

Abstract
Data from studies in animal models indicate that certain lipid metabolites, particularly diacylglycerol, ceramide, and acylcarnitine, disrupt insulin action. We evaluated the relationship between the presence of these metabolites in the liver (assessed by mass spectrometry) and hepatic insulin sensitivity (assessed using a hyperinsulinemic-euglycemic clamp with stable isotope tracer infusion) in 16 obese adults (body mass index, 48 ± 9 kg/m2). There was a negative correlation between insulin-mediated suppression of hepatic glucose production and intrahepatic diacylglycerol (r = −0.609; P =.012), but not with intrahepatic ceramide or acylcarnitine. These data indicate that intrahepatic diacylglycerol is an important mediator of hepatic insulin resistance in obese people with nonalcoholic fatty liver disease.
Keywords: Obesity, Lipid Intermediates, Lipid-Induced Insulin Resistance, NAFLD
One of the key functions of the liver is the production of glucose to provide fuel for the brain and other tissues that either require or prefer glucose as an energy substrate. However, impaired insulin-mediated suppression of hepatic glucose production increases plasma glucose concentration, and is involved in the pathogenesis of diabetes, the metabolic syndrome, and coronary heart disease.1 Obesity, particularly in conjunction with nonalcoholic fatty liver disease (NAFLD), is associated with hepatic insulin resistance.2-4 In fact, intrahepatic triglyceride (IHTG) content is a better predictor of hepatic insulin resistance than body mass index, percentage of body fat, and visceral fat mass.2-4 These observations suggest that the increase in IHTG itself is responsible for hepatic insulin resistance. However, hepatic steatosis resulting from a genetic defect in very low density lipoprotein–triglyceride secretion is not associated with abnormalities in hepatic insulin sensitivity,5,6 suggesting that increased IHTG content is a marker, but not a direct cause, of altered hepatic insulin action.
The mechanisms responsible for the inter-relationship among hepatic insulin resistance, obesity, and increased IHTG are not clear. Several intracellular lipid metabolites, particularly diacylglycerol (DAG), ceramides, and acylcarnitines, which are products of fatty acid metabolism, are purported to be involved in the pathogenesis of insulin resistance in skeletal muscle.7-9 It also is possible that the severity of liver cell injury and NAFLD activity contribute to hepatic insulin resistance independent of the amount of IHTG and lipid intermediates.10 The purpose of the present study was to evaluate whether the major putative lipid mediators of impaired insulin action (DAG, ceramides, and acylcarnitines)9,11 and histologic markers of liver injury and NAFLD activity are associated with hepatic insulin resistance in obese people.
Sixteen obese subjects (14 women and 2 men; age, 42.0 ± 10.8 y; body mass index, 48 ± 9 kg/m2; Supplementary Table 1), scheduled to undergo bariatric surgery at Barnes-Jewish Hospital in St Louis, MO, provided written informed consent and participated in this study, which was approved by the Washington University Human Research Protection Office. Potential participants who had diabetes, consumed ≥20 g/d of alcohol, or had any history of liver disease other than NAFLD were excluded. Hepatic insulin sensitivity was determined by using the hyperinsulinemic-euglycemic clamp procedure in conjunction with stable isotopically labeled glucose tracer infusion to assess insulin-mediated suppression of hepatic glucose production, as previously described.3 Liver tissue samples were obtained during surgery, by using a Tru-Cut needle (Cardinal Health, Dublin, OH) before gastric stapling, intestinal anastomosis, or band placement, to assess DAG, ceramide, and acylcarnitine contents and liver histology (Supplementary Materials and Methods). We also measured the circulating concentrations of several putative factors that can affect insulin action, including branched-chain amino acids (BCAA; leucine, isoleucine, and valine), tumor necrosis factor α, interleukin 6, and total adiponectin (Supplementary Materials and Methods).
Thirteen of our 16 subjects had NAFLD, whereas 3 had normal liver histology. The median intrahepatic DAG content was 0.75 (first quartile 0.59, third quartile 2.31) nmol/mg wet liver weight (range, 0.36 – 4.41 nmol/mg wet liver weight), mean intrahepatic ceramide content was 0.089 ± 0.021 nmol/mg wet liver weight (range, 0.044–0.119 nmol/mg wet liver weight), and median intrahepatic acylcarnitine content was 461 (338, 565) pmol/mg wet liver weight (range, 182–1139 pmol/mg wet liver weight). Insulin-mediated suppression of glucose rate of appearance (Ra) correlated negatively with intrahepatic DAG content (r = −0.609, P = .012) (Figure 1, top) but did not correlate with intrahepatic ceramides (total and individual species) (Figure 1, middle) or acylcarnitines (total or short-, medium-, and long-chain species) (Figure 1, bottom). Intra-hepatic DAG content correlated positively with the degree of microscopic steatosis (r = 0.789, P < .001) (Figure 2, top) and with IHTG content measured by using magnetic resonance spectroscopy in 10 of the 16 subjects (r = 0.709, P = .022), and the NAFLD activity score (r = 0.865, P < .001) (Figure 2, bottom). Insulin-mediated suppression of glucose rate of appearance (Ra) correlated negatively with the degree of steatosis (r = −0.533, P =.034), but did not correlate with other markers of liver pathology (grades of lobular inflammation, ballooning, and fibrosis stage). In addition, insulin-mediated suppression of glucose Ra did not correlate with total plasma BCAA (r = −0.145, P = .593); individual BCAA leucine, isoleucine, and valine (all P >.4); tumor necrosis factor α (r = −0.156, P = .563); interleukin 6 (r = −0.437, P = .091); or adiponectin (r = 0.366, P = .164).
Figure 1.
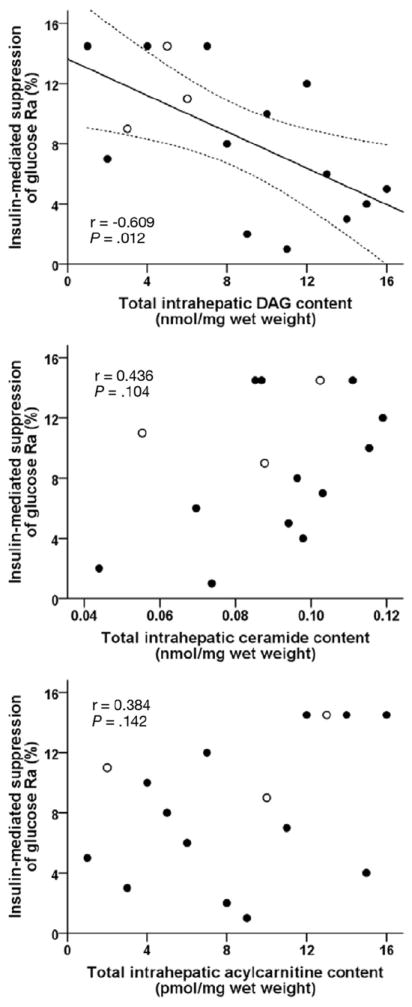
Relationship between hepatic insulin sensitivity, determined as insulin-mediated suppression of glucose Ra into plasma, and intrahepatic DAG (top), ceramide (middle), and acylcarnitine (bottom) contents. Open symbols represent subjects with no histologic evidence of steatosis. Data for DAG, acylcarnitines, and insulin-mediated suppression of glucose Ra are ranked (n = 16). Data for ceramides are from 15 subjects. Relationships were the same when individual species of DAG, ceramides, and acylcarnitines were analyzed separately.
Figure 2.
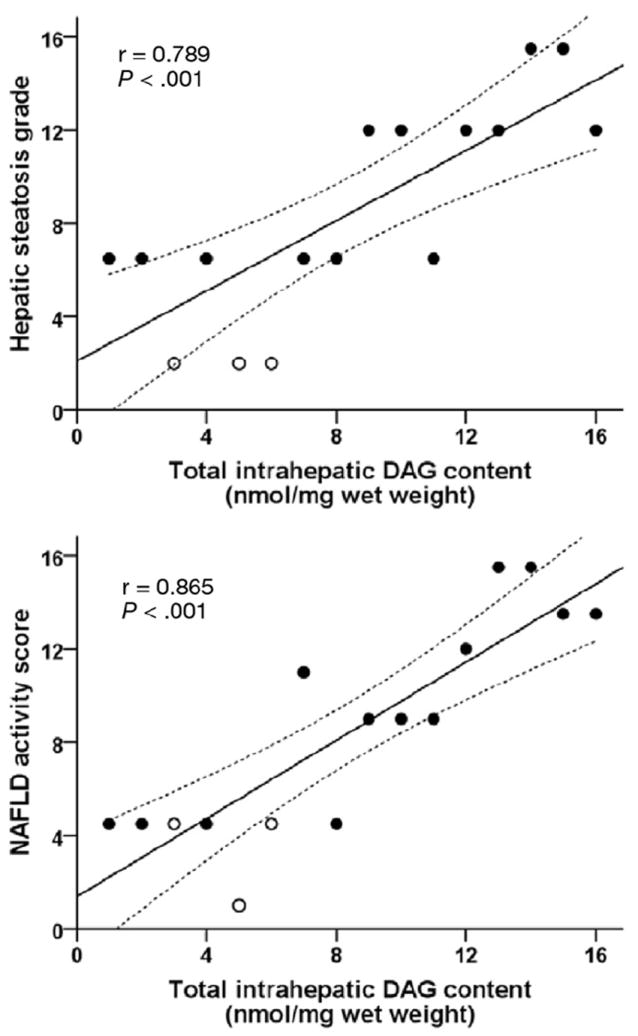
Relationship between intrahepatic DAG content and hepatic steatosis (top) and the NAFLD activity score (bottom). Open symbols represent subjects with no histologic evidence of steatosis. Data are ranked (n = 16).
These results support the notion that intrahepatic DAG is an important intracellular lipid mediator of hepatic insulin resistance in obese people with NAFLD, and likely contributes to the inverse relationship observed between IHTG content and hepatic insulin sensitivity.3,4 Our findings extend the results from a recent study that found intrahepatic DAG content was associated with the homeostasis model assessment of insulin resistance score,12 which provides a surrogate index of global insulin sensitivity.13 Our data suggest that the relationship between intrahepatic DAG and homeostasis model assessment of insulin resistance observed in that study was primarily owing to the effect of DAG on hepatic insulin sensitivity.
The potential mechanism responsible for DAG-induced insulin resistance has been elucidated from studies conducted in mouse models. These data have shown that experimentally increasing intrahepatic DAG content causes hepatic insulin resistance, whereas reducing or preventing an increase in intrahepatic DAG protects the liver from becoming insulin resistant.14-17 The amount of DAG in the liver correlates with the activation of protein kinase C,12,14-17 which inhibits insulin-mediated tyrosine phosphorylation of insulin receptor substrates 1 and 2.18,19 Therefore, intracellular DAG can impair insulin-mediated suppression of glucose production by direct effects on the insulin signaling cascade.
In conclusion, the results from the present study support an important role of intrahepatic DAG, but not intrahepatic ceramide or acylcarnitine, in the pathogenesis of hepatic insulin-resistant glucose metabolism in obese people with NAFLD. Moreover, our data help explain the direct relationship between IHTG content and hepatic insulin resistance observed previously.3,4 Even though intracellular triglycerides themselves likely are inert, the association between IHTG and DAG provides a potential mechanistic link between steatosis and hepatic insulin resistance.
Supplementary Material
Acknowledgments
The authors thank Freida Custodio, Jennifer Shew, and Dr Adewole Okunade for technical assistance; the staff of the Clinical Research Unit for their help in performing the studies; and the study subjects for their participation.
Funding
This study was supported by National Institutes of Health grants DK 37948, DK 56341 (Nutrition Obesity Research Center), RR024992 (Clinical and Translational Science Award), RR-00954 (Biomedical Mass Spectrometry Resource), DK089503 (Michigan Nutrition Obesity Research Center), and by a Scientist Development Grant from the American Heart Association (835140N).
Abbreviations used in this paper
- BCAA
branched-chain amino acids
- DAG
diacylglycerol
- IHTG
intrahepatic triglyceride
- NAFLD
nonalcoholic fatty liver disease
- Ra
rate of appearance
Footnotes
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi:10.1053/j.gastro.2012.03.003.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Kip KE, et al. Circulation. 2004;109:706–713. doi: 10.1161/01.CIR.0000115514.44135.A8. [DOI] [PubMed] [Google Scholar]
- 2.Fabbrini E, et al. Proc Natl Acad Sci U S A. 2009;106:15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korenblat KM, et al. Gastroenterology. 2008;134:1369–1375. doi: 10.1053/j.gastro.2008.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seppala-Lindroos A, et al. J Clin Endocrinol Metab. 2002;87:3023–3028. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 5.Amaro A, et al. Gastroenterology. 2010;139:149–153. doi: 10.1053/j.gastro.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visser ME, et al. Diabetologia. 2011;54:2113–2121. doi: 10.1007/s00125-011-2157-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeFronzo RA. Diabetologia. 2010;53:1270–1287. doi: 10.1007/s00125-010-1684-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shulman GI. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muoio DM. Biochim Biophys Acta. 2010;1801:281–288. doi: 10.1016/j.bbalip.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferreira DM, et al. Diabetologia. 2011;54:1788–1798. doi: 10.1007/s00125-011-2130-8. [DOI] [PubMed] [Google Scholar]
- 11.Nagle CA, et al. J Lipid Res. 2009;50(Suppl):S74–S79. doi: 10.1194/jlr.R800053-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumashiro N, et al. Proc Natl Acad Sci U S A. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace TM, et al. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 14.Neschen S, et al. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Choi CS, et al. J Biol Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D, et al. Cell Metab. 2010;11:402–411. doi: 10.1016/j.cmet.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jornayvaz FR, et al. Proc Natl Acad Sci U S A. 2011;108:5748–5752. doi: 10.1073/pnas.1103451108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erion DM, et al. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samuel VT, et al. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.