

Abstract
Background
Huntington’s disease (HD) is a dominantly inherited neurodegenerative condition characterized by dysfunction in striatal and cortical neurons. There are currently no approved drugs known to slow the progression of HD.
Objective
To facilitate the development of therapies for HD, we identified approved drugs that can ameliorate mutant huntingtin-induced toxicity in experimental models of HD.
Methods
A chemical screen was performed in a mouse HdhQ111/Q111 striatal cell model of HD. This screen identified a set of structurally related approved drugs (pizotifen, cyproheptadine, and loxapine) that rescued cell death in this model. Pizotifen was subsequently evaluated in the R6/2 HD mouse model.
Results
We found that in striatal HdhQ111/Q111 cells, pizotifen treatment caused transient ERK activation and inhibition of ERK activation prevented rescue of cell death in this model. In the R6/2 HD mouse model, treatment with pizotifen activated ERK in the striatum, reduced neurodegeneration and significantly enhanced motor performance.
Conclusions
These results suggest that pizotifen and related approved drugs may provide a basis for developing disease modifying therapeutic interventions for HD.
Keywords: Huntington’s disease, Drug Screening, Pizotifen, ERK Pathway, R6/2 HD mouse model
Introduction
Huntington’s disease (HD) is caused by a dominant mutation in the HD gene leading to expression of mutant huntingtin (Htt) protein containing an expanded polyglutamine tract. Htt is involved in a variety of cellular functions including vesicle transport, transcription, and energy metabolism [1,2]. Deletion of the Htt gene in mouse results in embryonic lethality indicating that it is required for development [3]. Wild-type huntingtin has approximately 20 glutamine repeats near its N-terminus. Mutant huntingtin causes disease when the number of polyglutamine repeats expands and exceeds 36 [4]. The inverse relationship between disease age-of-onset and CAG-repeat length indicates that polyglutamine expansion is a primary toxic feature of the mutant Htt [5]. HD typically develops in mid-life and has a course of 15–20 years. The most prominent debilitating symptoms include chorea, muscle wasting and psychiatric disturbances [6]. While a limited number of palliative treatments can alleviate cognitive and motor symptoms, there are currently no disease modifying therapies for HD [7].
A common downstream signaling pathway disrupted in HD is the extracellular signal-regulated-kinase (ERK) pathway (also known as the mitogen-activated protein kinase or MAPK pathway) [8]. BDNF-mediated ERK1/2 activation through the TrkB receptor is decreased in mutant Htt striatal cells and the reduction is associated with reduced survival. The alteration in signaling appears to be in part due to reduced expression of p52/p46 Sch docking proteins [9]. EGFR-induced ERK activation is disrupted in a Drosophila model of HD [10] and ERK activation is impaired after stimulation with EGF or NGF in PC12 cells expressing mutant huntingtin [11].
HD cell culture models are known to have enhanced susceptibility to cell death induced by serum withdrawal [12, 13, 14, 15, 16, 17]. We screened a library of 1120 approved drugs and pharmacologically active small molecules to identify compounds that could block loss of cell viability. We used a striatal cell line derived from a transgenic homozygous knock-in mouse where exon 1 of the Htt locus was replaced with an exon 1 containing 111 CAG repeats (STHdhQ111/Q111) [18]. This screen identified three structurally related approved drugs that rescued STHdhQ111/Q111 cells from loss of viability upon serum withdrawal. The three drugs, pizotifen, cyproheptadine, and loxapine, share a common tricyclic motif and are known to act as ligands for serotonin, histamine and dopamine receptors [19,20]. Examination of down-stream signaling events in STHdhQ111/Q111 cells treated with pizotifen showed that rescue of viability in these cells is dependent on transient activation of ERK1/2. In the R6/2 mouse model of HD [21], treatment with pizotifen improved motor function, ameliorated neurodegeneration, restored striatal area, and increased ERK2 activation in the striatum. These results suggest that pharmacological approaches directly activating ERK1/2 may represent an effective therapeutic strategy for HD and that pizotifen and related compounds may provide a basis for rapid development of disease-modifying therapies.
Materials and Methods
Chemical Screen
A chemical screen measuring ATP levels in STHdhQ111/Q111 cells after serum deprivation was performed in duplicate. The Prestwick Chemical library (Prestwick Chemical, Illkirch, FR) consisting of 1120 FDA-approved compounds was used to treat STHdhQ111/Q111 cells at 5 μM for 24 h in media with serum (DMEM, fetal bovine serum (FBS), penicillin and streptomycin (P/S), and G418 (Mediatech Inc., Manassas, VA). After 24 h, media was removed and serum-free media (SFM) with 5 μM compound was added for an additional 24 h. ATP levels were measured as per the manufacturer’s protocol using a luminescent ATP assay (ATPlite 1step, Perkin Elmer, Waltham, MA).
ATP Assays
Cells were plated (15,000 cells per well) in black clear-bottom collagen-coated 96-well plates (Becton Dickinson, Franklin Lakes, NJ). After 24 h, the indicated concentration of compound was added in 100 μl media (DMEM, 10% FBS, 1% P/S, 500 μg/ml G418) for 24 h. The media was removed and SFM with compound was added for an additional 24 h. ATP levels were measured using ATPlite 1step assay on a PerkinElmer 2030 Multilabel Plate ReaderVictor X3 (Perkin Elmer, Waltham, MA). For co-treatment with MEK Inhibitor U0126 and pizotifen, 30,000 cells were plated. After 24 h the media was removed and cells were pretreated with MEK inhibitor in SFM for 5 min. Following pretreatment, cells were incubated with pizotifen and MEK inhibitor for 24 h in SFM. Pizotifen malate, loxapine succinate, cyproheptadine hydrochloride were from Prestwick Chemical (Illkirch, FR). In experiments comparing STHdhQ111/Q111 to STHdhQ7/Q7 cells, both cell types were at identical passage numbers and cell lines were maintained in similar conditions. Although there is some difference in doubling time for the two cell types in serum-containing media, the experiments are carried out in SFM in which the cells do not grow.
Caspase Assay
STHdhQ7/Q7 and STHdhQ111/Q111 cells were plated and treated with compound as described above. The caspase activity was measured following the manufacturer’s protocol (Apo3/7 HTS, Cell Technology Inc., Mountain View, CA) with the following modifications. Cell media was discarded and 25 μl of 1X Apo 3 HTS lysis buffer was added to each well with 25 μl of DMEM. After gentle agitation for 8 min, samples (20 μl) were removed from the wells and the protein concentration was measured in duplicate (BCA Protein Assay Kit, Pierce, Rockford, IL). To the remaining 30 μl of sample in each well, 70 μl of caspase 3/7 detection reagent with DTT was added (final concentration was 1X caspase reagent and 15 mM DTT) and fluorescence was measured over time on a Fusion Universal Microplate Analyzer (Perkin Elmer, Waltham, MA).
Western Blot Analysis
STHdhQ7/Q7 and STHdhQ111/Q111 cells were incubated in SFM for 3 h followed by 15 min in compound (pizotifen 10 μM or inhibitor 10 μM). Cells were lysed in M-PER (Thermo Scientific, Rockford, IL) (manufacturer’s protocol) with protease inhibitor (Roche Diagnostics, Mannheim, Germany) and phosphatase inhibitor (Roche). Protein concentrations were measured with the BCA Protein Assay Kit (Pierce, Rockford, IL) and 10 μg of each sample was loaded per well on a NuPage 4–12% Bis-Tris gel (Invitrogen Corporation, Carlsbad, CA). Bands underwent volumetric analysis (ImageQuant, GE Healthcare, Piscataway, NJ). All the bands in each experiment (controls and samples constitute one complete experiment) were ran on the same gel and imaged on the same film to control for differences that would have occurred due to varying the exposure times. For the time course of ERK activation, cells were cultured in SFM for 3 h, then incubated in SFM with compound (10 μM) for the indicated amount of time. ERK1/2 antibody, phospho-ERK antibody (Thr202/Tyr204; Thr185 and Tyr187, Cell Signaling Technology, Danvers, MA) and β-tubulin 2.1 antibody (Sigma Aldrich, St. Louis, MO) were used per manufacturer’s protocol. Cells were pretreated with MEK inhibitor U0126 (Promega, Madison, WI) for 5 min before addition of pizotifen or DMSO.
Animal Housing
The R6/2 line of transgenic mice were obtained from Jackson Laboratories and housed at the Animal Facility of the Buck Institute for Research on Aging. Mice were group-housed in an enriched environment with igloo tunnels and unlimited access to food and water. Cages were also equipped with wood shavings, cotton nestlets and a filter top. Temperature, humidity and a 12 h light/dark cycle were tightly controlled. All experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals to minimize animal suffering and number for the present study. The Buck Institute for Research on Aging is an AAALAC international accredited institution (Unit #001070) and all procedures were approved by the Institutional Animal and Use Committee (A4213-01).
Genotyping
The presence of the mutation of interest carried by the R6/2 mice was confirmed by Polymerase Chain Reaction (PCR). DNA extraction from isolated tail snips of 3-week old mice was performed following the manufacturer’s protocol for the QIAGEN kit (Qiagen, Valencia, CA). DNA (0.5 μl) was amplified in a 25 μl PCR reaction consisting of 10% 10 × AM buffer [0.66 M Tris Base, 0.166 M (NH4)2SO4, 0.02 M MgCl2, 50 mg/ml BSA diluted in TE with a pH adjusted to 8.8], 10% DMSO and a working concentration of 0.2 μM for each primer [primer sequences: forward: 5′-CGC AGG CTA GGG CTG TCA ATC ATG CT-3′; reverse: 5′-TCA TCA GCT TTT CCA GGG TCG CCA T-3′], 0.4 mM of dNTPs and 1.5 Units of Taq Polymerase (Roche). PCR was conducted on an Eppendorf mastercycler with following cycling conditions: denaturation at 94°C 1 min (× 1 cycle), 94°C 30 s, 65°C 30 s, 72°C 1 min (× 35 cycles), final extension 72°C 5 min (× 1 cycle). Product size was verified on a 1.5% agarose gel.
Pizotifen treatment in HD mouse model
Hemizygous R6/2 mice were divided into a saline control and a pizotifen group. Pizotifen malate for mouse studies was obtained from British Pharmacopoeia Commission Laboratory, catalogue #411. Each group consisted of at least an n=12 or higher and treatment began at 6 weeks old. All groups received once daily intraperitoneal (IP) injections at 10 am ± 2 h. Control mice were treated with 0.9% saline. Two doses were tested for the pizotifen group: 7.5 mg/kg body weight and 10 mg/kg body weight. Pizotifen was diluted in 0.9% saline. The mice received pizotifen (0.75 mg/ml-1 mg/ml stock solution) and saline injections daily starting at 6 weeks of age.
Rotarod
Motor coordination was evaluated with the rotarod device (Rotamex 4/8, Columbus Instruments International). An initial 2-day baseline measurement was obtained from all age-matched groups prior to treatment at 5-weeks of age. Subsequently, each group consisting of at least an n=12 was tested on the rotarod performance approximately every 3 weeks for 3 days in a row. Measurements were obtained at 10 weeks of age. All rotarod sessions were divided into a training portion and three test trials. During the training session mice were accustomed to the device for 5 min at a constant speed (4 rpm). For each trial session the rotarod device was set to increase from 4–40 rpm over a 6 min period. The latency to fall from the apparatus was recorded and used to monitor motor coordination. There was at least a 30 min resting time between the training and each of the trial sessions.
Open Field Measurements
Spontaneous locomotor activity was monitored by the open field test. 10-week-old mice were placed in the center of square chambers (dimensions: 26″D × 25″H × 26″W) equipped with infrared photobeam sensors (E-63-12, Coulbourn Instruments, Whitehall, PA, USA) and camera coupled to TruScan99 Software (Version 2.02, Coulbourn Instruments, Whitehall, PA, USA). A set of 22 parameters in the floor and vertical plane were recorded for 10 min during the light cycle. We chose to analyze three parameters (number of movements, moving time, resting time) related to hyperactivity that are associated with HD behavioral phenotypes. Comparisons were performed using the 2-tailed Mann-Whitney non-parametric test.
Tissue Isolation and Western Blotting
Tissue from the cortex and striatum was isolated from mentioned thickness slices obtained using a standard grid device. The microdissected area was immediately stored at −80°C until use. Prior to liquid homogenization whole cortex preparations were resuspended in 1.5 ml and striatal samples in 0.5 ml of lysis buffer. Lysis buffer consisted of T-PER Solution (Thermo Scientific, #78510) supplemented with protease inhibitors (1 tablet/10 mL, Complete Mini, EDTA-free, Roche, #11836153001), phosphatase inhibitor cocktail I (Calbiochem PPI, #524624) 50 mM DTT, 3 mM Mg2Cl and 50 U/10 ml DNAse. Cell lysis was performed by mechanical dissociation with a 2 ml dounce homogenizer (30X pumps, rest 30 s, 30X pumps). Tissue lysates were sonicated with continuous 40 mA pulses for 3 × 30 s with a 30 s interval in between. Sonicated samples were centrifuged at 14,000 rpm and 4°C on a 5417 Eppendorf centrifuge for 20 min. Supernatant was collected for western blotting analysis. 20 μg of protein were prepared in a 1 × LDS buffer and 0.05 M DTT final concentration. Prior to electrophoresis samples were boiled for 10 min at 100°C. Proteins were separated in precast 4–12% Bis Tris gels by SDS-PAGE electrophoresis at 100 V for 1h. Protein transfer on a 0.45 μm nitrocellulose membrane was performed in NuPage transfer buffer containing 10% Methanol for 1 h at 350 mA for proteins 30–65 kDa. Protein transfer for over 80 kDa proteins was performed overnight at constant 20 V in NuPage Transfer buffer not containing Methanol. Membranes were blocked in 0.1% Tween 20 (TBS-T), 5% non-fat milk for 1 h at room temperature except for membranes probed for phospho antibodies, which were blocked in 3% BSA. Incubation with primary antibodies anti-rabbit phospho ERK (1:500, Cell Signaling, #4370), anti-rabbit ERK (1:500, Cell Signaling, #4695), anti-mouse GAPDH (1:1000, Fitzgerald, #10R-G109A), anti-rabbit D2DR (1:200, Santa Cruz, sc-5303), anti-rabbit DARPP-32 (1:250, Abcam, #ab40801), anti-rabbit GluR2/3 (1:500, Millipore), was performed in blocking solution at 4°C overnight. HRP coupled antibodies diluted in blocking solution (1 h incubation at room temperature) were used for secondary detection. Pierce ECL (Thermo Scientific) chemiluminescence was used to develop blots. ImageQuant TL (v2005, Amersham Biosciences) was used for densitometry analysis.
Volumetric and Striatal Area Measurements
Serial coronal paraffin embedded sections (7 μm thick) processed as described previously by a Leica RM2155 microtome and stained with cresyl violet were used for volumetric measurements of the ventricles and comparisons of the striatal areas between the different groups. Photomicrographs were obtained by a Nikon SMZ-U (ZOOM1:10) stereoscope and a Nikon Digital Camera (DXM1200) at the same magnification. A total of four sections at regular intervals spaced by about 10–13 sections were selected for analysis and matched for following rostrocaudal levels: (0.18 0.86, 0.5, 0.14 mm) coordinates AP from bregma (Paxinos & Watson, 1998). The perimeter of the ventricles and the striatum was traced manually and converted to surface area (μm2) by the program ImageJ 1.375 (Wayne Rasband, National Institute of Health, USA; Java 1.5.0–06). As anatomical landmarks for the striatum measurements served the borders of the corpus callosum, lateral ventricles and anterior commissure. Comparisons were performed with raw data or data normalized by hemisphere area. Average surface area of the striatum for each subject was estimated by summating the total striatal area of the four sections analyzed and dividing this value by the number of sections analyzed. For obtaining ventricular volumetric measurements, surface area was converted into volume by summating the surface area of the four sections analyzed multiplying by 36 μm to account for the thickness of the individual sections. The final ventricular volume value was obtained after adding volumes in between sections. To account approximately for volumes in the space between the four sections analyzed, the distance between sections was multiplied by the surface area of each successive section and added to the total sum value to obtain the final volumetric value for each individual ventricle.
Statistical Analysis
One-way ANOVA and Newman-Keuls multiple comparison statistical analysis tests were performed with Graphpad Prism software (La Jolla, CA) for drug screening and cell-based mechanism studies. Statistical significance is *p < 0.05, **p < 0.01 or ***p < 0.005. Data is plotted as mean values±SEM. For comparison of open field, the Mann Whitney test was used. Statistical differences of western blot densitometry, ventricular volume and striatal areas were determined by the one-way-ANOVA test (p < 0.05). The two-way ANOVA was used for statistical comparisons of rotarod. Survival data were analyzed using Kaplan-Meier survival curves (n =15 per treatment group).
Immunohistochemistry and Imaging
Following pentobarbital anesthesia mice were transcardially perfused with 4% paraformaldehyde and brains were paraffin-embedded, sectioned and deparaffinized with xylene. After antigen retrieval, sections were processed for immunohistochemical localization of DARPP-32 (1:250, Abcam, #ab40801) and phospho ERK (1:500, Cell Signaling, #4370) using primary antibodies diluted in 1% BSA overnight. Prior to primary antibody incubation samples were blocked in 10% goat serum in TBS. To remove non-specific binding, samples were washed 3 × in TBS (10min). Antigens were visualized with Alexa Fluor secondary antibodies (1:500). DAPI Prolong Gold was used as mounting medium after final 3 × washes in TBS to remove excess secondary antibodies. Images (dimensions) were obtained with a Nikon Digital Camera DXM1200 attached to a Nikon Eclipse E800 Fluorescence microscope equipped with filter cubes of following excitation and emission characteristics: Red; Excitation 515–560 Dichroic 565 Emission 572–642, Green; Ex 460–500 Dichroic 505 Em 510–560 and Blue; Ex 340–380 Dichroic 400 Em 435485. The computer system used for image capturing was ACT-1 (2.63, Nikon Corporation).
Results
A Chemical Screen in STHdhQ111/Q111 Cells Identifies Compounds that Restore ATP Levels
We have developed an assay for mutant huntingtin-mediated cell death in STHdhQ111/Q111 cells that measures loss of viability upon serum withdrawal [12,14]. After 24 hours of serum withdrawal, STHdhQ111/Q111 cells show a marked decrease in ATP levels while STHdhQ7/Q7 cells [18] are not significantly affected (Fig. 1A). Using the Prestwick Chemical Library consisting of 1120 approved drugs and pharmacologically active small molecules, we performed a screen to identify compounds that could enhance ATP levels in STHdhQ111/Q111 cells during serum withdrawal. We used a luminescence-based ATP assay to measure viability. STHdhQ111/Q111 cells were pretreated with 5 μM compound in media with serum. After 24 hours, the media was removed and serum-free media with 5 μM compound was added for an additional 24 hours. The screen was run in duplicate and each compound was normalized to same-plate DMSO controls. Compounds that increased ATP levels to 110% or greater as compared to controls were selected for retest. Thirty-three compounds met this criterion and of these, six retested as improving ATP levels in STHdhQ111/Q111 cells. We performed further examination of three of those compounds.
Figure 1. Compounds identified that increase ATP levels in STHdhQ111/Q111 cells after serum withdrawal.
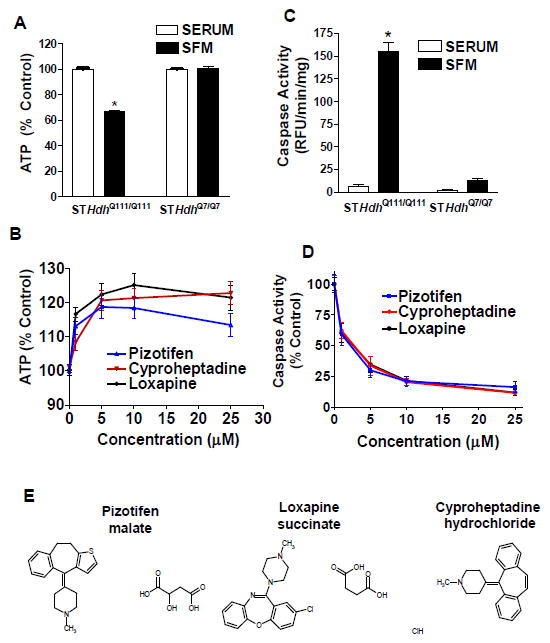
(A) An assay measuring ATP levels was developed to screen for compounds that reduce cell death after serum withdrawal in striatal mouse cells homozygous for mutant Htt (STHdhQ111/Q111). STHdhQ111/Q111 cells exhibit a decrease in ATP levels after 24 h of serum withdrawal. AT levels in STHdhQ7/Q7 cells are not affected by 24 h of serum withdrawal. Results were normalized to same-cell-type condition with serum. * indicates significant difference from all other conditions (n=24 biological replicates, mean±SEM). (B) ATP levels in STHdhQ111/Q111 cells after treatment with a dose-response of re-ordered compound for the top three screen hits (n=9–14 biological replicates, mean±SEM). (C) Comparison of caspase activity in STHdhQ111/Q111 and STHdhQ7/Q7 cells after 24 h serum withdrawal. * indicates significant difference from all other conditions in the figure (n=25 biological replicates, mean±SEM). (D) Dose-response measuring reduction in caspase activity after treatment with indicated compound in STHdhQ111/Q111 cells (n=18 biological replicates, mean±SEM). (E) Chemical structures of the hits.
Pizotifen, Cyproheptadine, and Loxapine Restore ATP Levels and Block Caspase Activation in STHdhQ111/Q111 Cells During Serum Withdrawal
Three top hits were selected for further study. As shown in Figure 1B, these drugs restored ATP levels in STHdhQ111/Q111 cells after serum withdrawal in a dose-dependent manner. The STHdhQ111/Q111 cells have significantly higher caspase-3/7 activity after 24 hours of serum withdrawal compared to the STHdhQ7/Q7 cells or compared to STHdhQ111/Q111 cells in serum (Figure 1C). We observed a slight increase in caspase activity in STHdhQ7/Q7 cells after serum withdrawal, but this was not significant. Caspase activation in the STHdhQ111/Q111 cells was significantly blocked by the addition of the pizotifen, cyproheptadine, or loxapine in a dose-dependent manner (Figure 1D, compound structures 1E).
Addition of pizotifen, cyproheptadine, or loxapine to the STHdhQ7/Q7 cells after serum withdrawal had no effect on ATP levels, indicating that the drugs do not have a general effect on viability but rather correct a deficit specific to the STHdhQ111/Q111 cells (Figure 2A). This activity also appears to be specific to STHdhQ111/Q111 cells challenged with serum withdrawal since the addition of the compound pizotifen to the STHdhQ111/Q111 cells in serum had no effect on ATP levels (Figure 2B). Treating the cells with combinations of pizotifen, cyproheptadine, and loxapine did not further increase ATP levels in a synergistic manner.
Figure 2. Compounds selectively rescue STHdhQ111/Q111 cells while having no effect on STHdhQ7/Q7.
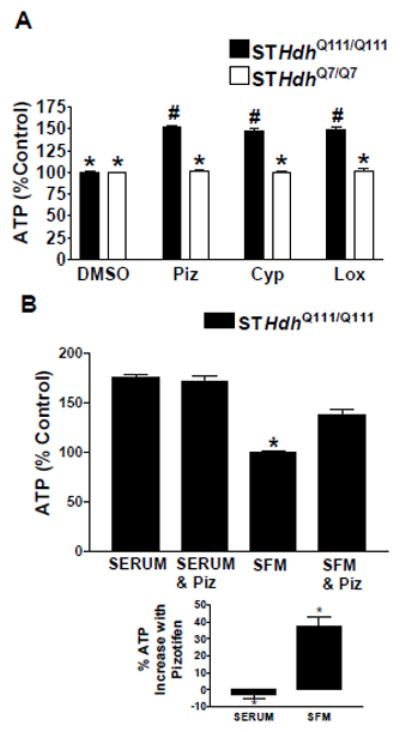
(A) Comparison of increases in ATP levels in STHdhQ111/Q111 and STHdhQ7/Q7 cells after treatment with 10 μM of compound in SFM. Cells were normalized to same-cell-type DMSO controls. * indicates significant difference from # (n=16 biological replicates, mean±SEM). (B) Comparison of treatment of STHdhQ111/Q111 cells with 10 μM pizotifen under conditions with and without serum. * indicates significant difference from all other conditions in the figure (n=11 biological replicates, mean±SEM). The % ATP increase after pizotifen treatment is shown to be significant in the bottom graph.
ERK1/2 is Activated in STHdhQ111/Q111 Cells Treated with Pizotifen, Cyproheptadine and Loxapine
To determine downstream signaling events and mechanisms of action, we measured the effect of these three drugs on ERK phosphorylation. A significant increase in the phosphorylation of ERK1 (p44) occurred after a 15-min treatment with pizotifen, cyproheptadine, or loxapine in both STHdhQ111/Q111 and STHdhQ7/Q7 cells (Figure 3A). A significant increase in phospho-ERK2 (p42) occurred after treatment with pizotifen and loxapine and an upward trend occurred after treatment with cyproheptadine in STHdhQ111/Q111cells. Pizotifen treatment significantly increased phospho-ERK2 in STHdhQ7/Q7 cells (Figure 3B).
Figure 3. Time course of pizotifen-induced activation of ERK.
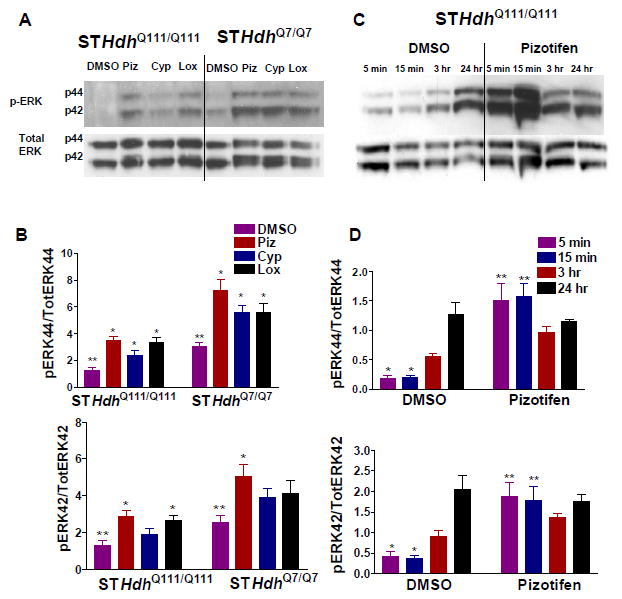
(A) Immunoblot showing increase in ERK phosphorylation in STHdhQ111/Q111 and STHdhQ7/Q7 cells after 15 min treatment with compound. (B) Quantification of the phospho-ERK/ Total ERK ratio for p44 (ERK1) and p42 (ERK2) bands in (A.) * indicates significant difference from ** for same-type cells. (n=6 biological replicates, mean±SEM). (C) Cells were treated in SFM with either pizotifen or DMSO for the time indicated on the figure. Representative western blots show the levels of phospho-ERK1/2 and total ERK1/2 levels. (D) Quantification of the phospho-ERK/ total ERK ratio for p44 (ERK1) and p42 (ERK2) bands in (C.) (n=3 biological replicates, mean±SEM). * indicates significant difference from ** for same-type cells.
We measured the time-course of pizotifen-induced ERK activation under conditions of serum withdrawal in STHdhQ111/Q111 cells and found significant increases in phospho-ERK1/2 after five and fifteen minutes of treatment (Figure 3C, D). This increase was no longer significant after 3 hours of treatment, indicating that a transient burst of ERK activity occurs within minutes of pizotifen treatment and subsides within a few hours. Phospho-ERK1/2 levels in DMSO control cells increased slowly with time and were approximately equal between pizotifen and control cells after 24 hours of treatment. The gradual increase in ERK activation alone does not confer protection to the STHdhQ111/Q111 cells during serum withdrawal. We did not detect any changes in the levels or localization of the Htt protein in these experiments indicating that the mechanism is not mediated by Htt clearance (data not shown).
Transient Phosphorylation of ERK1/2 is Necessary for Complete Rescue of STHdhQ111/Q111 Cells
To determine if transient pizotifen-induced ERK phosphorylation is necessary for rescue of mutant Htt toxicity, we blocked ERK phosphorylation by co-treatment with MEK1/2 inhibitor U0126. We observed significant phosphorylation in ERK1/2 after 15 minutes of pizotifen treatment, and this was completely abrogated by co-treatment with MEK inhibitor (Figure 4A, B). Inhibiting ERK1/2 phosphorylation in the presence of pizotifen prevented the rescue of STHdhQ111/Q111 cells in the ATP assay (Figure 4C).
Figure 4. MEK inhibitor prevents pizotifen rescue of STHdhQ111/Q111 cells.
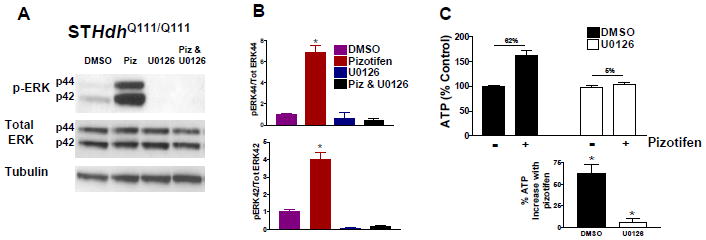
(A) Immunoblot showing increase in ERK phosphorylation in STHdhQ111/Q111 cells after 15 min treatment with pizotifen and MEK inhibitor in SFM. (B) Quantification of the phospho-ERK/Total ERK ratio for p44 (ERK1) and p42 (ERK2) bands in (A). * indicates significant difference from all other conditions in the figure (n=4 biological replicates, mean±SEM). (C) ATP levels of cells co-treated with pizotifen and MEK inhibitor for 24 hours in SFM. The % ATP increase after pizotifen treatment is shown to be significant in the bottom graph. * indicates significant difference from * (n=12 biological replicates, mean±SEM).
Investigation of the Therapeutic Target of Pizotifen
Pizotifen is a serotonin antagonist used to treat recurrent migraine [22]. It binds to over 30 receptors, including subtypes of the serotonin, dopamine, histamine, adrenoreceptor, and cholinergic receptors (for binding profile, see Young et. al. [19]). In addition, pizotifen has been shown to be a partial agonist of the 5-HT1A receptor. Agonism was reportedly blocked by the 5-HT1A antagonist WAY-100,635 [23]. WAY-100,635 also blocks the activation of ERK by several antipsychotic drugs that are agonists of the 5-HT1A receptor, as well as the activation of ERK through the 5-HT1A endogenous ligand serotonin [24]. Another 5-HT1A antagonist, NAD-299, exhibits a pharmacological profile similar to WAY-100,635 [25], with both WAY-100,635 and NAD-299 showing high selectivity for the 5-HT1A receptor (binding it with nanomolar affinities). However, it was noted that these compounds bind to a number of other receptors with micromolar affinities [26]. We hypothesized that these compounds could be useful tools for determining if the activation of ERK by pizotifen was occurring through the 5-HT1A receptor. To ascertain this, we attempted to block rescue of STHdhQ111/Q111 cells with these antagonists. We were unable to prevent rescue of the STHdhQ111/Q111 cells or the rapid activation of ERK by pizotifen when inhibiting the 5-HT1A receptor by pre-treating the cells with WAY-100,635 or NAD-299. We also attempted to either block or replicate the action of pizotifen with antagonists and agonists to a number of additional receptors pizotifen is known to interact with, but this also was unsuccessful (data not shown).
To determine if we had overlooked other active compounds from the Prestwick library, we retested risperidone, metergoline, ketanserin, spiperone, and promethazine. These drugs have overlapping targets with pizotifen and are known to act through serotonin receptors [20, 27, 28, 29, 30, 31, 32, 33]. One drug, promethazine, rescued STHdhQ111/Q111 cells (Figure 5). Promethazine is also a high affinity antagonist of the H1 receptor and has previously been reported to rescue an HD model [34]. Since cyproheptadine and promethazine are both antihistamines, we investigated whether the H1 receptor was involved in rescue by screening a panel of H1 antagonists. Cetirizine, diphenhydramine, ketotifen, desloratadine, and chlorphreniramine failed to rescue STHdhQ111/Q111 cells (Figure 6). This suggests that the activity of the drugs in the cell-based assay is not related solely to their common ability to block the H1 receptor.
Figure 5. Re-testing of compounds in the Prestwick library that are known ligands to serotonin receptors.
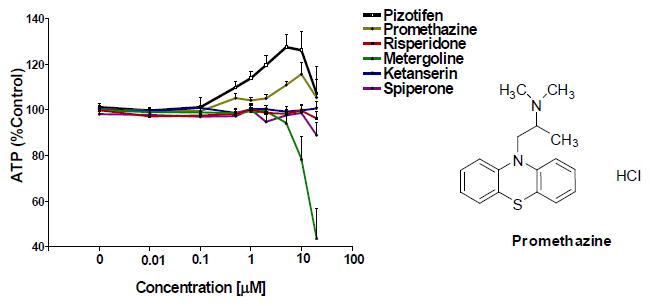
Compounds were re-ordered from Prestwick Chemical (Washington, DC). The structure of promethazine, which showed some ability to rescue, is to the right (n=8 biological replicates, mean±SEM).
Figure 6. Rescue of the STHdhQ111/Q111 cells is not related to antagonism of the H1 receptor.
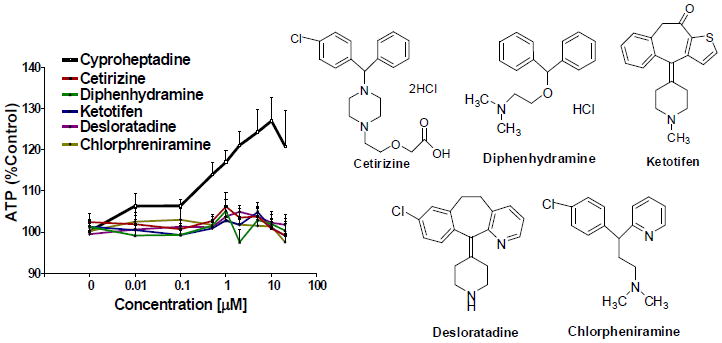
Testing of H1 receptor antagonists in the ATP assay (compounds from Sigma-Aldrich, St. Louis, MO) (n=6 biological replicates, mean±SEM).
Pizotifen Treatment Improves Motor Deficits in HD Transgenic R6/2 Mice
To evaluate potential therapeutic effects of pizotifen treatment in vivo, we measured motor function in HD transgenic R6/2 mice after treatment. Pizotifen (7.5 mg/kg or 10mg/kg) or saline was administered by daily IP injection starting at 6 weeks of age. The rotarod latency of the two randomized groups for treatment was the same at 5 weeks of age (Figure 7). Pizotifen improved rotarod performance at 10 weeks of age at both doses (7.5 mg/kg, or 10mg/kg) (Figure 7). We did not see a significant change in the mean weight (data not shown), lifespan (mean survival), or open field activity of the pizotifen-treated vs. untreated saline-treated HD transgenic R6/2 mice at 7.5 mg/kg (Tables 1, 2). The lack of effect on open-field behavior suggests that the improved rotarod performance is not simply due to acute effects on activity levels but instead to improved motor coordination.
Figure 7. Rotarod performance is improved in pizotifen-treated HD transgenic R6/2 mouse model.
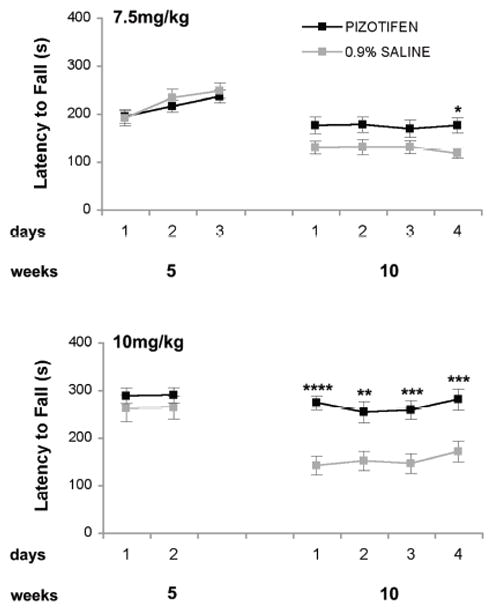
At 5 weeks of age prior to treatment the rotarod of the two groups were measured and are equal. At 10 weeks of age at all the indicated doses rotarod is significantly improved * p<0.05, ** p<0.005, *** p<0.0005, 2-way ANOVA analysis. (n=21 saline and n= 24 for pizotifen-treated R6/2 mice at 7.5 mg/kg; n=15 saline and n=13 pizotifen-treated R6/2 mice at 10mg/kg).
Table 1. Survival analysis of saline-treated and pizotifen-treated R6/2 transgenic mice.
Kaplan-Meier survival analysis revealed that survival of saline-treated (0.9%) and pizotifen-treated mice (7.5mg/kg) did not differ significantly (p=0.8694). A summary of sample size and median value corresponding to each group is provided on the table.
| Kaplan-Meier survival analysis (7.5mg/kg)
| ||
|---|---|---|
| 0.9% Saline | PIZOTIFEN | |
| sample size | 44 | 16 |
| median survival (days) | 87 | 88 |
| significance | p = 0.8694 | |
Table 2. Analysis of spontaneous locomotor activity in saline-treated and pizotifen-treated R6/2 transgenic mice.
Analysis using the non-parametric 2-tailed Mann-Whitney test did not show significant differences in number of movements, moving or resting time.
| 7.5mg/kg | number of movements | moving time (s) | resting time (s) | |||
|---|---|---|---|---|---|---|
|
| ||||||
| 0.9% saline | PIZOTIFEN | 0.9% saline | PIZOTIFEN | 0.9% saline | PIZOTIFEN | |
| mean | 841.73 | 721.04 | 175.95 | 145.48 | 424.04 | 454.51 |
| SD | 243.92 | 346.06 | 67.36 | 78.24 | 67.36 | 78.24 |
| standard error | 55.95 | 70.63 | 15.45 | 15.97 | 15.45 | 15.97 |
| n | 19 | 24 | 19 | 24 | 19 | 24 |
| 2-tailed Mann-Whitney | p = 0.4338 | p= 0.2874 | p= 0.2874 | |||
Pizotifen Corrects Signaling Defects and is Neuroprotective in HD Transgenic R6/2 mice
Mutant Htt causes the down-regulation of the dopamine- and cAMP-regulated 32-kDa phosphoprotein DARPP-32. DARPP-32 is involved in dopamine and serotonin signaling, is highly expressed in the striatum and prefrontal cortex and is a marker of neuronal dysfunction in HD mice [35,36]. By immunohistochemistry (IHC), we found that levels of DARPP-32 in the striatum of the HD transgenic R6/2 mice were reduced at 10 weeks of age qualitatively. Pizotifen treatment increased DARPP-32 staining in the striatum at this time point (Figure 8A). By immunoblot analysis, we observed reduced levels of DARPP-32 in the prefrontal cortex of the HD transgenic R6/2 mice at 10 weeks of age and pizotifen treatment increased DARPP-32 levels (Figure 8B, C). At 12-weeks of age, the pizotifen treatment resulted in a statistically significant increase in striatal DARPP-32 levels when compared to untreated R6/2 controls (Figure 8C).
Figure 8. DARPP-32 expression and striatal area are improved in the pizotifen-treated HD transgenic R6/2 mouse model.

(A) Immunohistochemistry of DARPP-32 in the striatum of NTG, R6/2 (saline) and R6/2 (pizotifen) mice (n=3 mice, representative image shown). (B, left panel) Western blotting of DARPP-32 and mGlu2/3 AMPA receptors in the cortex in wild-type, R6/2 (saline) and R6/2 (pizotifen) mice (n=3 mice mean±SEM). (B, right panel) Densitometry analysis of GluR2/3 protein levels normalized to GAPDH in the cortex of NTG, R6/2 and pizotifen treated mice. (C) Side by side comparison of densitometry analysis of DARPP-32 protein levels in the striatum (12 weeks old) and cortex (10 and 12 weeks old) of age matched NTG, R6/2 and pizotifen treated mice. (D, left panel) Striatal area of the NTG, R6/2 (saline) and R6/2 (pizotifen) mice. (Right panel) Examples of sections analyzed. Demarcation shows striatal area as it was measured. * p<0.05, ** p<0.005, *** p<0.0005, 1-way ANOVA (n=3 mice mean±SEM).
We evaluated several of the well-characterized receptors that are altered in HD including D2DR, GAD65/67, GluR2/3 and 5HT1B. The levels of mGluR2/3 in HD transgenic R6/2 mice at 10 weeks of age were slightly reduced and pizotifen treatment modestly increased levels of this receptor. However, this did not reach statistical significance (Figure 8B). D2DR, GAD65/67 and 5HT1B levels are also altered in the HD transgenic R6/2 mice, but pizotifen treatment did not normalize the levels (data not shown).
The HD transgenic R6/2 mice at 10 weeks of age have a pronounced reduction in striatal area [37]. We confirmed that the striatal area was reduced in the striatum of the HD transgenic R6/2 mice at 10 weeks of age and found that pizotifen treatment attenuated this reduction in striatal area (Figure 8D). Taken together, results demonstrating that pizotifen can partially normalize levels of DARPP-32 while increasing striatal area indicate that the correction of motor deficits in the R6/2 mouse by pizotifen is likely due to neuroprotective effects of the drug. We did not detect any changes in the levels of Htt protein or localization in the R6/2 mouse model suggesting that the mechanism is not mediated through clearance pathways (Figure 9, and data not shown).
Figure 9. Effects of pizotifen treatment on levels of huntingtin and phospho-S421 huntingtin in the cortex at 12-weeks.

Western blot analysis of the levels of huntingtin or phospho p421 huntingtin in the cortex of R6/2 mice (upper panel) for 12-week old NTG, saline and pizotifen treated mice. Densitometry analysis (lower panel) of the huntingtin and phospho p421 huntingtin levels normalized to GAPDH. (n=3 mice, mean±SEM).
ERK2 is Activated in the Striatum of HD Transgenic R6/2 MiceTreated with Pizotifen
Given our evidence that pizotifen promotes activation of ERK in STHdhQ111/Q111 cells in vitro and is required for rescue of toxicity, we evaluated ERK activation in vivo in the HD transgenic R6/2 mice. Treatment with pizotifen via IP injection was initiated at 6 weeks of age in HD transgenic R6/2 mice. At 10 weeks of age, we observed that levels of activated ERK2 (p42) were increased in the striatum of the pizotifen-treated HD transgenic R6/2 mice both by IHC and western blotting (Figure 10A, C). Interestingly, western blotting analysis showed that the levels of phospho-ERK1 (p44) are significantly reduced in the cortex of the HD transgenic R6/2 mice at 10 weeks of age with pizotifen treatment (Figure 10B). The activated ERK by IHC was demonstrated by an increased number of neurons staining positive for phospho-ERK qualitatively (Figure 10C). We did not observe ERK activation in GFAP positive cells (data not shown). These results show that pizotifen can activate striatal ERK1/2 in vitro and in vivo, suggesting that the motor and neuropathological improvements observed in the R6/2 mouse may be mechanistically related to the rescue observed in the cell model. The lower levels of phospho-ERK1 (p44) in the cortex may reflect distinct signaling pathways in these two regions of the brain in response to pizotifen.
Figure 10. ERK activation in HD transgenic R6/2 mouse model striatum.
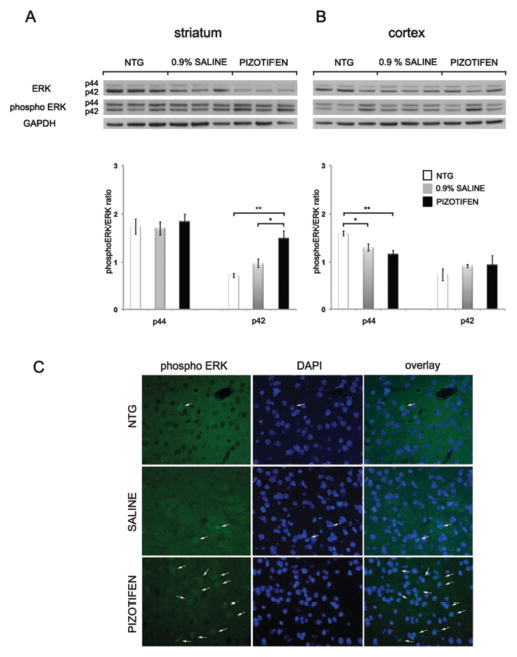
(A) Western blotting with ERK and phospho-ERK antibodies of the striatum and (B) cortex of NTG, R6/2 (saline) and R6/2 (pizotifen) mice (n=3 mice, mean±SEM). (C) Immunohistochemistry of phospho ERK1/2 in the striatum of NTG, R6/2 (saline) and R6/2 (pizotifen) mice (n=3 mice, representative image shown).
Discussion
We performed a screen of approved drugs and pharmacologically active small molecules to identify compounds that prevent mutant huntingtin-mediated cell death in STHdhQ111/Q111 cells. This screen identified three structurally related approved drugs (pizotifen, loxapine, and cyproheptadine) that increased ATP levels and decreased activation of caspase-3 in this cell-based HD model. Enhanced viability in cells treated with pizotifen is associated with transient (< 3 hours) activation of ERK. We further investigated the effects of pizotifen on HD models in the R6/2 transgenic mouse model of HD. When treated with pizotifen, motor function (as measured by rotarod performance) was significantly improved in the R6/2 mouse. This was accompanied by an increase in DARPP-32 protein expression and restoration of striatal area, suggesting that the effects of the drug were due in part to a neuroprotective mechanism in the mice. R6/2 mice show activation of ERK2 in the striatum in response to pizotifen treatment suggesting that the protective effects of pizotifen in mice are due in part to the activation of the ERK signaling pathway. However, the time course of the activation of ERK was not measured in vivo. Therefore, it is not known how the kinetics of ERK activation in our mouse study compares to that observed in our cell culture model. Additionally, it appears that the ERK pathway is activated in the striatum of R6/2 mice, while in the cortex it is inhibited. There are also changes in the total levels of ERK in vivo. Since pizotifen is known to act upon a number of receptors, the distinct signaling in the cortex and striatum during treatment may reflect differences in receptor expression levels or downstream signaling pathways in the two brain regions.
Our observations are in agreement with several recent publications suggesting that pharmacological activation of ERK could be a viable approach to HD therapy [38]. Activation ofERK was associated with rescue of the R6/2 mouse, PC12 cell, and Drosophila HD models after treatment with fisetin [39] and was partially responsible for rescue of N548 120Q HD cells by rotenone [15]. Rescue of death by cannabinoids in PC12 cells expressing mutant huntingtin was also reported to be dependent on ERK activation [40]. In neuronal cells, mutant Htt can activate the group I metabotropic receptor mGluR5, leading to increased activation of ERK and JNK and cell death. It was suggested that the activation of JNK is the primary cause of cell death and that the activation of ERK through mGluR5 is protective, although not protective enough to overcome the negative effects of JNK activation [41,42]. Therefore, even though the activation of mGluR5 is associated with excitotoxicity in HD and antagonists to the mGluR5 receptor have been shown to be therapeutic in the R6/2 model [43,44], the primary mechanism of rescue with these compounds may be unrelated to ERK inhibition.
It is known that the activation of ERK is crucial to maintaining normal cellular responses to growth factors [45]. In some HD models, the coupling of ERK activation to growth factor signaling is compromised [8,9,10,11] suggesting that treatments mimicking down-stream effects of growth factor signaling could be therapeutic. Cyproheptadine has recently been reported to rescue STHdhQ111/Q111 cells after serum withdrawal but the mechanism of action was not investigated [17]. Our results indicate that pizotifen and related compounds have pro-survival activities in HD models and should be investigated further as potential disease modifying therapies for HD. We provide data to support a model where pizotifen activates a known pro-survival pathway to rescue STHdhQ111/Q111 cells from serum withdrawal. We also provide evidence that pizotifen may block disease progression and neuropathology in HD transgenic R6/2 mice through a similar mechanism. However, pizotifen treatment did not rescue weight loss and increase lifespan in R6/2 mice. As the cause of death in the R6/2 mice is poorly understood, the specific mechanisms for how drugs such as HDAC inhibitors or growth factors are able to extend lifespan are uncertain. Perhaps this may be due to peripheral effects or distinct signaling pathways that are not engaged by pizotifen. However, we do show that pizotifen can ameliorate neurodegeneration and improve motor function (and thus presumably neuronal function) in a mouse model of HD. Future work will be to test pizotifen in a full-length HD mouse model and investigate pathways known to activate ERK in cell models to identify the precise target(s) of pizotifen relevant to Huntington’s disease.
Acknowledgments
This work was supported by CHDI, Inc., NIH T32 training grant AG000266 (LME, MRS), NIH U54 grant ULI DE019608 (REH, MRS), and The Buck Institute for Research on Aging. The authors declare no competing financial interests. We thank Mahru An, Carlotta Duncan, Sylvia Chen and Daniel Montora for mouse injections. We thank Cathy Vitelli for IHC staining.
Abbreviations
- HD
Huntington’s disease
- NTG
Nontransgenic
- SFM
Serum Free Media
- IHC
Immunohistochemistry
References
- 1.Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson CD, Davidson BL. Huntington’s disease: progress toward effective disease-modifying treatments and a cure. Hum Mol Genet. 2010;19:R98–R102. doi: 10.1093/hmg/ddq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 4.Warby SC, Montpetit A, Hayden AR, Carroll JB, Butland SL, et al. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet. 2009;84:351–366. doi: 10.1016/j.ajhg.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Djousse L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Genet A. 2003;119A:279–282. doi: 10.1002/ajmg.a.20190. [DOI] [PubMed] [Google Scholar]
- 6.Paulson HL, Albin RL. Huntington’s Disease: Clinical Features and Routes to Therapy. 2011. [PubMed] [Google Scholar]
- 7.Frank S, Jankovic J. Advances in the pharmacological management of Huntington’s disease. Drugs. 2010;70:561–571. doi: 10.2165/11534430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Roze E, Betuing S, Deyts C, Marcon E, Brami-Cherrier K, et al. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 2008;22:1083–1093. doi: 10.1096/fj.07-9814. [DOI] [PubMed] [Google Scholar]
- 9.Gines S, Paoletti P, Alberch J. Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J Biol Chem. 2010;285:21537–21548. doi: 10.1074/jbc.M109.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apostol BL, Illes K, Pallos J, Bodai L, Wu J, et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet. 2006;15:273–285. doi: 10.1093/hmg/ddi443. [DOI] [PubMed] [Google Scholar]
- 11.Song C, Perides G, Liu YF. Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem. 2002;277:6703–6707. doi: 10.1074/jbc.M110338200. [DOI] [PubMed] [Google Scholar]
- 12.Leyva MJ, Degiacomo F, Kaltenbach LS, Holcomb J, Zhang N, et al. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models. Chem Biol. 2010;17:1189–1200. doi: 10.1016/j.chembiol.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller JP, Holcomb J, Al-Ramahi I, de Haro M, Gafni J, et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron. 2010;67:199–212. doi: 10.1016/j.neuron.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varma H, Cheng R, Voisine C, Hart AC, Stockwell BR. Inhibitors of metabolism rescue cell death in Huntington’s disease models. Proc Natl Acad Sci U S A. 2007;104:14525–14530. doi: 10.1073/pnas.0704482104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varma H, Yamamoto A, Sarantos MR, Hughes RE, Stockwell BR. Mutant huntingtin alters cell fate in response to microtubule depolymerization via the GEF-H1-RhoA-ERK pathway. J Biol Chem. 2010;285:37445–37457. doi: 10.1074/jbc.M110.125542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gohil VM, Offner N, Walker JA, Sheth SA, Fossale E, et al. Meclizine is neuroprotective in models of Huntington’s disease. Hum Mol Genet. 2011;20:294–300. doi: 10.1093/hmg/ddq464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 19.Young R, Khorana N, Bondareva T, Glennon RA. Pizotyline effectively attenuates the stimulus effects of N-methyl-3,4-methylenedioxyamphetamine (MDMA) Pharmacol Biochem Behav. 2005;82:404–410. doi: 10.1016/j.pbb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, et al. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28:519–526. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- 21.Li JY, Popovic N, Brundin P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx. 2005;2:447–464. doi: 10.1602/neurorx.2.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stark RJ, Valenti L, Miller GC. Management of migraine in Australian general practice. Med J Aust. 2007;187:142–146. doi: 10.5694/j.1326-5377.2007.tb01170.x. [DOI] [PubMed] [Google Scholar]
- 23.Newman-Tancredi A, Conte C, Chaput C, Verriele L, Audinot-Bouchez V, et al. Agonist activity of antimigraine drugs at recombinant human 5-HT1A receptors: potential implications for prophylactic and acute therapy. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:682–688. doi: 10.1007/pl00005000. [DOI] [PubMed] [Google Scholar]
- 24.Cussac D, Duqueyroix D, Newman-Tancredi A, Millan MJ. Stimulation by antipsychotic agents of mitogen-activated protein kinase (MAPK) coupled to cloned, human (h)serotonin (5-HT)(1A) receptors. Psychopharmacology (Berl) 2002;162:168–177. doi: 10.1007/s00213-002-1043-0. [DOI] [PubMed] [Google Scholar]
- 25.Martin LP, Jackson DM, Wallsten C, Waszczak BL. Electrophysiological comparison of 5-Hydroxytryptamine1A receptor antagonists on dorsal raphe cell firing. J Pharmacol Exp Ther. 1999;288:820–826. [PubMed] [Google Scholar]
- 26.Johansson L, Sohn D, Thorberg SO, Jackson DM, Kelder D, et al. The pharmacological characterization of a novel selective 5-hydroxytryptamine1A receptor antagonist, NAD-299. J Pharmacol Exp Ther. 1997;283:216–225. [PubMed] [Google Scholar]
- 27.Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci. 2000;68:29–39. doi: 10.1016/s0024-3205(00)00911-5. [DOI] [PubMed] [Google Scholar]
- 28.Bonhaus DW, Weinhardt KK, Taylor M, DeSouza A, McNeeley PM, et al. RS-102221: a novel high affinity and selective, 5-HT2C receptor antagonist. Neuropharmacology. 1997;36:621–629. doi: 10.1016/s0028-3908(97)00049-x. [DOI] [PubMed] [Google Scholar]
- 29.Kennett GA. 5-HT1C receptor antagonists have anxiolytic-like actions in the rat social interaction model. Psychopharmacology (Berl) 1992;107:379–384. doi: 10.1007/BF02245165. [DOI] [PubMed] [Google Scholar]
- 30.Miranda F, Hong E, Sanchez H, Velazquez-Martinez DN. Further evidence that the discriminative stimulus properties of indorenate are mediated by 5-HT 1A/1B/2C receptors. Pharmacol Biochem Behav. 2003;74:371–380. doi: 10.1016/s0091-3057(02)01010-9. [DOI] [PubMed] [Google Scholar]
- 31.Muntasir HA, Takahashi J, Rashid M, Ahmed M, Komiyama T, et al. Site-directed mutagenesis of the serotonin 5-Hydroxytryptamine2c receptor: identification of amino acids responsible for sarpogrelate binding. Biol Pharm Bull. 2006;29:1645–1650. doi: 10.1248/bpb.29.1645. [DOI] [PubMed] [Google Scholar]
- 32.Fiorella D, Rabin RA, Winter JC. The role of the 5-HT2A and 5-HT2C receptors in the stimulus effects of hallucinogenic drugs. I: Antagonist correlation analysis. Psychopharmacology (Berl) 1995;121:347–356. doi: 10.1007/BF02246074. [DOI] [PubMed] [Google Scholar]
- 33.Fiorella D, Rabin RA, Winter JC. The role of the 5-HT2A and 5-HT2C receptors in the stimulus effects of m-chlorophenylpiperazine. Psychopharmacology (Berl) 1995;119:222–230. doi: 10.1007/BF02246164. [DOI] [PubMed] [Google Scholar]
- 34.Cleren C, Calingasan NY, Starkov A, Jacquard C, Chen J, et al. Promethazine protects against 3-nitropropionic acid-induced neurotoxicity. Neurochem Int. 2010;56:208–212. doi: 10.1016/j.neuint.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, et al. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci U S A. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Dellen A, Welch J, Dixon RM, Cordery P, York D, et al. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington’s disease mice. Neuroreport. 2000;11:3751–3757. doi: 10.1097/00001756-200011270-00032. [DOI] [PubMed] [Google Scholar]
- 37.Popovic N, Maingay M, Kirik D, Brundin P. Lentiviral gene delivery of GDNF into the striatum of R6/2 Huntington mice fails to attenuate behavioral and neuropathological changes. Exp Neurol. 2005;193:65–74. doi: 10.1016/j.expneurol.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Bodai L, Marsh JL. A novel target for Huntington’s disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington’s disease and its upregulation ameliorates pathology. Bioessays. 2012;34:142–148. doi: 10.1002/bies.201100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maher P, Dargusch R, Bodai L, Gerard PE, Purcell JM, et al. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Genet. 2011;20:261–270. doi: 10.1093/hmg/ddq460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scotter EL, Goodfellow CE, Graham ES, Dragunow M, Glass M. Neuroprotective potential of CB1 receptor agonists in an in vitro model of Huntington’s disease. Br J Pharmacol. 2010;160:747–761. doi: 10.1111/j.1476-5381.2010.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang SS, He J, Zhao DM, Xu XY, Tan HP, et al. Effects of mutant huntingtin on mGluR5-mediated dual signaling pathways: implications for therapeutic interventions. Cell Mol Neurobiol. 2010;30:1107–1115. doi: 10.1007/s10571-010-9543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ribeiro FM, Paquet M, Ferreira LT, Cregan T, Swan P, et al. Metabotropic glutamate receptor-mediated cell signaling pathways are altered in a mouse model of Huntington’s disease. J Neurosci. 2010;30:316–324. doi: 10.1523/JNEUROSCI.4974-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang TS, Tu H, Chan EY, Maximov A, Wang Z, et al. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiefer J, Sprunken A, Puls C, Luesse HG, Milkereit A, et al. The metabotropic glutamate receptor 5 antagonist MPEP and the mGluR2 agonist LY379268 modify disease progression in a transgenic mouse model of Huntington’s disease. Brain Res. 2004;1019:246–254. doi: 10.1016/j.brainres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 45.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]