligase in ER quality control. These experiments rely on the limited set of often artificial substrates that the E3 can be tested against. These tools provide a skewed view of substrate turnover at endogenous protein expression levels, as overexpression of the artificial substrate can modulate the landscape of the ER through activation of the UPR. Examples that approach more physiological levels of turnover include endogenous Class I MHC heavy chains [24,25], the turnover of immunoglobulin subunits [26] or degradation of proteins that undergo extensive processing in the ER such as the Hedgehog protein, which matures by autocatalytic cleavage, separating into an N-terminal signaling molecule and a C-terminal fragment that undergoes dislocation [27].
Connecting a particular E3 ligase complex with the turnover of specific ER-resident proteins tackles only one type of variability of ubiquitin-conjugation. Ubiquitin-conjugation to amino acid side chains other than lysine has been described for ER dislocation-induced ubiquitylation of TCRα [28], Class I MHC [29], as well as the NS-1 non-secreted immunoglobulin light chain [26]. Degradation of these substrates required the HRD1 ligase for ubiquitin-conjugation to serine, threonine and lysine residues. One can argue whether the identity of the acceptor amino acid plays a specific role, such as conferring susceptibility to hydrolysis of the linkage produced (amide versus thioester or hydroxy ester) or whether this is simply determined by its accessibility, caused by partial unfolding of the substrate protein. Attachment of ubiquitin may further modify the physical properties of the substrate [30], and help start it to unfold, which in return could free up preferred acceptor amino acids, if any.
Ubiquitylation is required for the dislocation reaction, but the exact role of ubiquitylation in this context remains ill-defined. Does it serve as a direct handle for extraction of the misfolded protein? Does it enable the recruitment of additional factors that continue and complete the dislocation reaction after its initial engagement? It is not clear whether p97 recruitment depends on mono-, poly- or multi-ubiquitylation of a substrate, nor do we know whether there is a single or several preferred ubiquitin-linkages. A possible complicating factor in interpretation of such experiments is the application of proteasome inhibitors, commonly used to visualize the presence of poly-ubiquitin adducts. While it is likely that poly-ubiquitin chains are present also in cells not treated with proteasome inhibitors, neither the extent nor dynamics of poly-ubiquitylation are immediately obvious in the untreated control setting. A K48-linked poly-ubiquitin tag was initially thought to determine proteasomal targeting, but a more complex picture emerged with the demonstrated involvement of all lysine linked ubiquitin-linkages in proteasomal targeting [31,32]. There may be a prominent role for K11-linked ubiquitin in ER quality control [31]. Furthermore, there is no evidence that poly-ubiquitin chains in vivo are homogeneous in linkage, leaving open the possibility of heterogeneity within a single or between multiple chains conjugated to a given substrate, thus rephrasing the question as linkage-dominant instead of linkage-specific.
Ubiquitin-linkage specificity is largely determined by the E2 enzyme; it was recently demonstrated how the E2 UBE2S specifically builds K11-linked ubiquitin chains [33]. Mammalian E2s currently implicated in dislocation, such as UBE2J1, UBE2J2 and UBE2G2 remain to be examined from the perspective of linkage type and the E3s they serve. Monoclonal antibodies that recognize specific ubiquitin linkages will be of considerable help [34,35]. The in vivo pairing of E2s with E3s remains enigmatic, and thus resolving the extent to which the promiscuity for E2–E3 pairings observed in vitro translates to the in vivo situation is a technical challenge.
Substrate extraction and deubiquitylation
Modification of a substrate with ubiquitin can recruit either of two multiprotein complexes that can extract the protein from the ER: p97 with its associated co-factors [12], and the 19S cap of the proteasome [36]. Although different in composition, the core of each complex consists of a ring-shaped, hexameric ATPase of the AAA family that can unfold polypeptides at the expense of ATP hydrolysis [4,12] (Figure2).
Figure 2. Protein dislocation and/or degradation.

Left panel: A proposed model of a dislocated protein that is ubiquitylated at the ER membrane and consequently engaged by p97 via NFD1/NPL4. A de-ubiquitylating enzyme cleaves ubiquitin to allow threading of the polypeptide through the central pore of p97. A hypothesized re-ubiquitylation step post-p97 then facilitates proteasomal targeting. Right panel: A poly-ubiquitin tag targets the protein for proteasomal degradation. The poly-ubiquitin chain is probably modified before it is finally removed to allow threading of the polypeptide through the central pore of the base of the 19S cap and into the proteolytic chamber of the 20S proteasome core particle.
Two different types of ubiquitin receptors allow substrate engagement by the proteasome, either integrated into the structure of the proteasome itself, such as Rpn10 and Rpn13, or in the form of shuttling factors such as Rad23, Dsk2 and Ddi1 (in yeast) [4]. Upon engagement, a de-ubiquitylating activity (Rpn11) associated with the cap of the proteasome removes ubiquitin from the substrate. This allows recycling of ubiquitin and ensures compliance with size constraints of the access portal to the proteasome, which can accommodate looped polypeptides but not those with a conjugated complex ubiquitin ensemble.
The 19S cap has been reported to associate with the Sec61 translocon. Moreover, the purified cap complex supports dislocation in vitro [36, 37, 38]. The proposed structure of the Sec61 translocon [39] includes a narrow pore that can accommodate only unstructured polypeptides (the folding status of dislocated proteins is discussed in Box 3), and therefore the functional relevance of this physical link requires further experimental support. Whereas one can see how the short alpha-helical ‘plug’ [39] in the Sec61 pore is displaced from the ER luminal side of the translocon upon protein translocation, it would presumably occlude the pore upon reverse passage. More appealing is the proposal that the proteasome can directly engage membrane proteins tagged with poly-ubiquitin[40], as has been suggested for turnover of Ubc6 in yeast. When proteasome function was selectively impaired at one of the chymotryptic sites (pre1-1), a distinct breakdown intermediate remained associated with the ER membrane, thought to represent the trans-membrane domain of Ubc6 severed from its digested cytosolic domain [41].
Box3: Outstanding questions.
-
-
Identity of dislocation channel(s): exclusively proteinaceous? Membrane discontinuities?
-
-
Which E2-E3 pairs are formed to facilitate dislocation and how promiscuous is such pairing (redundancy)?
-
-
Does dislocation require a preferred type of ubiquitin-linkage?
-
-
What is the folding status of misfolded ER proteins in the course of dislocation?
-
-
What is the role of cytosolic chaperones in dislocation, and which chaperones are involved?
-
-
Are there ER sub-compartments set aside for co-translational folding and modification reactions, physically and compositionally distinct from areas where quality control and dislocation occur?
p97 nucleates distinct multiprotein complexes implicated in diverse functions in the cell, ranging from dislocation and proteasomal targeting to cell cycle control and vesicular trafficking [12]. Adaptor proteins engage p97 and can either recruit or adapt it to a specific function. p97 engages ubiquitylated proteins via a dimeric adaptor consisting of NPL4 and UFD1. This dimer can engage ubiquitin and associates with the N-terminal domain of p97. Membrane-anchored auxiliaries such as VIMP [42], UBXD2 [43,44] and UBXD8 [45] may recruit this dislocation-competent complex to the ER membrane via their p97-interacting UBX-domain. p97 can directly interact with the ligase as shown for gp78 [40]. A hierarchy in p97 cofactors was described, such that FAF1 and UBXD7 only bind to p97 when in complex with the NPL4/UFD1 dimer, but neither engages p97 alone [44]. Of note, a p97-driven dislocation reaction has been described independently of NPL4/UFD1 [46]. It will be important to determine whether a binding hierarchy exists for the p97 complex implicated in dislocation, as this can provide insight into the timing of the dislocation reaction (Table 3 lists p97 cofactors involved in dislocation). Modulation of p97 co-factors can be regulated by phosphorylation or acetylation of p97 itself [47,48].
Table 3. p97 co-factors involved in mammalian dislocation.
| Protein | Function | Interaction Domain |
|---|---|---|
| UFD1/NPL4 [12] | Facilitates engagement of poly-ubiquitin proteins | UBX |
| UBXD2 (Erasin) [43] | p97 recruitment | UBX |
| UBXD8 [45] | p97 recruitment | UBX |
| YOD1 [13] | De-ubiquitylating activity at p97 | UBX |
| VIMP [42] | Recruits p97 to the ER membrane | Unknown |
| Peptide: N-glycosidase [100] | Enzymatic removal of N-linked glycans | PUB |
| Ataxin3 [15] | De-ubiquitylating activity at p97 | VCP binding motif (VBM) |
| SAKS1 [50] | Mediates ubiquitin interactions | UBX |
Akin to the proteasome, the hexameric pore of p97 can accommodate a polypeptide modified with ubiquitin [49]. There is some debate as to how p97 engages the polypeptide. One model suggests the protein enters via the D1 ring, and is threaded through the structure to exit via the D2 ring (Figure 2). This is contrasted with models where the polypeptide loops into the D2 ring alone, or where the hexamer could even dissociate to release the substrate [12]. Either model suggests similar space constraints, which is most relevant for the current discussion. It is unlikely that p97 can engage a protein bearing a poly-ubiquitin chain, exactly the type of chain that is thought to facilitate transfer to the proteasome with the help of shuttling factors. Indeed, DUBs participate in the mammalian dislocation reaction, in agreement with their function at the 19S cap of the proteasome. Expression of a catalytically inactive form of the ER-membrane anchored USP19 hampered dislocation of several substrates [14]. Also, the de-ubiquitylating enzyme Ataxin 3 associates with p97 and expression of a catalytically inactive mutant causes accumulation of poly-ubiquitylated substrates in association with p97 [15]. In a rather unconventional proposal, such poly-ubiquitin chains were deemed shielded from Ataxin 3 engagement by the p97 adaptor protein SAKS1, which could thus negatively regulate dislocation [50]. Furthermore, the de-ubiquitylating enzyme YOD1 is recruited to p97 via its UBX domain. Tampering with its function results in a near-complete blockade of dislocation [13]. Interference with dislocation/p97-associated de-ubiquitylating activity causes accumulation of misfolded proteins at a step prior to membrane extraction [13]. If de-ubiquitylating activity indeed were required to complete p97-catalyzed extraction, then such a blockade should be overcome by expression of a de-ubiquitylating enzyme that can engage these stalled poly-ubiquitinated dislocation intermediates. Exactly this was shown by co-expression of the catalytic domain of the Epstein Barr Virus large tegument protein BPFL1 (EBV DUB). Completion of the dislocation reaction thus relies on a de-ubiquitylating activity [16].
As proteasomal targeting requires ubiquitylation, it follows that substrates that undergo p97-mediated extraction, once de-ubiquitylated, require a second round of ubiquitin modification. Do p97-associated DUBs merely trim ubiquitin-chains or do they remove them completely? Polypeptides modified with mono-ubiquitin might pass through the central pore of p97 and then engage the proteasome directly, or do so after ubiquitin chain extension by either an E-3-mediated ligase reaction or via an E4-like activity (enzymes thought to engage and extend existing ubiquitin conjugates). The E4 Ufd2 has been linked to dislocation via Cdc48 in yeast [51]. In fact, soluble (dislocated) Ste6(p)* was observed in a system deprived of Ufd2 [52], where Ufd2 was suggested to increase the level of poly-ubiquitylation and thus facilitate proteasomal turnover, a suggestion that corroborates the model described above (Figure 2).
De-ubiquitylating activity at p97 opens the exciting possibility to modify the type of ubiquitin-linkage utilized, where one type of ubiquitin-build could induce extraction, followed by a switch to target the polypeptide to the proteasome.
Soluble in the cytosol
How the cell avoids aggregation of (partly) unfolded ER glycoproteins discharged into the cytosol is poorly understood. Hydrophobic protein domains find themselves exposed to an aqueous environment upon escape from the ER. As an example, when proteasomal proteolysis is blocked, Class I MHC products with their transmembrane segment fully intact occur as soluble intermediates in the cytoplasm of cells that express viral immunoevasins [24]. The cytoplasm houses an extensive chaperone network involved in the quality control of cytosolic proteins, with Hsp70 and Hsp90 its most famous family members [53]. Both have been proposed to triage the folding of complex membrane proteins, but there is limited direct evidence that ties them to quality control/dislocation of lumenal ER proteins.
One can visualize dislocated ER proteins in the cytosol by blocking proteasomal degradation, either via chemical (as just discussed for Class I MHC) or enzymatic means [16]. Expression of the catalytic domain of the large tegument-embedded ubiquitin-specific protease domain taken from Epstein-Barr virus, EBV-DUB, impairs proteasomal degradation by preemptively removing the ubiquitin-tag from substrates. In cells that express this EBV-DUB, dislocation of ER proteins continues, albeit at a reduced rate (as expected, if ubiquitylation is a prerequisite for the first step(s) in the dislocation pathway) and the misfolded ER-derived glycoprotein accumulates as a deglycosylated product in association with the cytosolic chaperone BAG6 (BAT3) [16]. In addition, an interaction with the TRICC/CCT complex was detected. BAG6 shuttles defective translation products for degradation to the proteasome [54,55], tying it to unfolded proteins. BAG6 does not merely associate with dislocation substrates when degradation is blocked, but is required to complete the dislocation reaction itself [56].
Engagement by chaperones, combined with a de-ubiquitylation step as an ER-resident protein exits from the ER, opens a window where substrates could deviate from the path to the proteasome. Such escape from proteolysis and the possibility of chaperone-mediated refolding could explain a set of observations where proteins (such as cholera toxin), formerly localized to the ER, escape to the cytoplasm and acquire their active conformation. Bacterial toxins may utilize dislocation machinery to reach the cytosol, be released from cytosolic chaperones – if they interact with them at all – and be allowed to reach their target for covalent modification (ADP-ribosylation, proteolysis, etc.).
Concluding remarks
Misfolded ER proteins are modified with ubiquitin, first, to complete their extraction and second, to mark them for destruction. The need for de-ubiquitylation ezymes manifests itself in p97-mediated extraction, and immediately prior to degradation by the proteasome, presumably to allow the unfolded polypeptide to access the pore of p97 or the proteasome. A clear understanding of this process will depend on detailed knowledge of the nature of the ubiquitin chains as they are constructed or trimmed (see Box3 for outstanding questions).
Box2: Folding status of dislocation substrates.
Translocation of proteins, as well as their threading into the central channel of the proteasome, presumably requires complete unfolding of the polypeptide chain. Whether this is also required for a misfolded protein to pass the ER membrane in the course of dislocation is still unclear. A substrate-GFP fusion protein shows no obvious loss of fluorescence while the protein undergoes dislocation [61], although this observation could be attributed to the β-barrel of GFP snapping back into shape once it reaches the cytosol. Also, DHFR-substrate fusion proteins are readily dislocated even in the presence of cell-permeable analogs of methotrexate that stabilize the DHFR moiety [62], albeit with slower kinetics [63]. These observations, while by no means conclusive, raise the possibility of a dislocon that can accommodate partially folded proteins, a suggestion not easily reconciled with the structures proposed for the Sec61 channel [39]. It is important to keep in mind that multi-domain proteins deemed misfolded in quality control may still have acquired fully folded domains. Furthermore, no protein with unfoldase activity (e.g. p97, 19S cap) has been detected on the luminal side of the ER, although a candidate in the form of the AAA ATPase TorsinA has been implicated in ER quality control [64]. The folding status of a protein undergoing dislocation across the ER membrane will remain a thorny issue until the identity of (a) putative channel(s) has been firmly established.
Acknowledgments
The authors are grateful to R. Ernst and C. Schlieker for continued scientific discussions. JHLC is a fellow of the Boehringer Ingelheim Fonds. LK and HLP receive grants from the NIH. We apologize for not being able to cite the work of all authors who contributed to this topic, due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braakman I, Bulleid NJ. Protein folding and modification in the Mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 2.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 3.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 4.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 6.Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- 7.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olzmann JA, Kopito RR. Lipid Droplet Formation Is Dispensable for Endoplasmic Reticulum-associated Degradation. J Biol Chem. 2011;286:27872–27874. doi: 10.1074/jbc.C111.266452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spandl J, Lohmann D, Kuerschner L, Moessinger C, Thiele C. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J Biol Chem. 2011;286:5599–5606. doi: 10.1074/jbc.M110.190785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klemm EJ, Spooner E, Ploegh HL. The dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and ER protein quality control. J Biol Chem. 2011 doi: 10.1074/jbc.M111.284794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel A, Walter P. Membrane lysis during biological membrane fusion: collateral damage by misregulated fusion machines. J Cell Biol. 2008;183:181–186. doi: 10.1083/jcb.200805182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stolz A, Hilt W, Buchberger A, Wolf DH. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011 doi: 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassink GC, Zhao B, Sompallae R, Altun M, Gastaldello S, et al. The ER- resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009;10:755–761. doi: 10.1038/embor.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst R, Claessen JH, Mueller B, Sanyal S, Spooner E, et al. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boutet SC, Disatnik MH, Chan LS, Iori K, Rando TA. Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell. 2007;130:349–362. doi: 10.1016/j.cell.2007.05.044. [DOI] [PubMed] [Google Scholar]
- 18.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 19.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 20.Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 23.Binette J, Dube M, Mercier J, Halawani D, Latterich M, et al. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology. 2007;4:75. doi: 10.1186/1742-4690-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, et al. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 25.Burr ML, Cano F, Svobodova S, Boyle LH, Boname JM, et al. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A. 2011;108:2034–2039. doi: 10.1073/pnas.1016229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimizu Y, Okuda-Shimizu Y, Hendershot LM. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol Cell. 2010;40:917–926. doi: 10.1016/j.molcel.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, et al. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagai T, Levy Y. Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc Natl Acad Sci U S A. 2010;107:2001–2006. doi: 10.1073/pnas.0912335107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, et al. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144:769–781. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39:477–484. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng W, Sergeyenko T, Zeng N, Brown JD, Romisch K. Characterization of the proteasome interaction with the Sec61 channel in the endoplasmic reticulum. J Cell Sci. 2007;120:682–691. doi: 10.1242/jcs.03351. [DOI] [PubMed] [Google Scholar]
- 38.Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, et al. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, et al. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 40.Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 41.Walter J, Urban J, Volkwein C, Sommer T. Sec61p-independent degradation of the tail-anchored ER membrane protein Ubc6p. EMBO J. 2001;20:3124–3131. doi: 10.1093/emboj/20.12.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 43.Liang J, Yin C, Doong H, Fang S, Peterhoff C, et al. Characterization of erasin (UBXD2): a new ER protein that promotes ER-associated protein degradation. J Cell Sci. 2006;119:4011–4024. doi: 10.1242/jcs.03163. [DOI] [PubMed] [Google Scholar]
- 44.Hanzelmann P, Buchberger A, Schindelin H. Hierarchical Binding of Cofactors to the AAA ATPase p97. Structure. 2011;19:833–843. doi: 10.1016/j.str.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 45.Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci U S A. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soetandyo N, Ye Y. The p97 ATPase dislocates MHC class I heavy chain in US2-expressing cells via a Ufd1-Npl4-independent mechanism. J Biol Chem. 2010;285:32352–32359. doi: 10.1074/jbc.M110.131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao G, Zhou X, Wang L, Li G, Schindelin H, et al. Studies on peptide:N- glycanase-p97 interaction suggest that p97 phosphorylation modulates endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A. 2007;104:8785–8790. doi: 10.1073/pnas.0702966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewens CA, Kloppsteck P, Forster A, Zhang X, Freemont PS. Structural and functional implications of phosphorylation and acetylation in the regulation of the AAA+ protein p97. Biochem Cell Biol. 2010;88:41–48. doi: 10.1139/o09-128. [DOI] [PubMed] [Google Scholar]
- 49.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22:451–462. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 50.LaLonde DP, Bretscher A. The UBX protein SAKS1 negatively regulates endoplasmic reticulum-associated degradation and p97-dependent degradation. J Biol Chem. 2011;286:4892–4901. doi: 10.1074/jbc.M110.158030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, et al. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 52.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER- associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Minami R, Hayakawa A, Kagawa H, Yanagi Y, Yokosawa H, et al. BAG-6 is essential for selective elimination of defective proteasomal substrates. J Cell Biol. 2010;190:637–650. doi: 10.1083/jcb.200908092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, et al. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature. 2011 doi: 10.1038/nature10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, et al. A ubiquitin ligase- associated chaperone holdase maintains polypeptides in soluble States for proteasome degradation. Mol Cell. 2011;42:758–770. doi: 10.1016/j.molcel.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 58.Schafer A, Wolf DH. Sec61p is part of the endoplasmic reticulum- associated degradation machinery. EMBO J. 2009;28:2874–2884. doi: 10.1038/emboj.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- 60.Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, et al. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 61.Fiebiger E, Story C, Ploegh HL, Tortorella D. Visualization of the ER-to- cytosol dislocation reaction of a type I membrane protein. EMBO J. 2002;21:1041–1053. doi: 10.1093/emboj/21.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tirosh B, Furman MH, Tortorella D, Ploegh HL. Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J Biol Chem. 2003;278:6664–6672. doi: 10.1074/jbc.M210158200. [DOI] [PubMed] [Google Scholar]
- 63.Bhamidipati A, Denic V, Quan EM, Weissman JS. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell. 2005;19:741–751. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 64.Nery FC, Armata IA, Farley JE, Cho JA, Yaqub U, et al. TorsinA participates in endoplasmic reticulum-associated degradation. Nat Commun. 2011;2:393. doi: 10.1038/ncomms1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kostova Z, Tsai YC, Weissman AM. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin Cell Dev Biol. 2007;18:770–779. doi: 10.1016/j.semcdb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 67.Kikkert M, Doolman R, Dai M, Avner R, Hassink G, et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- 68.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang H, Li Q, Shen Y, Sun A, Zhu X, et al. The ubiquitin ligase Hrd1 promotes degradation of the Z variant alpha 1-antitrypsin and increases its solubility. Mol Cell Biochem. 346:137–145. doi: 10.1007/s11010-010-0600-9. [DOI] [PubMed] [Google Scholar]
- 70.Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458:453–460. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- 71.Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ying Z, Wang H, Fan H, Wang G. The endoplasmic reticulum (ER)-associated degradation system regulates aggregation and degradation of mutant neuroserpin. J Biol Chem. 286:20835–20844. doi: 10.1074/jbc.M110.200808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shmueli A, Tsai YC, Yang M, Braun MA, Weissman AM. Targeting of gp78 for ubiquitin-mediated proteasomal degradation by Hrd1: cross-talk between E3s in the endoplasmic reticulum. Biochem Biophys Res Commun. 2009;390:758–762. doi: 10.1016/j.bbrc.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, et al. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell. 2008;19:1328–1336. doi: 10.1091/mbc.E07-06-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim SM, Acharya P, Engel JC, Correia MA. Liver cytochrome P450 3A ubiquitination in vivo by gp78/autocrine motility factor receptor and C terminus of Hsp70-interacting protein (CHIP) E3 ubiquitin ligases: physiological and pharmacological relevance. J Biol Chem. 285:35866–35877. doi: 10.1074/jbc.M110.167189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281:4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- 77.Zavacki AM, Arrojo EDR, Freitas BC, Chung M, Harney JW, et al. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29:5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lerner M, Corcoran M, Cepeda D, Nielsen ML, Zubarev R, et al. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol Biol Cell. 2007;18:1670–1682. doi: 10.1091/mbc.E06-03-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altier C, Garcia-Caballero A, Simms B, You H, Chen L, et al. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L- type channels. Nat Neurosci. 14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- 80.Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, et al. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol. 2009;186:685–692. doi: 10.1083/jcb.200906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JP, Brauweiler A, Rudolph M, Hooper JE, Drabkin HA, et al. The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol Cancer Res. 8:93–106. doi: 10.1158/1541-7786.MCR-08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maruyama Y, Yamada M, Takahashi K. Ubiquitin ligase Kf-1 is involved in the endoplasmic reticulum-associated degradation pathway. Biochem Biophys Res Commun. 2008;374:737–741. doi: 10.1016/j.bbrc.2008.07.126. [DOI] [PubMed] [Google Scholar]
- 83.Lu JP, Wang Y, Sliter DA, Pearce MM, Wojcikiewicz RJ. RNF170 Protein, an Endoplasmic Reticulum Membrane Ubiquitin Ligase, Mediates Inositol 1,4,5-Trisphosphate Receptor Ubiquitination and Degradation. J Biol Chem. 286:24426–24433. doi: 10.1074/jbc.M111.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neutzner A, Neutzner M, Benischke AS, Ryu SW, Frank S, et al. A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J Biol Chem. 286:8633–8643. doi: 10.1074/jbc.M110.197459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ron I, Rapaport D, Horowitz M. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet. 19:3771–3781. doi: 10.1093/hmg/ddq292. [DOI] [PubMed] [Google Scholar]
- 86.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, et al. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009;5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoshida Y, Chiba T, Tokunaga F, Kawasaki H, Iwai K, et al. E3 ubiquitin ligase that recognizes sugar chains. Nature. 2002;418:438–442. doi: 10.1038/nature00890. [DOI] [PubMed] [Google Scholar]
- 88.Kato A, Rouach N, Nicoll RA, Bredt DS. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc Natl Acad Sci U S A. 2005;102:5600–5605. doi: 10.1073/pnas.0501769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gong B, Chen F, Pan Y, Arrieta-Cruz I, Yoshida Y, et al. SCFFbx2-E3-ligase- mediated degradation of BACE1 attenuates Alzheimer's disease amyloidosis and improves synaptic function. Aging Cell. 9:1018–1031. doi: 10.1111/j.1474-9726.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshida Y, Tokunaga F, Chiba T, Iwai K, Tanaka K, et al. Fbs2 is a new member of the E3 ubiquitin ligase family that recognizes sugar chains. J Biol Chem. 2003;278:43877–43884. doi: 10.1074/jbc.M304157200. [DOI] [PubMed] [Google Scholar]
- 91.Guo X, Shen S, Song S, He S, Cui Y, et al. The E3 ligase Smurf1 regulates Wolfram syndrome protein stability at the endoplasmic reticulum. J Biol Chem. 286:18037–18047. doi: 10.1074/jbc.M111.225615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fry WH, Simion C, Sweeney C, Carraway KL., 3rd Quantity Control of the ErbB3 Receptor Tyrosine Kinase at the Endoplasmic Reticulum. Mol Cell Biol. 31:3009–3018. doi: 10.1128/MCB.05105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kanehara K, Xie W, Ng DT. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J Cell Biol. 188:707–716. doi: 10.1083/jcb.200907055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buck TM, Kolb AR, Boyd CR, Kleyman TR, Brodsky JL. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell. 21:1047–1058. doi: 10.1091/mbc.E09-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hughes BT, Nwosu CC, Espenshade PJ. Degradation of sterol regulatory element-binding protein precursor requires the endoplasmic reticulum- associated degradation components Ubc7 and Hrd1 in fission yeast. J Biol Chem. 2009;284:20512–20521. doi: 10.1074/jbc.M109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER- associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 98.Wang Q, Chang A. Substrate recognition in ER-associated degradation mediated by Eps1, a member of the protein disulfide isomerase family. EMBO J. 2003;22:3792–3802. doi: 10.1093/emboj/cdg378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee JH, Choi JM, Lee C, Yi KJ, Cho Y. Structure of a peptide:N-glycanase- Rad23 complex: insight into the deglycosylation for denatured glycoproteins. Proc Natl Acad Sci U S A. 2005;102:9144–9149. doi: 10.1073/pnas.0502082102. [DOI] [PMC free article] [PubMed] [Google Scholar]